

Abstract
The mammalian homolog of the basic helix-loop-helix transcription factor atonal-1 (Atoh1 or Math1) is required for development of cochlear hair cells that function as the mechanosensory cells required for audition. Forced expression of Atoh1 in cochlear-supporting cells may provide a way to regenerate hair cells and provide for a therapy for hearing loss. Additionally, Atoh1 is an inhibitor of proliferation and has further clinical applications in anticancer therapies. The goal of these experiments was to improve the method for Atoh1 expression by engineering a genetic construct that may be used in future translational applications. To address the poor control of Atoh1 expression in standard gene expression systems where Atoh1 is expressed constitutively at abnormally elevated levels, our aim was to engineer an inducible system whereby Atoh1 was upregulated by an inducer and downregulated once the inducer was removed. A further aim was to engineer a single genetic construct that allowed for conditional expression of Atoh1 independent of secondary regulatory elements. Here we describe a stand-alone genetic construct that utilizes the tamoxifen sensitivity of a mutated estrogen receptor (ER) ligand-binding domain for the conditional expression of Atoh1. The Atoh1-ER-DsRed construct is translated into an ATOH1-ER-DSRED fusion protein that remains sequestered in the cytoplasm and therefore rendered inactive because it cannot enter the nucleus to activate Atoh1 signaling pathways. However, application of 4-hydroxytamoxifen results in translocation of the fusion protein to the nucleus, where it binds to the Atoh1 enhancer, upregulates transcription and translation of endogenous ATOH1 and activates downstream Atoh1 signaling such as upregulation of the hair cell protein MYOSIN 7A. Removal of tamoxifen reverses the upregulation of endogenous Atoh1 signaling. This construct serves as an independent genetic construct that allows for the conditional upregulation and downregulation of Atoh1, and may prove useful for manipulating Atoh1 expression in vivo.
Introduction
One of the definitive genes for hair cell development is the mammalian homolog of the basic helix-loop-helix transcription factor atonal-1 (Atoh1 or Math1). Expression of Atoh1 in cochlear cells is both required and sufficient for hair cell genesis (Bermingham et al., 1999). Cells within the developing organ of Corti that express Atoh1 differentiate into hair cells (Helms et al., 2000), and Atoh1 is one of the earliest markers of hair cell differentiation. Additionally, Atoh1 knockout mice fail to develop hair cells (Isaka et al., 1999; Helms et al., 2000). Several studies have demonstrated that forced expression of the Atoh1 gene in cochlear-supporting cells is sufficient for hair cell genesis (Zheng and Gao, 2000; Kawamoto et al., 2003; Shou et al., 2003; Woods et al., 2004; Izumikawa et al., 2005; Jones et al., 2006; Gubbels et al., 2008; Kelly et al., 2012; Liu et al., 2012).
These initial experiments suggest that Atoh1 may be used therapeutically to treat hearing loss by regenerating lost hair cells. However, improvements to current expression systems are required to allow greater control over transgene expression. One limitation of commonly utilized gene therapies involves the regulation of the gene once it is infected into the host cell. Typically, the expression of transgenes is controlled by strong promoters such as the cytomegalovirus (cmv) or Simian vacuolating virus 40 (Sv40) promoters, resulting in constitutive transgene expression in artificial and uncontrolled levels. The expression levels exhibited by these promoters may produce results that do not represent endogenous gene activity. Additionally, constitutive gene expression systems may add a layer of uncertainty when interpreting the function of developmental genes such as Atoh1 that are upregulated and downregulated at certain times points during development (∼E14–E20 in murine cochleas) because the method does not allow for the downregulation of the desired gene.
Several genetic constructs exist that allow for induced or conditional expression of transgenes whereby gene expression is upregulated when a chemical inducer such as doxycycline (reviewed in Bertram and Hillen, 2008) or mifepristone (reviewed in Ngan et al., 2002) is added to the cellular environment, and gene expression is reduced upon removal of the inducing agent. Another commonly utilized inducible system is the Cre/ER-Lox system (Danielian et al., 1998), where gene expression is controlled by a fusion protein consisting of the Cre domain ligated to a tamoxifen-sensitive estrogen receptor (ER) ligand-binding domain (LBD) that selectively binds tamoxifen rather than endogenous estrogen (17β-estradiol) (Danielian et al., 1993; Littlewood et al., 1995). Although invaluable tools in basic research applications, none of these systems easily translates into clinical applications because they require multiple genetic constructs and complex crossbreading of transgenic animals to operate.
Here, we have engineered an independent construct that allows for conditional expression of Atoh1. In this system, Atoh1 expression is regulated by an ATOH1-ER fusion protein, which consists of the ATOH1 protein ligated to a tamoxifen-sensitive ER LBD (Danielian et al., 1993; Littlewood et al., 1995). The data show that in the absence of tamoxifen, the ATOH1-ER fusion protein is sequestered in the cytosol, where it remains quiescent. However, the addition of tamoxifen results in the localization of the fusion protein to the nucleus, activation of the 3′ Atoh1 promoter, transcription of Atoh1 mRNA, translation of ATOH1 protein, and expression of the downstream target MYOSIN 7A in cochlear cells. Removal of tamoxifen reverses the upregulation of endogenous Atoh1 signaling. This describes an independent genetic construct that enables conditional expression of Atoh1.
Materials and Methods
Generation of the Atoh1-ER-DsRed and control constructs
Three constructs were designed for these experiments (Fig. 1). First, a constitutively expressing positive control construct consisting of the Atoh1 sequence that was modified by polymerase chair reactions (PCR) cloning to include two consecutive flag-tag sequences (GATTACAAGGATGACGATGACAAG) preceding the start codon was engineered (cmv.Atoh1). Each of these sequences translates to a protein weight of approximately 1 kDa. A similar Atoh1 construct was engineered that contained an IRES-DsRed sequence (Atoh1-IRES-DsRed). The test construct was the inducible Atoh1-ER-DsRed transgene. For this construct, PCR cloning primers were designed so that (1) an EcoRI site was placed on the 3′ end and a Kozac sequence (CACC) was engineered upstream of the Atoh1 start codon; (2) the Atoh1 stop codon (TAG) was deleted; (3) this same flag-tagged Atoh1 sequence from above was linked to ER LBD by the sequence CTCGAGCCATCTGCTGGAGACATG; (4) the ER LBD stop codon (TAG) was deleted; (5) the mutated ER sequence was linked to DsRed by the sequence TCAGGATCTGGTTCAGGA; and (6) a NotI site was cloned on the 3′ end. The linker sequences were designed to translate into multiple proline sequences to provide an increased degree of freedom for the subunits of the fusion protein. The insert for the ER construct was amplified by a two-step PCR from template DNA (provided by A. McMahon, Harvard Medical School, Boston, MA) that has been mutated to limit endogenous 17β-estradiol binding at physiological concentrations (Danielian et al., 1993, 1998), and a DsRed plasmid obtained from a commercial vector (Clontech). Finally, a negative control (DsRed-ER) construct was engineered by PCR cloning primers designed so that (1) EcoRI site was placed on the 3′ end and a Kozac sequence (CACC) was placed upstream of the DsRed start codon; (2) the stop codon (TAG) for DsRed was deleted; (3) DsRed was linked to ER by the sequence TCAGGATCTGGTTCAGGATCCATG; and (4) the Not1 site was cloned onto the 3′ end.
FIG. 1.

Schematic of the genetic constructs used in these experiments. Consecutive flag-tag sequences were cloned upstream of the murine Atoh1 gene, and this construct was placed under control of a cytomegalovirus promoter (cmv.Atoh1) to serve as a positive control. The test construct consisted of this same flag-tagged Atoh1 construct ligated to a mutated ER LBD, which had been mutated to selectively bind 4-hydoxytamoxifen rather than endogenous estrogen, and a DsRed sequence to be used as a florescent marker to detect expression of the fusion protein. The three domains were separated by two linker sequences designed to code for triplet proline sequences in order to provide spatial separation and to reduce steric hindrance between moieties of the fusion protein. A negative control construct consisted of the DsRed sequence ligated to the ER LBD separated by one of the linker sequences described above. ATOH1, helix-loop-helix transcription factor atonal-1; CMV, cytomegalovirus; ER LBD, estrogen receptor ligand binding domain.
All of these constructs were subcloned into the multiple cloning site of the pcDNA3.1 (+) vector that employs a cmv promoter to drive gene expression. To accomplish this, inserts for Atoh1-ER-DsRed and DsRed-ER constructs were amplified by a two-step PCR from template DNA. AccuPrime Pfx SuperMix (Invitrogen) was used for the PCR amplification. The PCR products were gel-purified, digested with EcoRI and NotI, and purified with PureLink PCR Purification Kit (Invitrogen) as inserts. Next, 10 μg of pcDNA3.1 (+) was digested with EcoRI and NotI for 2 hr at 37°C. About 1 μl of calf intestinal alkaline phosphatase (Invitrogen) was added to the digestion solution and incubated at 37°C for 10 min. The digest was phenol-extracted, ethanol-precipitated, washed with 80% ethanol, and resuspended in sterile water. Ligations were performed by T4 DNA Ligase (Invitrogen), using fusion fragments as insert and pcDNA3.1 (+) as vector at a ratio of 3:1 (insert: vector). The ligations were transformed into TOP10 cells and equal volumes were plated on LB/Amp (100 μg/ml) plates. Sixteen colonies for each desired construct were picked for colony PCR with vector primers T7 and BGH reverse. Positive colonies were mini prepped with PureLink HQ Mini Plasmid DNA Purification Kit (Invitrogen), and verified by restriction digest with EcoRI and NotI. Positive clones were sequenced with vector primers T7 and BGH reverse and gene specific Atoh1-ER-DsRed (TTGTGTGCCTCAAATCCATC, CCTTACAAACCTACTACATACC) or DsRed-ER (CCCGTAATGCAGAAGAAGAC, GGTCAGTGCCTTGTTGGATG) sequencing primers to verify the cloning junctions and orientation. Glycerol stocks were then prepared from positive clones and stored at −80°C for further use.
Generation and electroporation of cochlear spheres
Cochlear-derived progenitor cells were generated and propagated floating aggregates (cochlear spheres) as previously described (Oshima et al., 2007) with the following modifications. Cochleas were isolated from litters of P0–P3 ROSA26-GFP mice, and the organs of Corti were dissected, pooled, trypsinized, triturated, and centrifuged. The pellet was resuspended in serum free media, filtered through a 70 μM cell strainer, and cultured for 5 days in this same medium supplemented with growth factors (10 ng/ml of fibroblast growth factor, insulin-like growth factor-1, epidermal growth factor and heparin sulfate). Floating aggregates were collected, centrifuged, and triturated by a 100 μl pipette, resuspended in 300 μl Optimem, and electroporated (8 pulses; 25 V; duration, 50 ms; interval, 100 ms with 2 mg/ml DNA in water, and incubated in 3:1 Fugene 6 overnight) using 50 μg of plasmid DNA. Spheres derived from the experimental (Atoh1-ER-DsRed) and control (cmv.Atoh1 and DsRed-ER) groups were expanded by culturing on 6-well plates for an additional 5 days in the same medium at 37°C, and then incubated in graded doses of 1 nM 4-hydoxy tamoxifen sulfate (tamoxifen) for 72 hr (N=10 for each dosage). Finally, spheres from each of these groups were centrifuged, adhered to glass coverslips by incubation for 2–4 hr at 37°C on glass coverslips coated with 1:1 poly-lysine/polyornithine, fixed in 4% paraformaldehyde for 20 min, washed three times in phosphate-buffered saline (PBS), and stored at 4°C for later analysis.
Generation, transfection, and analysis of House Ear Institute OC1 Spheres
House Ear Institute (HEI) cells obtained from cochlear-supporting cell explants obtained from the adult immortomouse (Kalinec et al., 2003) were cultured on a 3 mm dish, and transfected with the Atoh1-ER-DsRed construct (3:1 vector/Fugene 6) for 72 hr in proliferating conditions (39°C in DMEM+10% FBS). The cells were then trypsinized, washed in PBS, transferred into 6-well low-adherence culture dishes, and cultured at these same conditions for an additional 72 hr, which enabled the cells to proliferate as floating aggregates. Next, the spheres were cultured for 72 hr in differentiating conditions (33°C) in this same medium supplemented with either 1 nM tamoxifen (final concentration) or an equivalent volume of vehicle (100% ethanol). Individual spheres were collected from the cultures by a 1,000 μl pipette, incubated on glass coverslips coated with 1:1 polyornithine/polylysine and 10% FBS for 20 min or until they attached, fixed in 4% paraformaldehyde, washed in PBS, and processed for immunolabeling to the MYOSIN 7A protein. This was accomplished by incubating the cultures in a blocking solution of 0.1% triton-X 100 supplemented with 3% normal donkey serum for 1 hr, a 3×5 min PBS wash, incubation in a 1:200 dilution of anti-MYOSIN 7A antibodies (Proteus) for 1 hr, a 3×5 min PBS wash, incubation in 1:2,000 AlexaFluor 488 secondary antibody, a 3×5 min PBS wash, incubation in 1:200 dilution of TOPRO-3 (Invitrogen) nuclear label for 30 min, a 3×5 min PBS wash, and then mounting the coverslips. Spheres were imaged by a Zeiss LSM-510 Meta confocal microscope system and analyzed by Zeiss LSM510 operating software (Carl Zeiss) with filters to detect myosin 7a immunolabeling (FITC 499 nM), DsRed (Cy3 550 nM), and TOPRO-3 (Cy5 633 nM) signaling.
Nuclear translocalization assay
HEK cells were cultured until 50% confluent in 6-well culture dishes and then subjected to transfection using 3:1 target DNA to Fugene 6 Transfection Reagent (Roche). Cells were incubated for 24 hr, and then incubated with graded doses of tamoxifen for 5–7 days. Cells were then processed for cytosolic and nuclear fractionation (BioVision). The isolated nuclear fraction collected from each of 5 sample wells per condition was mounted to a coverslip, and average pixel density from 5 regions of interest (206.5×165.2 pixels at 20× magnification) was measured with a Cy3 (550 nM) filter on a Zeiss eipifluorescent microscope using MetaMorph software.
Luciferase assay
The Atoh1 3′ promoter region containing the beta-globin/LacZ fusion promoter (Helms et al., 2000) was subcloned from the J2XnGFP (from Jane Johnson, University of Texas Southwestern Medical School, Dallas, TX), cloned into the MCS of the pGL3-Promoter Luciferase Reporter Vector (Promega), and was stably expressed on a HEK cell line by selection to ampicillin. These cells were grown until 80% confluent on 6-well plates, and then transiently transfected with the cmv.Atoh1 control vector, the Atoh1-ER-DsRed construct, or a DsRed-ER negative control construct using 3:1 target DNA to Fugene 6 Transfection Reagent. All cells were also cotransfected with Renilla transfection controls. Cells were incubated for 72 hr in graded doses of tamoxifen, and then washed in PBS, lysed, and subjected to a Dual-Luciferase Reporter Assay (Promega). Firefly luciferase activity was measured in a manual TD-20/20 Luminometer (Turner Designs), normalized to Renilla controls, and multiplied by 1,000 to calculate arbitrary luminescence values.
RNA analysis
HEK cells were grown on 6-well plates until 80% confluent, and then were transiently transfected with either the Atoh1-ER-DsRed or cmv.Atoh1 construct as described above and incubated for 72 hr with different doses of tamoxifen. Next, total RNA was extracted by adding 1 ml Trizol reagent (Invitrogen) to each well for 5 min cells were scraped into a 1.5 ml tube (1 tube/well), incubated with 200 μl chloroform (in hood) for 2 min, and centrifuged for 20 min at 12,000×g at 4°C. Then the supernatant was collected in a new 1.5 ml tube, incubated with 1:1 equivalent volume of 2-propanol equal volume to supernatant, and centrifuged through the RNeasy mini kit columns at 8,000×g for 15 sec. RNA was eluted from the column by adding 700 μl RW1, centrifuging the column at 8,000×g 15 sec, adding two 500 μl washes of RPE2 and re-centrifuging at 8,000×g 15 sec, adding one spin to dry membrane (10,000×g, 1 min), and eluting the RNA by adding 45 μl RNAse-free water into new 1.5 ml tube and centrifuging a final time at 8,000×g for 15 sec.
For the reverse transcriptase PCR (RT-PCR) reaction, 45 μl of template RNA was added to a PCR tube and mixed with 20 μl 5× first-strand buffer, 11 μl 50 mM MgCl2, 5 μl dNTP (10 mM), 5 μl random primers (Invitrogen), 1.1 μl each of forward (aga tct aca tca acg ctc tgt c) and reverse primers (act ggc ctc atc aga gtc act g) designed to amplify 449 bp segment of the Atoh1 cDNA, and 13 μl dH2O for a total reaction volume of 100 μl. The hexamers were incubated at 25°C for 10 min, the RT reaction consisted of 37°C for 60 min, and RT incubation was 95°C for 5 min, held at 4°C, and stored on ice until run on 1% agarose gels for analysis.
For quantitative PCR (qPCR) analysis, HEK cells were cultured on 48-well plates, transfected with the Atoh1-ER-DsRed construct, and incubated in graded doses of tamoxifen (4 wells each for each concentration) for 72 hr. Positive control wells were transfected with cmv.Atoh1 in the absence of tamoxifen. After 72 hr, total RNA was extracted and stored as described above. Meanwhile, a PCR tube containing 300 μl of qPCR Master Mix (Invitrogen) and 300 μl dH2O was vortexed, and then the solution was divided into 5 tubes (120 μl each). About 6 μl of template cDNA was added to each tube, which were then divided into two wells in which 3 μl of Atoh1 or 18s control probes was added in a 96-well plate (TempPlateIII PCR plate; USA Scientific) (18s standard in column 1, Atoh1 in column 4), mixed by pipeting, and split by adding 20 μl from column 1 to columns 2 and 3 and then adding 20 μl from column 4 to columns 5 and 6. The 96-well plate was covered with optically clear film, and bubbles on the bottom of the wells were shaken away. The qPCR was run on an ABI 7700 Real-Time PCR machine (Azco Biotech, Inc.). ΔCt values were calculated by subtracting Atoh1 cycle number from 18s cycle number (threshold=0.1 logFluorescence) for each condition, and normalized to values obtained from transfected cells incubated without tamoxifen to yield ΔΔCt values, which were then used to calculate fold change. The mean fold change (±SEM) for five repeated samples conducted in three independent experiments was then normalized to zero tamoxifen levels, and then analyzed for statistical significance described below.
Western blot analysis
HEK cells were grown to 80% confluence in 10-well Corning culture plates, transiently transfected with the Atoh1-ER-DsRed construct using 3:1 target DNA to Fugene 6 Transfection Reagent, and incubated with graded doses of tamoxifen for 72 hr at 37°C. Control samples were similarly transfected with either DsRed-ER (negative control) or a positive control vector (cmv.Atoh1). For tamoxifen withdrawal experiments, HEK cells were similarly transfected with either the Atoh1-ER-DsRed or cmv.Atoh1 construct and cultured in 1 μM tamoxifen for 48 hr; the medium was removed, washed, and replaced with a medium lacking tamoxifen; and the cells were further cultured for 24, 72, or 96 hr. Control groups (0 tamoxifen and cmv.Atoh1-transfected cells) were incubated for 72 hr in vehicle, rather than tamoxifen, and further treated and maintained as the 24 hr group above.
Cytosolic and nuclear fractions were collected by harvesting trypsinized-EDTA-adherent cells, centrifugation at 500×g for 5 min, washing cells by resuspension in PBS, recentrifugation at 500×g for 3 min, removing the supernatant, and adding 400 μl of ice-cold Cytoplasmic Extraction Reagent I (NE-PER Nuclear and Cytoplasmic Extraction Reagents, Thermo Scientific) to the dry pellet. The pellet was then vortexed for 15 sec and incubated on ice for 10 min, and 22 μl of ice-cold Cytoplasmic Extraction Reagent II was added and incubated on ice for 1 min. The suspension was vortexed again for 5 sec and centrifuged for 5 min at 16,000×g, and the supernatant was collected as the cytoplasmic fraction. The pellet containing the nuclei was resuspended in 200 μl of ice-cold Nuclear Extraction Reagent, vortexed for 15 sec every 10 min for 40 min in ice, and centrifuged for 10 min at 16,000×g, and the supernatant was collected as the nuclear fraction. Total protein from each fraction was measured (BioRad) and diluted to a final concentration of 50 μg/μl.
Next, 1:10 NuPage Sample Reducing Agent and 1:4 NuPage LDS sample buffer (both from Invitrogen) were added to the recovered supernatant, which was then boiled at 100°C for 5 min. Thirty-microgram samples were then loaded on NuPage 10% bis-tris gel (Invitrogen) (120 V for 45 min), and then transferred (0.5 A for 1 hr on ice using a Mini Trans-Blot Electrophoretic Transfer Cell; BioRad) to a polyvinylidene difluoride membrane that was blocked in TBS-T (50 mM Tris, 150 mM NaCl, 0.05% Tween 20, HCl to pH 7.6) supplemented with 10% Carnation dry milk on a shaker for 1 hr at room temperature, and then incubated in ATOH1 antibody (1:50, mouse, Developmental Studies Hybridoma Bank), in TBS-T overnight on a shaker at 4°C. The next day, membranes were washed 3 times for 10 min in TBS-T, incubated in horseradish peroxidase-conjugated goat antimouse secondary antibody (Jackson Immunoresearch) diluted 1:10,000 in TBS-T supplemented with 1% milk for 1 hr at room temperature, washed 3 times for 10 min, incubated in 0.1 ml/cm2 Amersham ECL Western Block Detection Kit (GE Healthcare) for 5 min at room temperature, and then analyzed by a ChemiDoc XRS illumination system equipped with a charge-coupled device camera (BioRad). Protein size was estimated by simultaneous electrophoresis of a 10–250 kDa Kaleidoscope Standard (BioRad). To control for loading differences, membranes were stripped by an agitated 15 min incubation in TBS-T supplemented with 1% sodium dodecyl sulfate, and then reprobed as described by murine monoclonal anti-β-actin antibody (Sigma) diluted 1:2,000 or mouse monoclonal anti-PCNA (Santa Cruz) diluted 1:1,000 in TBS-T.
Statistical analysis
Unless otherwise noted, all experiments were repeated three times. Their mean values and standard error of the mean were calculated and analyzed for significance by an unpaired two-tailed Student's t-test with an alpha of 0.05.
Results
Tamoxifen-dependent nuclear translocalization of the ATOH1-ER-DSRED fusion protein
HEK cells transfected with the Atoh1-ER-DsRed construct constitutively expressed an ATOH1-ER-DSRED fusion protein that was detected by the fluorescence of the DsRed moiety. As can be seen by fluorescence microscopy, in the absence of tamoxifen, the ATOH1-ER-DSRED fusion protein remained sequestered within the cytosol of transfected cells (Fig. 2). Incubation of transfected cells in increasing doses of tamoxifen resulted in a translocation of the DsRed signal from the cytoplasm to the nucleus as measured by co-localization with the nuclear label DAPI. To quantify the nuclear translocation of the ATOH1-ER-DSRED fusion protein, HEK cells were transfected with this construct, incubated in graded doses of tamoxifen for 72 hr, and subjected to nuclear fractionation, and their isolated nuclei were pooled and mounted on coverslips for analysis of DsRed fluorescence. The results showed that control groups transfected with DsRed-ER exhibited a low level of background 568 nM fluorescence (194.7±0.27 mean arbitrary units [au]) in the absence of tamoxifen. Groups of cells transfected with Atoh1-ER-DsRed and not exposed to tamoxifen exhibited an insignificant increase in 568 nM fluorescence (392.0±68.35 au; p=0.16), which suggested that a low level of nuclear translocalization of the ATOH1-ER-DSRED fusion protein is present in the absence of tamoxifen. Incubating the cultures in 1 nM (848.0±29.33 au; p<0.05) and 1 μM (3,200.9±156.95 au; p<0.05) tamoxifen for 72 hr resulted in a significant increase in 568 nM fluorescence in the nuclear fraction, which demonstrates a tamoxifen-dependent increase in translocation of ATOH1-ER-DSRED to the nucleus. However, incubation with 100 μM tamoxifen for 3 days was toxic to the cells, and the remaining nuclei contained 568 nM fluorescence that was statistically equivalent to groups receiving no tamoxifen (597.8±40/46; p=0.07). Removal of tamoxifen from the culture medium resulted in an increase in cytoplasmic DsRed signal after 72 hr, at which time there was both cytoplasmic and nuclear labeling of the DsRed signal. Complete cytoplasmic localization of the fusion protein, including nuclear exclusion to basal levels, occurred within 2 weeks of tamoxifen washout (see below).
FIG. 2.

Tamoxifen-induced dose-dependent localization of the ATOH1-ER-DSRED fusion protein to the nucleus. Top: In the absence of tamoxifen, HEK cells transfected with the Atoh1-ER-DsRed construct exhibited expression of the DsRed signal in the cytoplasm. Addition of tamoxifen for 72 hr resulted in the co-localization of the DAPI-labeled nuclei (blue) and DsRed signals, which demonstrated a dose-dependent translocation of this fusion protein into the nucleus. Scale bars=20 μM. Graph: Subcellular fractionation of transfected HEK cells indicates that in the absence of tamoxifen, there is a low level accumulation of DsRed fluorescence in the nucleus similar to DsRed-ER-transfected controls in the absence of tamoxifen. Increasing doses of tamoxifen results in an increase in the DsRed signal in the nuclear fraction, indicating that tamoxifen causes a nuclear translocation of the ATOH1-ER-DSRED fusion protein. *Significant difference (p<0.05) from 0 tamoxifen. ●Significant difference (p<0.05) from 1 mM tamoxifen.
To determine the optimal tamoxifen concentration required for ATOH1-ER-DSRED nuclear translocation, this construct was transfected into spheres generated from the cochleas of ROSA26-GFP mice, which express EGFP in all cochlear cell types. Transfected spheres were incubated in graded doses of tamoxifen and imaged for a decrease in diffuse cytoplasmic expression and an increase in punctate localization of the ATOH1-ER-DSRED fusion protein, which is suggestive of nuclear localization of the DsRed signal, at daily intervals for 3 days. Using this method, the minimal effective dose was empirically determined to be 1 nM tamoxifen for 2–7 days (Fig. 3). These results were analogous to similarly treated HEK cells grown in culture (Fig. 4B).
FIG. 3.

Optimal tamoxifen concentration and incubation time for nuclear localization of the ATOH1-ER-DSRED fusion protein. The Atoh1-ER-DsRed construct was electroporated into cochlear spheres generated from ROSA26-GFP mice (green) and incubated with graded doses of tamoxifen. Top left: In the absence of tamoxifen, the ATOH1-ER-DSRED fusion protein (red) is expressed diffusely in the cytoplasm of transfected cells. Top right: Addition of 1 nM tamoxifen for 48 hr results in a decrease in diffuse cytosolic DsRed signaling and increase in punctate localization of the ATOH1-ER-DSRED fusion protein after 48 hr, which is suggestive of nuclear localization (DsRed fluorescence=red). The table describes dose and temporal effects of tamoxifen on punctate expression of the fusion protein. The yellow box highlights minimal effective conditions (+, nuclear localization in <90% of cells; −/+, ∼50% nuclear localization; −, >10% nuclear localization). Scale bars=10 μM.
FIG. 4.

Tamoxifen-induced ATOH1-ER-DSRED fusion protein activation of the 3′ Atoh1 promoter. (A) HEK cells were stably transfected with a 3′Atoh1enhancer/promoter.luciferase construct, transiently transfected with either the Atoh1-ER-DsRed or DsRed-ER control construct, incubated for 72 hr in increasing doses of tamoxifen, and then lysed and subjected to luciferase assay. (B) HEK cells transfected with the DsRed-ER construct exhibited a tamoxifen-dependent nuclear translocation of DsRed fluorescence similar to the Atoh1-ER-DsRed construct shown in Fig. 2. (C) Luciferase assays indicated that HEK cells transfected with DsRed-ER control constructs exhibited no significant change in 3′Atoh1enhancer/promoter.luciferase activity. HEK cells transfected with Atoh1-ER-DsRed exhibited a significant (*p<0.05) increase in luciferase activity in the presence of tamoxifen, indicating a tamoxifen-dependent binding of the ATOH1-ER-DSRED fusion protein to the 3′ Atoh1 promoter region.
Tamoxifen-dependent binding of the ATOH1-ER-DSRED fusion protein to the 3′ Atoh1 promoter
Since ATOH1 acts as a feed-forward autoregulatory transcription factor, whereby it positively regulates its own expression (Helms et al., 2000), the ability of the ATOH1-ER-DSRED fusion protein to upregulate endogenous Atoh1 gene expression was examined. To test this, the Atoh1-ER-DsRed transgene was transiently transfected into a HEK cell line engineered in our laboratory that stably expressed a luciferase reporter gene under control of the 3′ Atoh1 promoter region (Helms et al., 2000).
Tamoxifen induced binding to the promoter region of the Atoh1 gene measured by luciferase binding assay (Fig. 4). The lowest effective dose to elicit 3′ Atoh1 promoter activity was 1 nM tamoxifen. On average, 1 μM tamoxifen yielded higher yet not statistically different luciferase activity than the minimal effective dose of 1 nM. Similarly, there was no significant difference in 3′ Atoh1 promoter activity between cultures transfected with the Atoh1-ER-DsRed transgene and cultured in tamoxifen, and cultures transfected with the positive control cmv.Atoh1 construct. In contrast, HEK cells transfected with the DsRed-ER-negative control construct exhibited a nuclear translocation of the DsRed signal at increased concentrations of tamoxifen (Fig. 4B), but failed to exhibit a significant tamoxifen-dependent increase in 3′ Atoh1 promoter activity.
Tamoxifen-dependent upregulation of Atoh1 RNA
Next, the ability of this construct to upregulate Atoh1 RNA expression in HEK cells was analyzed by both RT-PCR and qPCR (Fig. 5). To test this, HEK cells were transfected with either Atoh1-ER-DsRed or cmv.Atoh1 control constructs, incubated in tamoxifen for 72 hr, and assayed for expression of Atoh1 RNA. The results showed that there were no significant differences in Atoh1 RNA expression between untransfected HEK cells (FC=0.69±0.29) and HEK cells transfected with the Atoh1-ER-DsRed construct in the absence of tamoxifen (normalized to FC=1.0), which suggested that the construct is inactive in the absence of tamoxifen. However, incubation of transfected cells in 1 nM of tamoxifen resulted in a 4.19 (±1.31)-fold increase in Atoh1 RNA expression. This was significantly less than the 7.8 (±0.48)-fold increase observed from cmv.Atoh1-transfected controls. HEK cells transfected with the DsRed-ER control vector exhibited no significant tamoxifen-dependent change in Atoh1 RNA expression (data not shown).
FIG. 5.

Tamoxifen-induced ATOH1-ER-DSRED fusion protein expression of Atoh1 RNA. HEK cells were transiently transfected with either the Atoh1-ER-DsRed or control construct, incubated with different doses of tamoxifen, and then assayed for Atoh1 RNA expression. Top: RT-PCR suggests that incubation in tamoxifen results in increased Atoh1 mRNA levels. Bottom: Quantitative PCR demonstrated that increased doses of tamoxifen resulted in increased Atoh1 mRNA expression. *Significantly different (p<0.05) from untransfected controls; ♦significantly different from 1 μM tamoxifen (p<0.05).
Tamoxifen-dependent upregulation and downregulation of endogenous ATOH1 protein
Next, we used Western analysis to determine whether the Atoh1-ER-DsRed construct was capable of inducing translation of the ATOH1 protein. To accomplish this, HEK cells were transfected with either test or control transgenes, incubated with graded doses of tamoxifen for 72 hr, and either cytosolic or nuclear protein was extracted and analyzed for ATOH1 translation by Western blot analysis. HEK cultures that were transfected with Atoh1-ER-DsRed exhibited both anti-FLAG and anti-ATOH1 immunolabeling to a ∼110 kDa protein corresponding to the predicted molecular weight of the ATOH1-ER-DSRED fusion protein (denoted as band a in Fig. 6). This band was absent in HEK cells transfected with both control vectors. The 110 kDa band observed in cytoplasmic fractions of Atoh1-ER-DsRed-transfected cells was strongest in the absence of tamoxifen and decreased as the dose of tamoxifen increased. This further demonstrated that increasing doses of tamoxifen resulted in decreased cytoplasmic accumulation of the ATOH1-ER-DSRED fusion protein (Fig. 4B). Correspondingly, increased doses of tamoxifen resulted in increased levels of this same fusion protein in the nuclear fraction, which suggested that increased doses of tamoxifen resulted in a nuclear accumulation of the ATOH1-ER-DSRED fusion protein.
FIG. 6.

Reversible tamoxifen-induced ATOH1-ER-DSRED fusion protein expression of the endogenous ATOH1 protein. HEK cells were transiently transfected with the Atoh1-ER-DsRed construct and incubated with different doses of tamoxifen for 72 hr, and cytosolic or nuclear protein was collected and processed for Western blot analysis for ATOH1 expression. Positive control samples were transfected with a cmv.Atoh1 construct consisting of consecutive flag-tag sequences, and negative control samples were transfected with the DsRed-ER construct. (A) Ani-ATOH1 labeling of the cytosolic fraction isolated from HEK cells transfected with either Atoh1-ER-DsRed or cmv.Atoh1 and incubated with 1 μM tamoxifen for 72 hr illustrates the relative expression between the 110 kDa ATOH1-ER-DSRED fusion protein (a), 47 kDa flag-tagged ATOH1 protein (b), and 45 kDa endogenous Atoh1 protein (c). (B) The top row shows that increased concentrations of tamoxifen resulted in decreased anti-FLAG immunolabeling of the ∼110 kDa ATOH1-ER-DSRED fusion protein in the cytosolic fraction, and increased immunolabeling of this fusion protein in the nuclear fraction. Labeling for the 110 kDa fusion protein is absent in the cytosolic fraction of cmv.Atoh1 and DsRed-ER-transfected controls. However, immunolabeling for the 47 kDa flag-tagged ATOH1 protein is present in the cytosolic and nuclear fraction at equivalent levels of cells transfected with the cmv.Atoh1 construct. Middle row: Immunolabeling the cytosolic fraction with anti-ATOH1 shows a 47 kDa band corresponding to the transfected flag-tagged cmv.Atoh1 construct (b), and a 45 kDa signal corresponding to endogenous Atoh1 (c). Since ATOH1 is autoregulatory, cmv.Atoh1-transfected cells exhibit an increased intensity of endogenous 45 kDa immunolabeling over the low level ATOH1 expression seen in DsRed-ER-transfected cells. In cells transfected with the Atoh1-ER-DsRed construct, increased tamoxifen levels corresponded with increased immunolabeling of the 45 kDa endogenous ATOH1 protein, and no labeling to the 47 kDa flag-tagged ATOH1 signal is observed. The letters a, b, and c correspond to the positions denoted in (A). Bottom row: Immunolabeling to cytosolic β-actin is shown for a control for gel loading. (C) The cytosolic and nuclear fractions of HEK cells transfected with either Atoh1-ER-DsRed or cmv.Atoh1 were isolated 0, 24, 72, or 96 hr after 72 hr incubation in 1 mM tamoxifen and compared with untreated samples. In the absence of tamoxifen, there was a relatively large concentration of anti-ATOH1 immunolabeling of the 110 kDa fusion protein in the cytosolic fraction compared with the nuclear fraction. Immediately after tamoxifen washout (0 hr), there was an increase in nuclear anti-ATOH1 immunolabeling for both the 110 kDa fusion protein and the endogenous 45 kDa protein that decreased over time to baseline levels at 72 hr. The decrease in the nuclear immunolabeling to the 110 kDa fusion protein corresponded to an increase in cytoplasmic immunolabeling of the 110 kDa fusion protein over this same time. Control cells transfected with cmv.Atoh1 and incubated for 72 hr in the absence of tamoxifen failed to exhibit labeling to the 110 kDa fusion protein in any condition. The letters a, b, and c correspond to the positions denoted in (A).
The results also showed that HEK cells transfected with the positive control cmv.Atoh1 vector exhibited ATOH1 immunolabeling at approximately 47 kDa (Fig. 6, band b). This band is heavier than the 45 kDa band (Fig. 6, band c) expected form the endogenous ATOH1 protein because of the repeat flag-tag sequences added to the control vector. In these positive control experiments, an increase in endogenous 45 kDa ATOH1 immunolabeling over background levels is also present, which supports the theory that Atoh1 exhibits positive autoregulation. Negative control cultures that were transfected with equivalent concentration of the DsRed-ER plasmid failed to exhibit immunolabeling to the ATOH1-ER-DSRED fusion protein and flag-tagged Atoh1, and exhibited endogenous ATOH1 immunolabeling equivalent to background levels observed in nontransfected HEK cells (data not shown). HEK cells that were transfected with Atoh1-ER-DsRed exhibited a tamoxifen-dependent increase in cytosolic ATOH1 immunolabeling at 45 kDa, which suggested that the increased nuclear concentration of the ATOH1-ER-DsRed fusion protein corresponded with increased concentrations of endogenous ATOH1 in the cytoplasm (Fig. 4B). Incubation of either control vector in 1 μM tamoxifen did not alter the intensity of the 45 kDa ATOH1 band, which suggested that tamoxifen by itself had no effect on endogenous ATOH1 translation.
Finally, we sought to determine whether the withdrawal of tamoxifen resulted in an inactivation of Atoh1-ER-DsRed signaling. To test this, we transfected HEK cells with either the Atoh1-ER-DsRed or cmv.Atoh1 control constructs, incubated them in 1 μM tamoxifen for 72 hr (a negative control group was cultured in the absence of tamoxifen), isolated the nuclear and cytosolic fractions, and assayed for anti-ATOH1 immunolabeling of the 110 kDa ATOH1-ER-DSRED fusion protein, flag-tagged ATOH1 of the control vector, or the endogenous ATOH1 protein (Fig. 6C). In the absence of tamoxifen, the 110 kDa ATOH1-ER-DSRED fusion protein was more highly expressed in the cytosolic fraction than in the nuclear fraction (Fig. 6C, band a). After 72 hr in tamoxifen (0 hr), there was an increase in nuclear ATOH1-ER-DSRED fusion protein and the endogenous 45 kDa ATOH1 protein (Fig. 6C, band c), consistent with an upregulation of endogenous Atoh1. Furthermore, the observed increase in nuclear fusion protein and endogenous ATOH1 decreased over time to baseline levels by 72 hr and corresponded to increased cytoplasmic ATOH1-ER-DSRED fusion protein over this same time. These results demonstrated that the tamoxifen-dependent upregulation of ATOH1 by the Atoh1-ER-DsRed construct is reversible, because removal of tamoxifen from the medium resulted in a return of the ATOH1-ER-DSRED fusion protein from the nucleus to the cytoplasm and corresponded to a downregulation of endogenous ATOH1 expression to basal levels.
Tamoxifen-dependent activation of signaling downstream of Atoh1
Next, we sought to determine whether tamoxifen-induced activation of the Atoh1-ER-DsRed construct was also sufficient for activation of endogenous Atoh1 signaling pathways. To test this, we transfected HEI OC1 cells with this Atoh1-ER-DsRed construct, and measured the expression of MYOSIN 7A, which is a downstream target of Atoh1 signaling. First, we transfected proliferating HEI OC1 cells with the Atoh1-ER-DsRed construct, and then we cultured these cells as floating aggregates in differentiating conditions (39°C) in either the presence or the absence of 1 μM tamoxifen for 3 days, before fixing them and measuring immunofluorescence to MYOSIN 7A. We found no significant difference in the numbers of DsRed-positive cells (p=0.07) between the groups receiving tamoxifen or not, which suggests similar transfection efficiencies between these two groups (Fig. 7). However, the transfected cells that were incubated with 1 μM tamoxifen exhibited decreased diffuse cytoplasmic DsRed labeling accompanied with increased punctate DsRed Fluorescence that colabeled with the nuclear dye TOPRO, demonstrating a nuclear translocation of the ATOH1-ER-DSRED fusion protein. Furthermore, cells incubated in 1 nM tamoxifen exhibited a significant increase (p<0.001) in MYOSIN 7A immunolabeling (79%;±5%) compared with transfected cells incubated in vehicle alone (26%;±6%).
FIG. 7.

Tamoxifen-induced ATOH1-ER-DSRED fusion protein upregulation of MYOSIN 7a in spheres derived from inner ear tissue. HEI OC1 cells were transfected with the Atoh1-ER-DsRed construct in proliferating conditions, and then grown as floating aggregates in differentiating conditions in the presence of either tamoxifen or vehicle. Transfected aggregates cultured for 72 hr in the absence of tamoxifen exhibited cytoplasmic DsRed signaling (top left image) and low levels of MYOSIN 7a immunolabeling (bottom chart). Similarly, transfected aggregates cultured in 1 nM tamoxifen for 72 hr exhibited a nuclear localization of the DsRed signal (top right image) and a significant (*p<0.05) increase in the number of MYOSIN 7a-labeled cells (bottom chart). Scale bars=10 μM.
Finally, this construct was electroporated into neonatal organs of Corti isolated from transgenic mouse pups that express GFP under control of the 3′ Atoh1 promoter (Lumpkin et al., 2003), and the organ explants were cultured in the medium supplemented with I nM tamoxifen or equivalent volumes of vehicle for 3–5 days (Fig. 8). These results indicate that in the absence of tamoxifen, electroporated cells exhibited diffuse cytoplasmic DsRed fluorescence, indicating that the ATOH1-ER-DSRED fusion protein was sequestered to the cytoplasm. However, organs of Corti incubated in 1 nM tamoxifen exhibited decreased diffuse cytoplasmic DsRed fluorescence and increased punctate DsRed fluorescence that colabeled with a punctate GFP signal in 97% (±3%) of the electroporated cells in the region of the greater epithelial ridge. This suggests that the fusion protein binds to endogenous Atoh1 promoter in the organ of Corti explants. Further immunolabeling directed to the flag-tag sequence preceding the Atoh1 moiety of the ATOH1-ER-DSRED fusion protein in this tissue showed that 87% (±4%) of the electroporated cells in the GER region also exhibited MYOSIN 7A immunolabeling.
FIG. 8.

Electroporation of the Atoh1-ER-DsRed construct into cultured organs of Corti. Organs of Corti were isolated from neonatal pups that express a nuclear tagged GFP under control of the 3′ Atoh1 promoter, electroporated with the Atoh1-ER-DsRed construct, and cultured for an additional 3–5 days. (A–C) Electroporated cells exhibited diffuse cytoplasmic labeling in organs of Corti cultured in the absence of tamoxifen. (D–G) Organs of Corti cultured in 1 nM tamoxifen exhibited punctate signaling that co-localized with punctate GFP signaling. (H) Immunolabeling of the flag-tag sequence of the ATOH1-ER-DSRED fusion protein (red) colabels for MYOSIN 7a immunostaining (green).
Discussion
Here we describe the molecular signaling of an independent genetic construct where endogenous ATOH1 can be conditionally upregulated once tamoxifen is added to the cell medium or cochlear environment, and can be downregulated once the tamoxifen is removed (Fig. 9). Cells transfected with this construct constitutively express an ATOH1-ER-DSRED fusion protein (Fig. 1) (Danielian et al., 1993, 1998; Littlewood et al., 1995). In the absence of tamoxifen, the ATOH1-ER-DSRED fusion protein remains sequestered within the cytosol because the ER moiety is inactivated by heat shock protein-90 (HSP90) (Danielian et al., 1993) (Fig. 2). The addition of tamoxifen to the transfected cells results in the release of HSP90, and a dose-dependent localization of the ATOH1-ER-DSRED fusion protein to the nucleus (Figs. 2–8). Once in the nucleus, the ATOH1 moiety of the fusion protein binds to the 3′ enhancer/promoter region of the Atoh1 gene (Helms et al., 2000) (Fig. 4) and upregulates the expression of Atoh1 mRNA (Fig. 5) and protein (Fig. 6B) in a dose-dependent manner. Further, removal of tamoxifen from the medium results in a reversal of this effect: return of the ATOH1-ER-DSRED fusion protein from the nucleus to the cytoplasm and downregulation of endogenous ATOH1 expression to basal levels (Fig. 6C). Finally, the Atoh1 expression was sufficient for upregulation of MYOSIN 7A, which suggests that this model upregulated Atoh1 in sufficient concentrations to elicit downstream signaling.
FIG. 9.
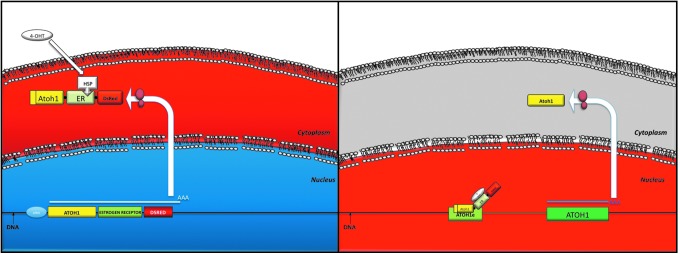
Independent construct for tamoxifen-dependent expression of Atoh1. Left: The cmv promoter drives constitutive expression of the ATOH1-ER-DSRED fusion protein, which is sequestered in the cytosol by HSP90 binding to the ER LBD, rendering it inactive. 4-OHT competes with HSP90 and allows the ATOH1-ER-DSRED fusion protein to translocate to the nucleus (right), where it binds to the endogenous 3′ Atoh1 promoter region and expresses endogenous Atoh1 signaling in a feed-forward mechanism. Withdrawal of tamoxifen results in a nucleus-to-cytoplasm translocation of the ATOH1-ER-DSRED fusion protein and inactivation of endogenous Atoh1 signaling. The Atoh1-ER-DsRed construct can be placed under control of different promoters to confer cell-specific expression and/or packaged into viral particles for in vivo infection of a stand-alone inducible Atoh1 expression construct. 4-OHT, 4-hydroxytamoxofen.
In cells that normally express this gene, Atoh1 is typically expressed at low levels, and a small fold change in endogenous Atoh1 upregulation is sufficient to measure physiological effects (Bossuyt et al., 2009; VanDussen and Samuelson, 2010). In contrast, the use of strong promoters in forced expression models, such as the cmv promoter used here, can result in unnaturally elevated levels of Atoh1. In the model presented here, the cmv promoter drives the expression of the ATOH1-ER-DSRED fusion protein at a high level. However, in the absence of tamoxifen, the fusion protein is sequestered in the cytosol and rendered inactive by HSP90 because it cannot enter the nucleus to activate Atoh1 expression pathways. The presence of tamoxifen allows for the fusion protein to translate into the nucleus, bind to endogenous Atoh1 response elements, and upregulate endogenous ATOH1 at either moderate levels (see anti-Atoh1 immunolabeling in Fig. 6 band c, 1 nM tamoxifen in Atoh1-ER-DsRed-transfected cells) or elevated levels observed in forced expression models (compare 1 μM tamoxifen in the next lane of the same figure with band b of the cmv.Aoh1-transfected cells). Therefore, tamoxifen concentrations can be titrated to modulate the levels of endogenous expression of ATOH1 protein in cells transfected with the Atoh1-ER-DsRed construct. Although the absolute protein concentrations are not presented here, the Western data suggest that high doses of tamoxifen can induce the Atoh1-ER-DsRed construct to upregulate endogenous Atoh1 at levels approximating levels observed in constitutive cmv.Atoh1 expression.
The significance of this inducible and independent construct for Atoh1 expression is that it has potential translational applications. The advantage over constitutively expressing systems is the ability to control the temporal and quantitative expression of Atoh1. Furthermore, the stand-alone aspect provides an advantage over standard inducible systems such as, doxycycline (Moutier et al., 2003) or mifepristone (Ngan et al., 2002) systems, which require extensive crossbreeding of transgenic animals and cannot be applied to potential human gene therapies. By contrast, this single Atoh1-ER-DsRed construct, or similar constructs lacking the DsRed moiety, can be packaged into viral particles and injected into the cochleas of deafened animals, and Atoh1 can be induced by either local or oral administration of tamoxifen. In this scenario, withdrawal of tamoxifen would downregulate Atoh1. Therefore, this independent construct can induce Atoh1 expression without the need for coexpression of secondary regulatory elements required in complex doxycycline or mifepristone models.
Additionally, this construct may be used to limit the ectopic expression of Atoh1-generated hair cells observed in many gene therapy models (Zheng and Gao, 2000; Kawamoto et al., 2003; Woods et al., 2004; Gubbels et al., 2008). One reason for the observed ectopic expression of Atoh1-induced cochlear hair cells is the indiscriminate infection of Atoh1 into cochlear cells. Typically, Atoh1 is either packaged into viral particles or directly electroporated into tissue, both of which infect cochlear cells randomly. Further experiments are underway aiming to limit ectopic hair cell expression by placing the Atoh1-ER-DsRed construct under the control of cochlear-supporting cell-specific promoters such as PROX1 (Bermingham-McDonogh et al., 2006), and TAK1 (Parker et al., 2011), which are selectively expressed in the cochlear-supporting cells that reside beneath the cochlear hair cells and are the desired cell type of for hair cell replacement in neonatal and adult mammals, respectively. In this case, even though many cell types could be virally infected with the transgene, the numbers of ectopic hair cells would be limited because only those cell types adjacent to the hair cells would express Atoh1-ER-DsRed. Current experiments incorporating these modifications of this construct are underway and could lead to an independent translational delivery system where Atoh1 can be expressed in a temporal, quantitative, and cell-specific locations within the organ of Corti.
A similar construct lacking the DsRed moiety has been previously described (Woods et al., 2004). When electroporated into organs of Corti derived from E13 embryos, it was reported that 50% of the cells in Kölliker's organ transfected with their ATOH1-ER fusion protein expressed hair cell markers (myosin 6 and phalloidin) under similar conditions. Our hair cell conversion rate of 87% represents an improvement over this result, and may be attributed to differences between these studies such as choice of hair cell marker (myosin 6 vs. myosin 7a), the age of the explant (E13 vs. P1), or the physical structure of the genetic construct, which bears further mention. The aforementioned study used an ATOH1 moiety that was linked directly to the ER moiety. The construct presented in this article includes two sequences (CTCGAGCCATCTGCTGGAGACATG and TCAGGATCTGGTTCAGGA) aimed to provide a degree of freedom between the moieties of the ATOH1-ER-DSRED fusion protein. Therefore, it could be that the spacing sequences included in this construct results in a more active fusion protein. In support of this theory, a cmv.Atoh1-ER construct engineered by the same linker between ATOH1 and ER moieties produced results that were not significantly different from the data presented here (data not shown).
Acknowledgments
The authors would like to thank their research team, including Kevin Jiang at the Massachusetts Eye and Ear Infirmary and Ashley Galetta, and Caitlin Simmons of Emerson College for their dedication. This work was supported by the National Institute of Deafness and Other Communicative Disorders (R03DC010065 [M.A.P.]; RO1DC007174 [A.E.]; P30DC05209 [Massachusetts Eye and Ear Infirmary Core Support for Hearing Research]).
Author Disclosure Statement
No competing financial interests exist.
References
- Bermingham N.A., Hassan B.A., Price S.D., et al. (1999). Math1: an essential gene for the generation of inner ear hair cells. Science 284, 1837–1841 [DOI] [PubMed] [Google Scholar]
- Bermingham-McDonogh O., Oesterle E.C., Stone J.S., et al. (2006). Expression of Prox1 during mouse cochlear development. J. Comp. Neurol. 496, 172–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram R., and Hillen W. (2008). The application of Tet repressor in prokaryotic gene regulation and expression. Microb. Biotechnol. 1, 2–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossuyt W., Kazanjian A., De Geest N., et al. (2009). Atonal homolog 1 is a tumor suppressor gene. PLoS Biol. 7, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielian P.S., White R., Hoare S.A., et al. (1993). Identification of residues in the estrogen receptor that confer differential sensitivity to estrogen and hydroxytamoxifen. Mol. Endocrinol. 7, 232–240 [DOI] [PubMed] [Google Scholar]
- Danielian P.S., Muccino D., Rowitch D.H., et al. (1998). Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 8, 1323–1326 [DOI] [PubMed] [Google Scholar]
- Gubbels S.P., Woessner D.W., Mitchell J.C., et al. (2008). Functional auditory hair cells produced in the mammalian cochlea by in utero gene transfer. Nature 455, 537–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms A.W., Abney A.L., Ben-Arie N., et al. (2000). Autoregulation and multiple enhancers control Math1 expression in the developing nervous system. Development 127, 1185–1196 [DOI] [PubMed] [Google Scholar]
- Isaka F., Ishibashi M., Taki W., et al. (1999). Ectopic expression of the bHLH gene Math1 disturbs neural development. Eur. J. Neurosci. 11, 2582–2588 [DOI] [PubMed] [Google Scholar]
- Izumikawa M., Minoda R., Kawamoto K., et al. (2005). Auditory hair cell replacement and hearing improvement by Atoh1 gene therapy in deaf mammals. Nat. Med. 11, 271–276 [DOI] [PubMed] [Google Scholar]
- Jones J.M., Montcouquiol M., Dabdoub A., et al. (2006). Inhibitors of differentiation and DNA binding (Ids) regulate Math1 and hair cell formation during the development of the organ of corti. J. Neurosci. 26, 550–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinec G.M., Webster P., Lim D.J., and Kalinec F. (2003). A cochlear cell line as an in vitro system for drug ototoxicity screening. Audiol. Neurootol. 8, 177–189 [DOI] [PubMed] [Google Scholar]
- Kawamoto K., Ishimoto S.-I., Minoda R., et al. (2003). Math1 gene transfer generates new cochlear hair cells in mature guinea pigs in vivo. J. Neurosci. 23, 4395–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly M.C., Chang Q., Pan A., et al. (2012). Atoh1 directs the formation of sensory mosaics and induces cell proliferation in the postnatal mammalian cochlea in vivo. J. Neurosci. 32, 6699–6710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood T.D., Hancock D.C., Danielian P.S., et al. (1995). A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 23, 1686–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Dearman J.A., Cox B.C., et al. (2012). Age-dependent in vivo conversion of mouse cochlear pillar and Deiters' cells to immature hair cells by Atoh1 ectopic expression. J. Neurosci. 32, 6600–6610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumpkin E.A., Collisson T., Parab P., et al. (2003). Math1-driven GFP expression in the developing nervous system of transgenic mice. Gene Expr. Patterns 3, 389–395 [DOI] [PubMed] [Google Scholar]
- Moutier R., Tchang F., Caucheteux S.M., and Kanellopoulos-Langevin C. (2003). Placental anomalies and fetal loss in mice, after administration of doxycycline in food for tet-system activation. Transgenic Res. 12, 369–373 [DOI] [PubMed] [Google Scholar]
- Ngan E.S., Schillinger K., DeMayo F., and Tsai S.Y. (2002). The mifepristone-inducible gene regulatory system in mouse models of disease and gene therapy. Semin. Cell Dev. Biol. 13, 143–149 [DOI] [PubMed] [Google Scholar]
- Oshima K., Grimm C.M., Corrales C.E., et al. (2007). Differential distribution of stem cells in the auditory and vestibular organs of the inner ear. J. Assoc. Res. Otolaryngol. 8, 18–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker M.A., Jiang K., Kempfle J.S., et al. (2011). TAK1 expression in the cochlea: a specific marker for adult supporting cells. J. Assoc. Res. Otolaryngol. 12, 471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou J., Zheng J.L., and Gao W.Q. (2003). Robust generation of new hair cells in the mature mammalian inner ear by adenoviral expression of Hath1. Mol. Cell Neurosci. 23, 169–179 [DOI] [PubMed] [Google Scholar]
- VanDussen K.L., and Samuelson L.C. (2010). Mouse atonal homolog 1 directs intestinal progenitors to secretory cell rather than absorptive cell fate. Dev. Biol. 346, 215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods C., Montcouquiol M., and Kelley M.W. (2004). Math1 regulates development of the sensory epithelium in the mammalian cochlea. Nat. Neurosci. 7, 1310–1318 [DOI] [PubMed] [Google Scholar]
- Zheng J.L., and Gao W.Q. (2000). Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat. Neurosci. 3, 580–586 [DOI] [PubMed] [Google Scholar]