

Abstract
RNA silencing is an established method for investigating gene function and has attracted particular interest because of the potential for generating RNA-based therapeutics. Using lentiviral vectors as an efficient delivery system that offers stable, long-term expression in postmitotic cells further enhances the applicability of an RNA-based gene therapy for the CNS. In this review we provide an overview of both lentiviral vectors and RNA silencing along with design considerations for generating lentiviral vectors capable of RNA silencing. We go on to describe the current preclinical data regarding lentiviral vector-mediated RNA silencing for CNS disorders and discuss the concerns of side effects associated with lentiviral vectors and small interfering RNAs and how these might be mitigated.
Introduction
Determining the function of genes and the roles they play in both the healthy CNS and in CNS diseases or disorders remains a key challenge for the neuroscience field. RNA silencing has emerged as a novel method to investigate gene function both in vitro and in vivo. Extensive research into the RNA-silencing pathway has elucidated the mechanism by which endogenous microRNAs (miRNAs) and exogenous small interfering RNAs (siRNAs) silence gene expression. After the identification of genes involved in a dominant CNS disease, generating a gene therapy approach that will efficiently and sustainably silence the expression of a target gene in vivo will require an efficient and safe delivery system. Similar efficiency may be required if the goal is genetic analysis.
Lentiviral vectors represent one of the most attractive delivery systems for gene therapy to the CNS and possess several advantages over other viral vector systems. First, they have been demonstrated to efficiently transduce postmitotic cells including CNS neurons in vitro and in vivo (Naldini et al., 1996a,b; Blomer et al., 1997; Thomas et al., 2003; Naldini, 2011) and have been effective in several animal models of CNS diseases (Ralph et al., 2005; Raoul et al., 2005; Singer et al., 2005; Sapru et al., 2006; Alves et al., 2008; Piedrahita et al., 2010; Horvath et al., 2011; Zou et al., 2011; Lv et al., 2012; Sun et al., 2012). Second, lentiviral vectors have been reported to have low cytotoxicity and immunogenicity (Naldini et al., 1996b; Abordo-Adesida et al., 2005; Schambach et al., 2013). Third, they have a relatively large transgene capacity of approximately 8–9 kb (Zufferey et al., 1998). Fourth, long-term transgene expression has been demonstrated in the CNS (Naldini et al., 1996b; Blomer et al., 1997; Thomas et al., 2003; Schambach et al., 2013). Fifth, they can be pseudotyped with a number of different envelope proteins to modify their tissue and cell tropism (Watson et al., 2002; Wong et al., 2004). Sixth, they can be made integration-deficient, significantly reducing the risk of insertional mutagenesis (Philippe et al., 2006; Yáñez-Muñoz et al., 2006).
Review
Lentiviral vector overview
A schematic showing the basic structure of an HIV-1 virion is shown in Fig. 1. After wild-type HIV enters a cell, the single-stranded RNA (ssRNA) genome is reverse transcribed into a double-stranded DNA genome, using cis-acting regulatory sequences at the 5′ and 3′ ends (Fig. 2A). The region 5′ to the gag gene, known as the gag leader sequence (GLS) contains the primer binding site (PBS) which is an 18 nucleotide sequence complementary to the cellular tRNALys,3 primer (one of the three major tRNALys isoacceptors found in mammalian cells) used to initiate reverse transcription (Das et al., 1994). The 5′ and 3′ genome ends contain an R region (R), which is a short sequence that forms a direct repeat at each end of the lentiviral RNA genome. The R region is involved in the initiation of proviral transcription and also contains the polyadenylation signal AAUAAA (Valsamakis et al., 1991). The 5′ end of the lentiviral RNA genome contains the U5 region, a unique noncoding region, which is the first part of the genome to be reverse transcribed. The 3′ end of the lentiviral RNA genome also has a unique noncoding region, the U3 region, that contains the promoter elements responsible for transcription of the proviral genome (Dull et al., 1998; Ramezani and Hawley, 2002). After reverse transcription of the single-stranded RNA, the double-stranded DNA product formed contains an identical U3, R, U5 sequence at the 5′ and 3′ ends (Fig. 2A). These are known as the long terminal repeats (LTRs) and play a central role in integration and viral gene expression. Transcription of the provirus initiates at the 5′ end of the R region within the 5′ LTR, whereas polyadenylylation occurs at the 3′ end of the R region within the 3′ LTR. The 3′ LTR contains additional upstream elements located within the U3 region, which are necessary for efficient polyadenylylation (Valsamakis et al., 1991). The viral genome also contains a central polypurine tract (cPPT) and central termination sequence (cTS), which are essential for initiating and terminating the central plus-strand DNA synthesis during reverse transcription (Charneau et al., 1992) and forming the central DNA flap that facilitates nuclear import (Zennou et al., 2000). Before integration the newly synthesized linear viral DNA undergoes nuclear translocation via the cellular nuclear import pathway as part of a nucleoprotein complex known as the preintegration complex (PIC) (Farnet and Haseltine, 1991). Once in the nucleus the viral IN integrates the double-stranded linear viral DNA into the host cell genome to form the provirus. The viral IN mediates this event by performing two-nucleotide staggered cuts at each end of the viral DNA and at the integration sites in the host cell genome, catalyzing insertion of the viral DNA into the host cell genome (Craigie, 2001). However, integration is not an efficient process and a considerable amount of viral DNA is also converted into episomal circular DNA (Li et al., 2001; Wu, 2008). Other important cis-acting elements are the encapsidation sequence (ψ), located in the GLS and required for packaging of the viral genome during particle assembly (Lever et al., 1989; Kaye et al., 1995), and toward the 3′ genome end the Rev response element (RRE), the sequence of which is essential for the export of the viral RNA out of the nucleus.
FIG. 1.

Basic structure of the wild-type HIV-1 virus. The diagram illustrates the key structural features of the wild-type HIV-1 virus. The surface glycoprotein (SU, gp120) and transmembrane glycoprotein (TM, gp41) are responsible for binding to the target cell and initiating cell entry. The matrix proteins (MA, p17) separate the outer lipid envelope from the inner capsid core (CA, p24). The capsid forms a cone-shaped inner core containing the condensed ribonucleoprotein complex, which is composed of two copies of the single-stranded viral RNA genome and nucleocapsid protein (NC, p7) that facilitate reverse transcription. The inner core also contains several viral enzymes including reverse transcriptase (RT, p66/p51), integrase (IN, p31), and protease (PR, p10) that are essential for viral replication. Color images available online at www.liebertpub.com/hgtb
FIG. 2.

Structure of the HIV-1 proviral genome and the third-generation lentiviral vector system. (A) The HIV-1 proviral genome contains nine genes and several cis-acting sequences. (B) To reduce the risk of generating replication-competent viruses, essential viral genes are provided in trans and separated onto three helper plasmids, which prevents them being incorporated into the vector genome. Third-generation lentiviral vectors are generated with four plasmids: the packaging plasmid, envelope plasmid, Rev plasmid, and transfer plasmid. (C) The transfer plasmid, which carries the viral vector genome, contains only essential cis-acting viral sequences. In the example shown the vector would encode a CMV-driven transgene and an H1-driven shRNA cassette. In this plasmid the 5′ LTR U3 region is replaced by a heterologous promoter such as the RSV promoter, whereas on the 3′ LTR the U3 region bears a deletion that removes transcriptional activity; after reverse transcription in the transduced cell the LTRs on the double-stranded DNA viral genome have the structure of the plasmid 3′ LTR. CMV, cytomegalovirus promoter; cPPT, central polypurine tract; cTS, central termination sequence; dLTR, long terminal repeat from which 400 bp in the U3 region has been deleted; GLS, gag leader sequence; ψ, encapsidation sequence; LTR, long terminal repeat; Poly-A, polyadenylylation signal; RRE, Rev response element; RSV, Rous sarcoma virus promoter; shRNA, short hairpin RNA; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element. Color images available online at www.liebertpub.com/hgtb
Wild-type HIV-1 has a single-stranded RNA genome containing nine genes: however, to produce functional lentiviral vectors only three genes (gag, pol, and env), common to all retroviruses, are essential. The polyproteins encoded by these genes are cleaved to form multiple protein subunits. The Gag polyprotein is cleaved by the viral protease to form structural proteins including the matrix (MA), capsid (CA), and nucleocapsid (NC) proteins that are important for the formation of the viral particles and packaging of the viral genome (Peng et al., 1989). The Pol polyprotein is also cleaved by the viral protease to form the viral enzymes reverse transcriptase (RT), protease (PR), and integrase (IN), which are essential for the reverse transcription of the viral genome, virion maturation, and integration into the host cell genome (Debouck, 1991). The Env polyprotein is cleaved by cellular proteases to form the transmembrane (TM) and surface (SU) glycoproteins (Fig. 1), which are inserted into the plasma membrane and are incorporated into the virion during viral budding (Stein and Engleman, 1990). TM and SU are vital for binding and entry into cells and determine the viral tropism, which can be manipulated by the process of pseudotyping. Lentiviral vectors are often pseudotyped with the G glycoprotein of vesicular stomatitis virus (VSV-G), which provides a broad tropism including postmitotic CNS neurons (Burns et al., 1993; Naldini et al., 1996b; Kwon et al., 2007).
Lentiviruses also express a number of other genes through differential splicing of the full-length viral mRNA (Ramezani and Hawley, 2002). The tat and rev genes encode the regulatory proteins trans-activator of transcription (Tat) and regulator of expression of viral proteins (Rev), which are involved in the trans-activation of gene expression and nuclear export of viral mRNA (Daly et al., 1989; Keen et al., 1996; Kay et al., 2001). Rev facilitates nuclear export of unspliced viral RNAs, using the existing cellular pathway, by forming a complex with the cellular nuclear export factor exportin-1 and binding to the RRE located in the env gene of the viral RNA (Fischer et al., 1995). The remaining four genes vif, vpr, vpu, and nef are not essential for viral replication, have been associated with the pathogenesis of HIV-1 (Cullen, 1991), and are omitted from lentiviral vectors.
Concerns over the safety of lentiviral vectors have been raised, regarding the potential to generate replication-competent viruses (RCVs) and insertional mutagenesis after viral genome integration. However, third-generation lentiviral vector systems incorporate several important safety features to improve biosafety and their therapeutic potential, and in common with other lentiviral vectors do not encode any viral genes (Dull et al., 1998; Kim et al., 1998). The essential coding genes are separated onto different DNA plasmids and provided in trans in the vector-producing cells to prevent reconstitution by recombination into an RCV (Lever et al., 1989; Kay et al., 2001). Diagrams showing the structure and organization of the four plasmids required to generate a third-generation replication-defective lentiviral vector are shown in Fig. 2B and C. The transfer plasmid encoding the transgene or short hairpin RNA (shRNA) contains only the essential cis-acting regulatory sequences. The nonessential viral coding genes such as vif, vpr, vpu, and nef have been removed and it has been demonstrated that the tat gene can also be removed without a substantial reduction in viral yield, provided the U3 region in the 5′ LTR of the packaging plasmid is replaced with a promoter such as the Rous sarcoma virus (RSV) or cytomegalovirus (CMV) promoter (Dull et al., 1998) (Fig. 2C). The essential viral coding genes such as gag, pol, env, and rev that encode important enzymes and structural proteins are then provided in trans on three separate plasmids, which include a packaging plasmid encoding gag and pol, an envelope plasmid encoding the VSV-G env, and a Rev plasmid encoding rev (Fig. 2B).
To further increase biosafety, third-generation lentiviral vectors can also be made self-inactivating (SIN). This involves deleting a 400-bp region from the U3 region in the 3′ LTR, which during reverse transcription of the viral RNA genome serves as a template for the U3 regions in both the LTRs (denoted as dLTR; Fig. 2C). The consequence of this deletion is that the transcriptional activity of the 5′ LTR is virtually abolished, preventing the production of full-length vector RNA in transduced target cells and further reducing the risk of generating an RCV (Miyoshi et al., 1998; Zufferey et al., 1998). In addition, SIN vectors have also been shown to reduce the activation of adjacent oncogenes (Miyoshi et al., 1998). Reconstitution of the 3′ U3 region by homologous recombination with the intact U3 region on the 5′ LTR cannot occur as the 5′ U3 region was previously replaced with a heterologous promoter to allow tat removal (see above; Fig. 2C). SIN lentiviral vectors require an internal RNA polymerase II (Pol II) promoter for transgene expression or Pol III promoter for shRNA expression (Fig. 2C); however, it has been shown that this does not result in a substantial reduction in viral titer, and SIN lentiviral vectors have successfully been used to transduce neuronal tissue in vivo (Dull et al., 1998; Miyoshi et al., 1998; Zufferey et al., 1998).
Integration-deficient lentiviral vectors
Lentiviral vectors provide long-term expression by integrating their genome into the host cells (Fig. 3A). However, the specific integration sites cannot be predicted and it has been shown that lentiviral vectors preferentially integrate into active gene loci, albeit with patterns that are less mutagenic than those observed with other retroviral vectors (Schroder et al., 2002; Schambach et al., 2013). This has the potential to result in insertional mutagenesis and the activation of proto-oncogenes (Baum and Fehse, 2003; Baum et al., 2006) and has been highlighted in studies showing the development of cancers in both mouse models and human clinical trials with various retroviral vectors (Li et al., 2002; Hacein-Bey-Abina et al., 2003; Themis et al., 2005).
FIG. 3.
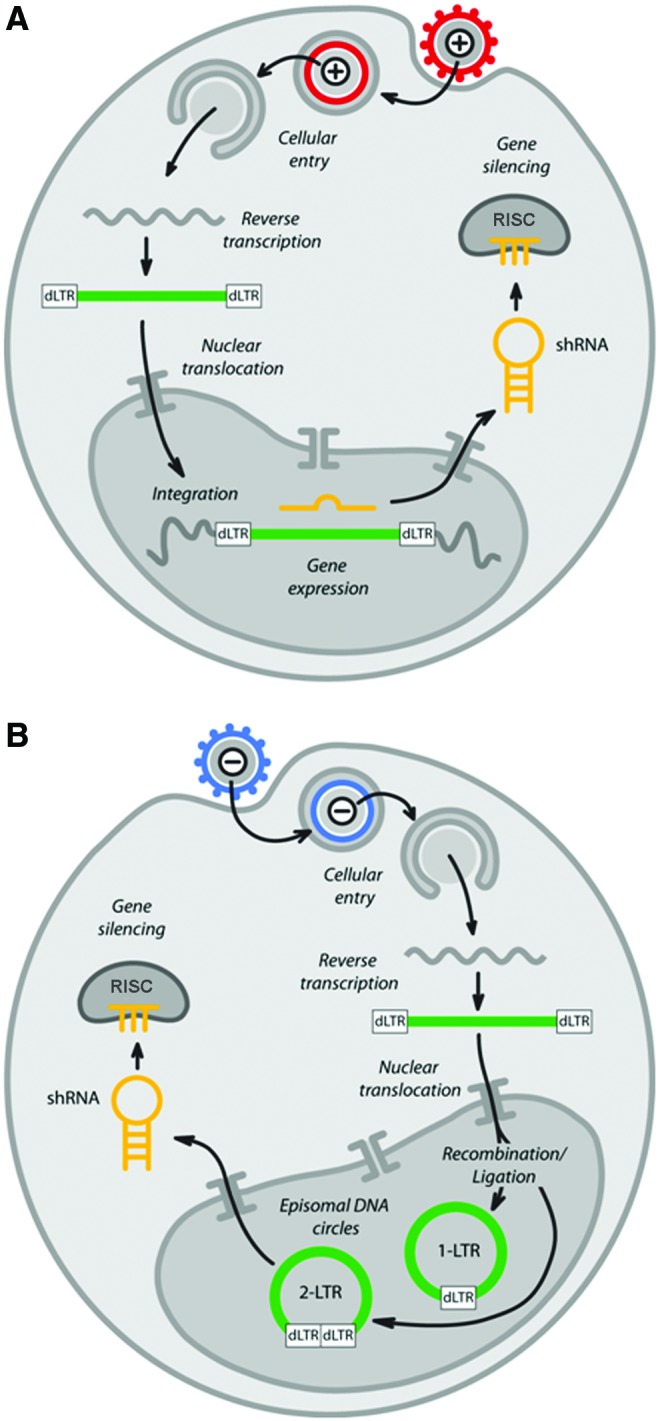
Fate of the lentiviral genome after transduction by integration-proficient and deficient lentiviral vectors. (A) Integration-proficient vectors pseudotyped with VSV-G are thought to enter host cells via the endosomal pathway. Once inside the cytosol the RNA genome is reverse transcribed in a multiprotein complex known as the preintegration complex (PIC). The PIC is then thought to translocate to the nucleus via the nuclear pore complex whereupon the viral vector DNA is integrated into the host cell DNA. The transcriptional activity of the viral vector LTR is minimal because of extensive deletions in the U3 region. Instead, shRNA expression proceeds from the internal RNA polymerase III (Pol III) promoter. The shRNA is then exported to the cytosol, where the loop is removed by Dicer and the antisense strand is loaded into the RNA-induced silencing complex (RISC) complex. (B) Integration-deficient vectors gain access to the cell the same way, whereupon reverse transcription and nuclear translocation proceed normally. However, the PIC contains a mutant integrase, thereby inhibiting integration. Instead, the viral vector DNA forms double-stranded circles after self-ligation or recombination of the LTRs (partial deletion of the U3 region in self-inactivating vectors, denoted as dLTR). This is then followed by shRNA expression and gene silencing as normal. Color images available online at www.liebertpub.com/hgtb
To circumvent this, integration-deficient lentiviral vectors (IDLVs) can be generated by mutating the integrase-coding sequence in the viral pol gene. These types of integrase mutations can the grouped into two classes (Engelman, 1999; Philpott and Thrasher, 2007; Wanisch and Yáñez-Muñoz, 2009): First, class II mutations that disrupt multiple cellular functions of integrase including viral assembly, nuclear translocation, and reverse transcription and therefore reduce the amount of viral DNA produced as well as integration. These mutations are unsuitable for generating integration-deficient lentiviral vectors. Second, class I mutations, which are commonly created by changing amino acids in the catalytic active site of IN, which specifically affect integration without reducing viral DNA production and can therefore be used to produce integration-deficient lentiviral vectors. Class I mutations are most commonly generated by replacing one of the three amino acids that make up the catalytic triad (D64, D116, and E152 for HIV-1 IN) (Philpott and Thrasher, 2007; but see also Philippe et al., 2006, for mutation of the 262RRK motif ). Specifically, pseudotyped lentiviral vectors carrying an Asp-64→Val change at the catalytic site of integrase have been demonstrated to reduce the rate of integration in vitro to 1:1000–1:10,000 compared with wild-type vector, and to significantly reduce integration in vivo (Leavitt et al., 1996; Yáñez-Muñoz et al., 2006). The residual integration is thought to be due to recombination events rather than to integrase activity (Nightingale et al., 2006), but it has not been formally discounted that it is mediated by integrase but with noncanonical features due to the mutations (Wanisch and Yáñez-Muñoz, 2009). Alternatively, mutating or deleting the U3 and U5 attachment sites (att) located within LTRs of the viral genome can also significantly reduce integration (Masuda et al., 1995, 1998; Brown et al., 1999; Nightingale et al., 2006). Integrase binds to the att sites and cleaves the 3′-terminal dinucleotide at both ends of the viral genome, which is essential for the attachment and integration of the viral genome into the host DNA (Masuda et al., 1998; Philpott and Thrasher, 2007). However, mutations in the att sites are not as efficient at attenuating integration as class I IN mutations (Wanisch and Yáñez-Muñoz, 2009).
Failure to integrate into the host genome results in increased formation of episomal circular DNA molecules. Two forms of episomal circular DNA can be generated: circular DNA containing a single LTR, which is formed by homologous recombination between the two LTRs, or circular DNA containing two LTRs that is formed by nonhomologous end-joining of the two LTRs (Farnet and Haseltine, 1991; Li et al., 2001) (Fig. 3B). Crucially, it has been shown that these episomal DNA circles are transcriptionally active (Stevenson et al., 1990; Cara et al., 1996; Wu, 2004).
Integration-deficient lentiviral vectors can be produced at high titers and efficiently transduce both mitotic and postmitotic cells in vitro and in vivo and express enhanced green fluorescent protein (eGFP) at a level comparable to vectors incorporating wild-type integrase (Loewen et al., 2003; Saenz et al., 2004; Philippe et al., 2006; Yáñez-Muñoz et al., 2006; Rahim et al., 2009; reviewed by Wanisch and Yáñez-Muñoz, 2009; Peluffo et al., 2012). As expected, only transient expression was observed in mitotic cells as the vector became diluted by successive cell cycles (being nonreplicating as well as nonintegrating) (Philippe et al., 2006). In contrast, robust stable in vivo expression of eGFP was observed after 30 days in postmitotic cells including striatal and hippocampal neurons, for up to 8 weeks in motor neurons and interneurons after intraspinal injection, and for at least 9 months in the retinal pigment epithelium (Philippe et al., 2006; Yáñez-Muñoz et al., 2006; Peluffo et al., 2012). We and other groups have also shown efficient transduction of corticospinal and rubrospinal neurons, with eGFP expression visible in their fibers in the spinal cord (Philippe et al., 2006; Rahim et al., 2009; Hutson et al., 2012b). This is especially exciting as both of these tracts provide important models for investigating therapies for spinal cord injury (SCI), and the demonstration that they can be efficiently transduced using integration-deficient lentiviral vectors provides the potential for future studies to deliver genes or siRNAs that may promote regeneration and functional recovery after a SCI. Integration-deficient lentiviral vectors have the potential to be a safe and efficient means of transducing neurons in vivo, allowing the investigation of novel gene therapies to CNS disorders.
RNA silencing overview
RNA silencing is a process of posttranscriptional gene regulation, whereupon the expression of a target gene is silenced by small noncoding RNAs (Elbashir et al., 2001a; Zamore, 2001). Two mechanisms of RNA silencing have been described and depend on the complementarity between the small noncoding RNA and the mRNA of the targeted gene: low complementarity results in translational inhibition and mRNA degradation whereas high complementarity results in the selective catalytic cleavage of the mRNA, known as RNA interference (RNAi). There are two main types of small noncoding RNAs that can mediate RNA silencing: first, micro-RNAs (miRNAs), which are endogenously expressed, ∼21- to 24-nucleotide, single-stranded RNAs that usually bind multiple target mRNAs with partial complementarity. They have been demonstrated to mediate gene silencing by direct cleavage (if bound with high complementarity) or translational inhibition and mRNA deadenylation and degradation after P-body relocalization (Rev et al., 2003; Doench and Sharp, 2004; Jakymiw et al., 2005; Liu et al., 2005; Sen and Blau, 2005; Engels and Hutvagner, 2006; Eulalio et al., 2009a; Kulkarni et al., 2010). Second, small interfering RNAs (siRNAs), which are exogenously derived, ∼21- to 24-nucleotide, single-stranded, target-specific RNAs that are designed to bind with perfect complementarity to a single target mRNA and therefore used to mediate RNAi (Tuschl et al., 1999; Elbashir et al., 2001a,b). siRNAs with 19 nucleotides or fewer can also successfully mediate RNAi (Brummelkamp et al., 2002; Ge et al., 2010a,b) although they are thought to be processed by a different mechanism than the longer siRNAs (Siolas et al., 2005; Ge et al., 2010a,b). However, both miRNAs and siRNAs have been reported to mediate RNA silencing by either RNAi or translational inhibition (Doench et al., 2003; Zeng et al., 2005).
Hundreds of miRNAs have been identified within the human genome, with approximately 1000 predicted to exist. They are thought to control the activity of ∼30% of all protein-coding genes, participate in the regulation of numerous cellular processes, and potentially underlie certain human disorders (Bentwich et al., 2005; Filipowicz et al., 2008). The nervous system has been shown to express numerous miRNAs with a wide range of functions in both developing and mature neurons. In developing neurons miRNAs have been shown to play a role in neuronal patterning, cell specification, and axon path-finding and in mature neurons they have been linked to modulating synaptic plasticity and axon regeneration, and the dysregulation of miRNA activity could play a role in several nervous system diseases including Alzheimer's disease, Huntington's disease, Parkinson's disease, fragile X syndrome and Tourette's syndrome (Krichevsky et al., 2003; Kosik, 2006; Christensen and Schratt, 2009; Eacker et al., 2009; Strickland et al., 2011; Lerch et al., 2012).
The first step in the miRNA biogenesis pathway (shown on the right side of Fig. 4) is the expression of genes encoding large ∼300- to 1000-nucleotide primary miRNAs (pri-miRNAs) that are generally transcribed by endogenous RNA polymerase II (Pol II) and can contain one or more stem-and-loop hairpin structures (Denli et al., 2004; Lee et al., 2004; Kosik, 2006). In the nucleus pri-miRNAs are recognized and cleaved by the RNase III enzyme Drosha and the cofactor DiGeorge syndrome critical region gene 8 (DGCR8), resulting in the release of an ∼70-nucleotide hairpin termed a precursor miRNA (pre-miRNA) that contains a two-nucleotide 3′ overhang and several bulges and mismatches (Lee et al., 2003; Zeng and Cullen, 2005; Han et al., 2006). The pre-miRNA stem binds to the nuclear export molecule exportin-5 (Exp5) and its GTP-bound cofactor RAN and is actively transported into the cell cytoplasm (Yi et al., 2003; Zeng and Cullen, 2004).
FIG. 4.
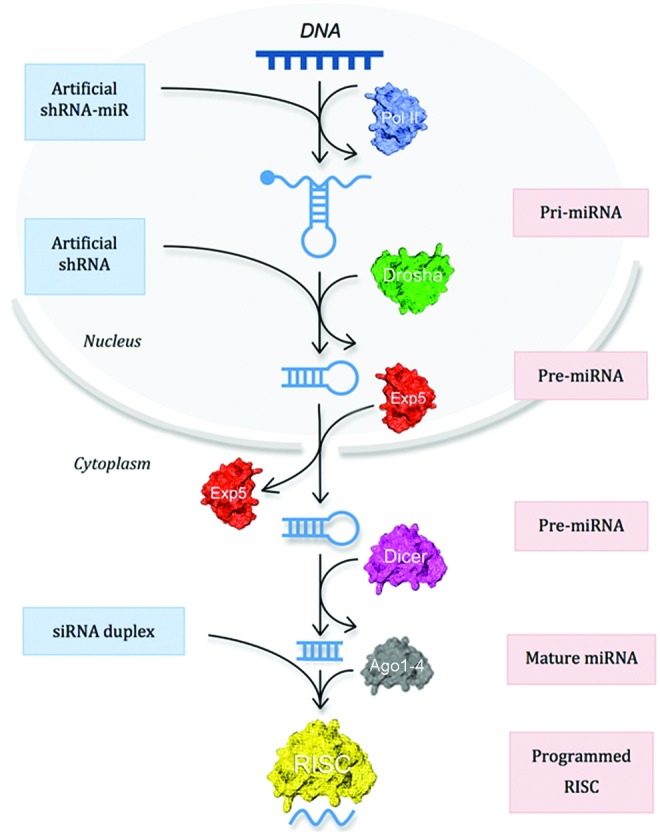
The mammalian RNA silencing pathway. The diagram summarises the major features of the RNA silencing pathway in mammalian cells. The pink boxes on the right represent the endogenous miRNA pathway, while the blue boxes on the left indicate the points at which the endogenous silencing pathway can be subverted to produce knockdown of specified target mRNAs. Artificial miRNA-like constructs, known as shRNA-miRs, are transcribed from RNA Pol II or III promoters and enter the miRNA pathway at the start. Artificial shRNAs are generally expressed from RNA Pol III promoters and enter the miRNA pathway at the Dicer stage. Lastly, conventional ∼21–23 nucleotide siRNAs only need to be unwound in the cytoplasm before they enter the RISC to mediate gene silencing. Color images available online at www.liebertpub.com/hgtb
In the cytoplasm the two-nucleotide 3′ overhang is recognized by another RNase III enzyme, Dicer, which contains a double-stranded RNA (dsRNA)-binding domain, an RNA helicase domain, a Piwi/Argonaute/Zwille (PAZ) domain, and two RNase III domains (Bernstein et al., 2001). Dicer binds and cleaves the pre-miRNA, removing the hairpin loop and leaving an miRNA duplex (Bernstein et al., 2001). The miRNA duplex then undergoes strand selection, in which the strand with the less thermodynamically stable 5′ end, known as the antisense or guide strand, is incorporated into the RNA-induced silencing complex (RISC). The other strand, known as the sense or passenger strand, is released and degraded (Cullen, 2006). The structure of a typical miRNA duplex has ∼6–8 nucleotides at the 5′ end of the guide strand, which is known as the seed region, and is fully complementary to a region on the 3′ UTR of the target mRNA (Lewis et al., 2003; Birmingham et al., 2006). The central ∼9–12 nucleotides of the miRNA are usually noncomplementary to the target mRNA and form a bulge that prevents the mRNA from being cleaved by the RISC and undergoing RNAi, resulting instead in translational inhibition (Zeng et al., 2003; Doench and Sharp, 2004; Engels and Hutvagner, 2006; Filipowicz et al., 2008). The nucleotides at the 3′ end of the miRNA are usually complementary to those at the 5′ end, and it has been shown that mismatches to the target mRNA in this region do not affect RNA silencing (Engels and Hutvagner, 2006).
In Drosophila, strand selection and loading into the RISC involve at least Dicer and a dsRNA-binding interactor, R2D2, which makes up the RISC loading complex (RLC). R2D2 is thought to bind and sense the thermodynamic properties of the miRNA duplex and subsequently direct Dicer to the 5′ end of the less thermodynamically stable strand, which is then loaded into the RISC as the guide strand (Tomari et al., 2004; Engels and Hutvagner, 2006). Two mammalian orthologs of the dsRNA-binding protein R2D2 have been described: HIV-1 TAR (trans-activation response element) RNA-binding protein (TRBP) and PACT (protein activator of Protein Kinase R) (Chendrimada et al., 2005; Lee et al., 2006). In addition, interactions between the two-nucleotide 3′ overhang of the miRNA duplex and the PAZ domain of Argonaute, a protein component of the RISC, is also thought to be involved in strand selection (Lingel et al., 2003). The RISC is an RNA-binding multiprotein complex with sequence-specific RNA cleavage activity (Engels and Hutvagner, 2006). One important protein family that has been shown to incorporate into RISC are the Argonautes, which are thought to be essential for the catalytic degradation of the target mRNA (Hammond et al., 2001; Liu et al., 2004). Argonaute proteins contain two key domains, a PAZ domain and a PIWI domain (Hutvagner and Simard, 2008; Wang et al., 2008). The PAZ domain can bind to ssRNA or dsRNA with 3′ overhangs (Lingel et al., 2003) and the PIWI domain contains an RNase-H subdomain, which is thought to be responsible for the nuclease activity of RISC (Song et al., 2004). Mammals express four Argonaute proteins (Ago1, Ago2, Ago3, and Ago4); however, in mammals only Argonaute-2 (Ago2) has been shown to have catalytic activity and therefore only an RISC that incorporates Ago2 can mediate RNAi (Liu et al., 2004). After binding of the miRNA to the complementary site on the target mRNA, Ago2 cleaves the mRNA in the center of the base-paired region. However, if there are mismatches in the central region, which is the case for many miRNAs that tend not to be fully complementary, then the target mRNA will not be cleaved by Ago2 and instead translation will be inhibited (Elbashir et al., 2001c; Zeng et al., 2003; Doench and Sharp, 2004; Engels and Hutvagner, 2006; Filipowicz et al., 2008). Another core component of the RISC is the glycine-tryptophan protein of 182 kDa (GW182) protein, which interacts with and acts downstream of the Argonaute proteins (Eulalio et al., 2009b; Krol et al., 2010). GW182 has been shown to interact with cytoplasmic poly(A)-binding protein (PABPC), which in turn recruits and binds the deadenylase complex CAF1-CCR4-NOT. This complex can remove the poly(A) tail from mRNA, thereby making it liable to degradation (Behm-Ansmant et al., 2006; Braun et al., 2011; Chekulaeva et al., 2011; Fabian et al., 2011). GW182 proteins therefore play an important role in the deadenylation and degradation of mRNAs by the RISC (Giraldez et al., 2006; Wu et al., 2006; Eulalio et al., 2009b).
The miRNA biogenesis pathway can be exploited to generate siRNAs (shown on the left side of Fig. 4). There are three entry points within the miRNA pathway. First, synthetic siRNA duplexes can be transfected into cells, which become incorporated into RISC, and knock down expression of the target gene (Elbashir et al., 2001a). Synthetic siRNA duplexes can be designed to be fully complementary to the target mRNA and therefore mediate RNAi, or conversely they can be designed to be identical to an endogenous miRNA duplex, incorporating mismatches, and therefore mediate translation inhibition (Doench et al., 2003; Zeng et al., 2003). However, the key disadvantage to using synthetic siRNA duplexes is that because they are not expressed from a DNA vector, only transient knockdown is achieved, so repeated application of the siRNA can be required (Cullen, 2005). It has also been demonstrated that siRNA duplexes can elicit an interferon (IFN) response, resulting in the upregulation of interferon-stimulated genes (ISGs), a global inhibition of gene expression, and cytotoxicity (Sledz et al., 2003; Sledz and Williams, 2004; Read et al., 2009; Rossi, 2009; Suggate et al., 2009).
Second, short hairpin RNAs (shRNAs) mimicking the structure of endogenous pre-miRNAs can be designed, cloned, and expressed under the control of an RNA Pol III promoter such as the H1 or U6 promoter (Brummelkamp et al., 2002; Paddison et al., 2002). The DNA expression vectors can then be transfected or transduced into cells, whereupon the cassette is transcribed to yield the shRNA in the nucleus. The expression from an RNA Pol III promoter means that the shRNA lacks a poly(A) tail and requires the addition of five thymidine nucleotides at its 3′ terminus. Termination of transcription occurs after the second of the five thymidine nucleotides, generating an shRNA with a two-nucleotide 3′ overhang consisting of two uridine residues, which mimics the pre-miRNA structure (Brummelkamp et al., 2002). The shRNA then joins the miRNA biosynthesis pathway and is exported to the cytoplasm, where it undergoes Dicer cleavage before incorporation of the siRNA into the RISC. A separate class of short shRNAs (sshRNAs) with a stem length of 19 nucleotide base pairs or fewer and that are not cleaved by Dicer have also been described; however, the exact mechanism by which they are processed is still not clear (Siolas et al., 2005; Ge et al., 2010a,b). The main advantage of shRNAs and sshRNAs is that they can be stably expressed in transfected/transduced cells (Brummelkamp et al., 2002). However, there are still disadvantages: high-level expression from the RNA Pol III promoter can cause off-target silencing and may cause cellular toxicity through saturating the endogenous RNA-silencing machinery (Yi et al., 2005; Davidson and Boudreau, 2007) or by evoking an IFN response (Bridge et al., 2003; Jackson et al., 2003; Fish and Kruithof, 2004; McBride et al., 2008; Boudreau et al., 2009; Read et al., 2009). In addition, RNA Pol III promoters are difficult to regulate and do not offer cell-specific expression (Silva et al., 2005).
Third, artificial pri-miRNAs, termed shRNA-miRs or miR-embedded shRNAs, can be designed and cloned into DNA expression vectors under the control of RNA Pol II or III promoters. RNA Pol II promoters can be used because shRNA-miRs go through all the steps of the endogenous miRNA biogenesis pathway including the cleavage by Drosha, which generates a two-nucleotide 3′ overhang and therefore does not require an RNA Pol III promoter to generate it (Silva et al., 2005; Zeng and Cullen, 2005; Zeng et al., 2005). The structure of shRNA-miRs has often been modeled on the extensively studied endogenously expressed human miR-30 miRNA (Zeng et al., 2002, 2003, 2005; Silva et al., 2005). There have been conflicting results when comparing the efficiency of shRNAs and shRNA-miRs: the latter have been shown to generate a higher level of siRNA production compared with shRNAs (Silva et al., 2005) and produce a more potent knockdown of the targeted mRNA (Dickins et al., 2005; Silva et al., 2005). However, other studies have reported that shRNAs provide the highest level of expression, siRNA production, and target knockdown, although it was later reported that this may also result in cytotoxicity in vitro and fatality in vivo because of saturation of the endogenous RNA-silencing machinery and activation of an IFN response (Grimm et al., 2006; Li et al., 2007; Boudreau et al., 2008, 2009; McBride et al., 2008). One recent study comprehensively compared siRNA production, processing, and target mRNA knockdown after siRNA sequences predicted to be identical were embedded in shRNA or shRNA-miR scaffolds (Maczuga et al., 2013). This study demonstrated that although siRNA production from shRNA-miRs was considerably lower than shRNAs, the shRNA-miR scaffold provided a more sustained and effective knockdown of the targeted mRNA in vivo. They also used next-generation sequencing to determine whether identical siRNAs were processed from the shRNA and shRNA-miR scaffolds. Surprisingly, they demonstrated that the endogenous RNAi machinery did not generate the predicted siRNA from either the shRNA or shRNA-miR scaffolds, leaving doubts over the currently accepted cleavage sites and design rules. The shRNA scaffold gave rise to a more heterogeneous pool of siRNAs, suggesting that shRNA-miRs that undergo both Drosha and Dicer cleavages may be more likely to generate the expected siRNA (Maczuga et al., 2013). Taken together, these studies demonstrate that shRNA-miRs, which undergo full processing, are not expressed at such high levels, do not saturate the endogenous RNA-silencing machinery, or invoke a strong IFN response are therefore more suitable as an RNA-based therapy. shRNA-miRs also have the advantage of being able to be expressed from cell type-specific and inducible RNA Pol II promoters (Rao and Wilkinson, 2006).
Design considerations for lentiviral vector-mediated RNA silencing
Designing an effective siRNA sequence is vital for efficient knockdown of a targeted gene. Although numerous siRNA design algorithms are now available, generating an effective siRNA sequence can still be problematic and unpredictable. This most likely reflects accessibility issues for the siRNA because of the tertiary structure of the target mRNA (Krueger et al., 2007). Some studies have suggested that as little as 10–25% of siRNA sequences recommended by their algorithm, against a given target, is capable of silencing gene expression by >90% (Reynolds et al., 2004). In addition, different sequences exhibit variable levels of specificity, with some more likely to produce off-target effects than others (Jackson et al., 2003; Birmingham et al., 2006; Fedorov et al., 2006).
Initial design paradigms were primarily concerned with structural and chemical features of siRNA molecules such as duplex length, backbone chemistry, and the base composition and length of the 3′ overhangs (Elbashir et al., 2001a,d). Using an empirical approach, Elbashir and co-workers demonstrated that 21-nucleotide duplexes with symmetrical UU or TT 3′ overhangs produced the most efficient silencing effect (Elbashir et al., 2001c). In comparison, sequences bearing AA, CC, or GG overhangs were 2- to 4-fold less effective. Furthermore, evaluation of the sequence itself suggested that certain positions on the guide strand, such as the nucleotide sequence surrounding the cleavage site, are particularly important for efficient silencing. Mismatches in this region are poorly tolerated compared with positions near the 5′ end of the strand. Other studies have shown that duplexes as long as 29 nucleotides in length can mediate efficient gene silencing (Paddison et al., 2002). Furthermore, similarly long constructs lacking the prototypical 3′ overhangs have also been shown to be effective (Kim et al., 2005). Longer siRNAs have the advantage that they can be transfected at lower concentrations without altering silencing efficiency, although they are more prone to stimulating the interferon response (Reynolds et al., 2006). Despite these reports, symmetrical, 21-nucleotide duplexes bearing dinucleotide 3′ overhangs remain the most common choice.
Subsequently, a number of studies demonstrated that highly functional siRNAs exhibit lower internal energy at the 5′ end of the guide strand (Khvorova et al., 2003; Schwarz et al., 2003). In other words, there is a higher percentage of weaker A-U base pairs in this region, making the duplex easier to separate. This is known as thermodynamic asymmetry and is thought to promote loading of the guide strand as opposed to the passenger strand into the RISC. Statistical analysis of experimentally validated siRNAs suggests that this phenomenon correlates strongly with siRNA functionality (Amarzguioui and Prydz, 2004; Chalk et al., 2004; Reynolds et al., 2004). Reynolds and co-workers evaluated 180 siRNAs targeting either the firefly luciferase or human cyclophilin B mRNA (90 sequences each) and showed that sequences containing at least three A-U base pairs between positions 1 and 7 of the guide strand are better silencers (Reynolds et al., 2004). Similar approaches have also demonstrated that moderate to low G-C content and an absence of repeats or palindromes also correlate well with improved knockdown efficiency (Elbashir et al., 2002; Chalk et al., 2004; Reynolds et al., 2004). If the G-C content is too high, separation of the duplex becomes more difficult; if it is too low then the base pairing between the siRNA and the target mRNA is less stable. Most highly functional siRNAs possess a G-C content of 36–52% (Reynolds et al., 2004). siRNAs that contain internal repeats or palindromes can form fold-back, hairpin-like structures instead of forming siRNA duplexes (Kirchner et al., 1998; Reynolds et al., 2004). The formation of these secondary structures can be predicted by evaluating the melting temperature (Tm) of the siRNA. Sequences with high Tm have a greater propensity to form secondary structures. Highly functional siRNAs tend to exhibit lower Tm (Amarzguioui and Prydz, 2004; Chalk et al., 2004; Reynolds et al., 2004). Preference for certain bases at specific positions was also shown to improve selection of effective sequences. These include an A at position 1 of the guide strand, a C at position 19, a higher frequency of A-U base pairs between positions 1 and 7, and an A/U at position 10. The first three rules are likely to govern the thermodynamic properties of the duplex whereas the latter suggests that the RISC preferentially cleaves after an A or U residue.
None of the current design algorithms are foolproof and they can still return siRNA sequences exhibiting low silencing efficiency and at the same time miss highly functional candidates. Predesigned and validated siRNA sequences can now be purchased from certain proprietors. Notwithstanding manufacturer claims, it is advisable to screen a minimum of three siRNA sequences for each gene before carrying out experiments; we typically screen four or five single siRNAs (or pools of four siRNAs).
Lentiviral vector-mediated RNA silencing for in vivo models of CNS disorders
Lentiviral vectors have been demonstrated to efficiently transduce CNS neurons and mediate RNA silencing in the brain and spinal cord in vivo, where they have been successfully used to ameliorate a number of animal models of CNS diseases and disorders.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder resulting in progressive muscle weakness and atrophy due to the selective degeneration and death of motor neurons in the brain and spinal cord. Mutations in the superoxide dismutase-1 (SOD1) gene can produce novel toxic properties of the SOD1 protein resulting in motor neuron death. More than 100 unique SOD1 mutations have been described, which account for approximately 20% of familial ALS cases. Attenuating the expression of SOD1 therefore represents an attractive therapeutic approach. In two of the first studies to use lentiviral vector-mediated RNAi in vivo for a CNS disorder, silencing the expression of SOD1 in a mouse model of familial ALS was shown to increase motor neuron survival, improve motor performance, and successfully delay onset and slow down progression of the disease (Ralph et al., 2005; Raoul et al., 2005).
Parkinson's disease belongs to a set of neurodegenerative disorders known as synucleinopathies and is characterized by the progressive loss of midbrain dopaminergic neurons and the formation of cytoplasmic inclusions known as Lewy bodies, which are a hallmark of these disorders. The primary structural component of Lewy bodies is α-synuclein (SNCA); the misregulation and overexpression of SNCA lead to α-synuclein accumulation in neurons, which is thought to be neurotoxic. Knocking down the expression of α-synuclein is a promising therapeutic strategy for reducing the buildup and neurotoxicity of α-synuclein. Robust silencing of human α-synuclein, using lentiviral vector-mediated RNAi, was demonstrated in the human dopaminergic cell line SH-SY5Y and in neurons in the rat striatum after overexpression of the human α-synuclein gene (Sapru et al., 2006). In Parkinson's disease the neural circuitry that controls motor function changes as a consequence of the dopaminergic neuronal degeneration. These changes are in part driven by an increase in γ-aminobutyric acid (GABA) production due to the transcriptional upregulation of the gene encoding GABA-producing enzyme glutamate decarboxylase 1 (GAD1), also known as GAD67. Knocking down the expression of GAD67 therefore represents a good target to normalize the increased activity of these GABAergic neurons. Horvath and colleagues used lentiviral vectors expressing shRNA-miRs to successfully knock down the expression of GAD67 in a rat model of Parkinson's disease and thereby normalize the neuronal activity that is increased because of dopamine loss (Horvath et al., 2011).
Alzheimer's disease is a progressive neurodegenerative disease characterized by neuronal death that results in the gross atrophy of the cortex and certain subcortical areas. Neuronal death is thought to occur as a consequence of accumulations of neurotoxic insoluble plaques composed of β-amyloid protein and intracellular neurofibrillary tangles composed of the microtubule-associated protein tau. The neurotoxic β-amyloid is processed from amyloid precursor protein via sequential cleavages by β- and γ-secretases. The major β-secretase involved in β-amyloid production is β-site APP (amyloid precursor protein) cleaving enzyme-1 (BACE1), making it a prime target for RNA silencing. In another early study to use lentiviral vector-mediated RNA silencing for a CNS disorder, knockdown of BACE1 attenuated APP cleavage and β-amyloid production and furthermore reduced the neurodegeneration and behavioral deficits in a mouse model of Alzheimer's disease (Singer et al., 2005). Using an alternative strategy aimed at reducing the tau-based pathology, one study by Piedrahita and colleagues showed beneficial effects of knocking down cyclin-dependent kinase-5 (CDK5). Restricting the phosphorylation of tau can prevent its aggregation into aberrant neurofibrillary tangles, and CDK5 is a key kinase thought to play a role in tau phosphorylation and pathology. Silencing CDK5 using lentiviral vectors expressing siRNAs reduced tau phosphorylation and the number of neurofibrillary tangles in a mouse model of Alzheimer's disease (Piedrahita et al., 2010).
Early-onset torsion dystonia is an autosomal dominant genetic disorder usually affecting children, characterized by painful uncontrollable muscle contractions that slowly progress throughout the body, leaving the patient in a debilitated state. These symptoms are not the result of neuronal loss but of neuronal dysfunction and are caused by a loss of function mutation in the DYT1 gene that encodes the protein torsinA. TorsinA is an AAA ATPase, that is, an adenosine triphosphatase (ATPase) associated with diverse cellular activities; this family of ATPases is involved in a great range of cellular processes. Mutant torsinA is thought to act through a dominant negative effect, sequestering wild-type torsinA to the nuclear envelope. Specifically knocking down the mutant torsinA and restoring the function of wild-type torsinA may therefore present an effective therapy for torsion dystonia. Lentiviral vector-mediated RNAi was shown to effectively knock down the mutated torsinA in vitro, rescuing cells from its dominant negative effect and restoring the normal distribution of wild-type torsinA (Gonzalez-Alegre et al., 2005).
Machado–Joseph disease (MJD) or spinocerebellar ataxia type 3 (SCA3) is an autosomal, dominantly inherited neurodegenerative disorder and a member of the polyglutamine (PolyQ) repeat disease family. The neuropathological features of MJD include the degeneration and death of neurons in the cerebellum, pons, striatum and substantia nigra, which generates symptoms including a progressive ataxia and loss of coordination in the arms and legs, abnormal gait, spasticity, ophthalmoplegia, nystagmus, dysarthria, and dystonia (Sudarsky and Coutinho, 1995). MJD is caused by a genetic mutation resulting in the expansion of trinucleotide CAG repeats in the coding region of the ATXN3 gene. This results in the expression of a toxic, gain-of-function mutant ataxin-3 protein that accumulates in the nucleus, forming intranuclear inclusion bodies that lead to neuronal degeneration and death. Wild-type ataxin-3 plays an important role in ubiquitin-mediated proteolysis (Doss-Pepe et al., 2003). A favorable therapeutic strategy would therefore selectively silence the expression of the mutant but not the wild-type allele, thereby maintaining the normal expression and function of wild-type ataxin-3. This has been achieved by targeting an shRNA to a single-nucleotide polymorphism (SNP) at the 3′ end of the CAG expansion. This SNP is in linkage disequilibrium with the disease-causing CAG-expanded allele and could provide a treatment for 70% of patients with MJD. After overexpression of the human mutated ATXN3 gene, lentiviral vector-mediated silencing of the mutated ATXN3 transcript was demonstrated to be both selective and efficient in human 293T cells in vitro and in the rat brain in vivo (Alves et al., 2008). Furthermore, silencing the mutated ATXN3 gene significantly decreased the number and size of toxic intranuclear inclusions and reduced the number of degenerating neurons in vivo (Alves et al., 2008).
Huntington's disease (HD) is also an autosomal, dominantly inherited neurodegenerative disorder and a member of the PolyQ repeat disease family. HD is caused by a CAG repeat expansion mutation in the huntingtin (HTT) gene, resulting in an abnormally long polyglutamine tract in the huntingtin protein. Accumulation of the mutant HTT protein in striatal and cortical neurons leads to nuclear aggregation, the formation of intranuclear inclusion bodies, and the dysfunction of multiple cellular processes including transcription regulation, cell metabolism, and neurotransmission that result in neurodegeneration and cell death. HD is invariably fatal, with patients usually presenting symptoms during mid-adult life. The neuronal loss in the striatum and cortex leads to a progressive loss of muscle coordination, cognitive decline, and psychiatric problems. One study aimed to enhance neuronal viability in an in vitro model of HD by modifying the expression of genes thought to be important in striatal neuron signaling and that are dysregulated in HD. Using lentiviral vectors expressing shRNAs they demonstrated that knocking down the expression of regulator of G-protein signaling-2 (RGS2) or Ras homolog enriched in striatum-2 (RASD2) is neuroprotective in primary striatal neurons that are overexpressing the mutant HTT protein (Seredenina et al., 2011). Alternatively, the use of a similar strategy to the one mentioned previously for MJD, whereby SNPs found only in the mutant HTT allele are targeted for knockdown by siRNAs, holds great promise as an effective therapeutic strategy for HD. Two studies have shown that allele-specific siRNAs targeting heterozygous SNPs can distinguish between the mutant and wild-type HTT and could be used to specifically knock down the mutant HTT in three-quarters of patients with HD (Lombardi et al., 2009; Pfister et al., 2009). Although allele-specific siRNAs have been shown to effectively knock down the mutant HTT allele, while preserving the wild-type allele in cell lines derived from patients with HD (van Bilsen et al., 2008; Hu et al., 2009, 2010; Lombardi et al., 2009; Pfister et al., 2009; Fiszer et al., 2011), future preclinical studies are needed that can evaluate this strategy in transgenic animal models of HD that express both the human wild-type and mutant alleles of the HD gene. Delivery of siRNAs to an animal model will require the use of an effective and therapeutically relevant delivery system of which integration-deficient lentiviral vectors should be a principal candidate.
However, although the use of RNAi to target SNPs holds promise as a therapeutic strategy for MJD, HD, and other PolyQ diseases, the prevalence of targetable SNPs amid the affected population can still be a limiting factor.
Neuropathic pain is a pathophysiological condition that can occur as a result of injury to the peripheral or central nervous system. Symptoms include spontaneous pain, mechanical and thermal hyperalgesia, and pain in response to innocuous stimuli (allodynia). Causes include direct trauma, where the nerve is physically cut or crushed, as well as neuropathies resulting from disorders such as diabetes mellitus, viral infections or drug treatments, such as chemotherapy. The damage to peripheral nerves results in the release of pain-related inflammatory mediators and can stimulate significant changes in global gene expression (Wang et al., 2002; Xiao et al., 2002; Tegeder et al., 2006). This includes up- or downregulation of a wide range of ion channels, receptors, neuropeptides, enzymes, and growth factors, some of which have been shown to increase the excitability of sensory neurons and lead to the development of ectopic or aberrant activity such as central sensitization and facilitation of the pain state. Work from our laboratories has shown that both integrating and integration-deficient lentiviral vectors can efficiently transduce CNS and peripheral nervous system (PNS) neurons in vitro and in vivo including motor neurons, interneurons, and dorsal root ganglion (DRG) neurons (Peluffo et al., 2012). In this study we further go on to demonstrate that both integrating and integration-deficient lentiviral vectors expressing shRNAs can mediate efficient knockdown of transient receptor potential cation channel subfamily V member-1 (TRPV1) in DRG neurons in vitro. TRPV1 is a polymodal ion channel that plays an important role in nociception, particularly as an integrator of inflammatory stimuli (Julius and Basbaum, 2001).
Two other studies have used similar strategies to attenuate neuropathic pain by knocking down genes involved in the development of central sensitization (Zou et al., 2011; Sun et al., 2012). In these studies lentiviral vectors and shRNAs were used to knock down the expression of protein kinase C isoform-γ (PKCγ) or nuclear factor-κB (NF-κB). PKCγ is highly expressed in the CNS and is involved in intracellular signal transduction. It has previously been shown to be activated in persistent pain states and has been implicated in the development of central sensitization (Malmberg et al., 1997; Martin et al., 2001). Lentiviral vector-mediated RNAi successfully knocked down PKCγ expression in neuronal cultures in vitro and the spinal cord in vivo and attenuated mechanical allodynia and thermal hyperalgesia for >6 weeks, using the chronic constriction injury model of neuropathic pain (Zou et al., 2011). NF-κB expression and activation have been shown to be upregulated after sciatic nerve injury (Ma and Bisby, 1998), and it is known that NF-κB plays a major role in the expression of multiple pain-related mediators including several proinflammatory cytokines (Baeuerle and Henkel, 1994; Makarov, 2000). After intrathecal injection of lentiviral vectors, shRNA-mediated silencing of NF-κB attenuated the chronic constriction injury-induced expression of the proinflammatory cytokines tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) and additionally reduced mechanical allodynia and thermal hyperalgesia for >4 weeks (Sun et al., 2012). Together these studies demonstrate that lentiviral vector-mediated knockdown of factors that contribute to central sensitization and persistent pain states is a viable strategy to alleviate and manage neuropathic pain.
After a CNS injury, adult neurons demonstrate a limited regenerative response, which is due to a number of factors including a limited intrinsic growth state and an inhibitory environment containing multiple growth-inhibitory molecules. One strategy to enhance axonal regeneration after injury is to attenuate the neurons' response to growth-inhibitory molecules by knocking down the receptors to which they bind. A study by Lv and colleagues (2012) employed lentiviral vectors and RNAi to knock down the reticulon-4 receptor (RTN4R), also known as Nogo receptor-1 (NgR1); a major component of the Nogo receptor complex that binds multiple growth-inhibitory molecules. After a spinal cord injury, NgR1 knockdown resulted in increased numbers of neuronal fibers in and around the lesion site and improved hind limb motor function compared with controls (Lv et al., 2012). In work from our own group we employed a similar strategy to attenuate the response of neurons to the inhibitory CNS environment. RNA silencing was used to knock down the expression of leucine-rich repeat and immunoglobulin domain containing-1 (LINGO-1), another essential component of the NgR complex. Using a potentially therapeutically applicable delivery system, cerebellar granular neurons (CGNs) were efficiently transduced by integration-deficient lentiviral vectors encoding shRNAs targeting LINGO-1. LINGO-1 mRNA expression was shown to be significantly knocked down; however, neurite outgrowth was not enhanced on a growth-inhibitory substrate in vitro. Further investigation demonstrated that when high lentiviral vector concentrations were used or when the lentiviral vectors encoded an shRNA, an interferon response was elicited that may have been responsible for a component of the observed knockdown and cytotoxicity, which may in part explain why neurite outgrowth was not enhanced after LINGO-1 knockdown (Hutson et al., 2012a).
In addition to knocking down molecules in neuronal receptor complexes, RNA silencing also has the potential to posttranscriptionally attenuate the expression of genes encoding other axon growth-inhibitory proteins, thereby enhancing regeneration after an SCI. Studies have demonstrated enhanced growth of DRG and retinal ganglion cell (RGC) axons on an inhibitory CNS myelin substrate in vitro after RNAi-mediated silencing of NgR1, p75NTR (also known as nerve growth factor receptor; NGFR), and ras homolog family member A (RHOA) (Ahmed et al., 2005; Suggate et al., 2009). RNA silencing of the ubiquitin ligase Cdh1-APC has been shown to increase neurite outgrowth in postnatal CGNs on permissive and inhibitory myelin substrates in vitro (Konishi et al., 2004; Stegmuller et al., 2006; Kim and Bonni, 2007). RNAi has also been used as a technique to investigate the role of regeneration-associated genes in regulating neurite outgrowth. RNA silencing of the transcription factors SMAD family member-1 (Smad1) and SRY-box containing gene-11 (Sox11) has been demonstrated to reduce the neurite outgrowth of adult DRG neurons cultured in vitro (Jankowski et al., 2006; Zou et al., 2009). In addition, transient knockdown of Sox11, using siRNA duplexes, reduced the regeneration of peripheral nerves from DRG neurons in vivo (Jankowski et al., 2009). Conversely, overexpression of the regeneration-associated gene Kruppel-like factor-4 (KLF4) has been shown to reduce neurite outgrowth, suggesting KLF4 as a potential target for RNA silencing (Moore et al., 2009).
These preclinical studies present encouraging proof of concept that lentiviral vector-mediated RNAi-based therapies can improve both pathological and behavioral outcomes in animal models of CNS disorders, indicating that similar gene therapy approaches may become a viable therapeutic option in the future. However, concerns regarding siRNA-associated side effects, including sequence-specific and -nonspecific off-target effects and the induction of an immune response, remain key obstacles in development and clinical application of siRNA-based therapies.
Side effects associated with lentiviral vectors and siRNAs
Lentiviral vectors have generally been demonstrated to be safe and well tolerated in the CNS, with transduced neurons appearing healthy and an absence of a significant immune response (Naldini et al., 1996a,b; Blomer et al., 1997; Abordo-Adesida et al., 2005). However, data from our own and other laboratories suggest that lentiviral vectors themselves may contribute to the induction of an IFN response and a reduction in neuronal viability (Bauer et al., 2008; Hutson et al., 2012a). IFNs are a class of immunomodulatory cytokines produced and secreted by multiple cell types including neurons, which protect against viral infection (Samuel, 2001; Delhaye et al., 2006). The production of IFNs leads to the activation of ISGs, causing a global inhibition of gene expression and ultimately cell death. The induction of an IFN response has been previously reported for lentiviral vectors; whereby the transduction of cortical neurons was shown to elevate the levels of cleaved caspase-3 and enhance the expression of the ISG 2′,5′-oligoadenylate synthase-1 (OAS1), which was then further increased by the expression of an shRNA (Bauer et al., 2008). The data in a paper from our laboratory are in agreement with this study and show that at high vector concentrations, lentiviral vectors themselves can induce the expression of OAS1 and an IFN response in CNS neurons, resulting in the downregulation of off-target genes and cytotoxicity. Furthermore, when the lentiviral vectors encoded an shRNA, a further increase in cytotoxicity and ISG expression was observed (Hutson et al., 2012a). These results strongly suggest that the vector concentration should be titrated to determine the lowest multiplicity of infection (MOI) that can be used to produce effective knockdown and the required functional effect (Ulusoy et al., 2009; Grimm, 2011). How lentiviral vectors trigger an IFN response and neuronal cytotoxicity is not clear. It may result from the presence of the Gag protein, as it has been previously shown that injection of a chimeric HIV-1 Env–Gag fusion protein is neurotoxic and can augment excitotoxic damage in the CNS (Barks et al., 1993). Alternatively, the concentration protocol used in vector production often includes an ultracentrifugation step leading to residual immunoreactive molecules such as serum components being present in the vector stock, which could stimulate an IFN response (Bao et al., 2009).
Owing to the highly specific target recognition between the siRNA and the target mRNA, which can distinguish sequences differing by a single nucleotide, siRNAs were perceived to be ideal specific tools for studying gene function. However, a number of studies have now raised concerns about siRNA specificity, the potential for off-target and nonspecific effects, and in some cases lethal toxicity (Jackson et al., 2003; Saxena et al., 2003; Semizarov et al., 2003; Scacheri et al., 2004; Bilanges and Stokoe, 2005; Ehlert et al., 2010; Grimm et al., 2010; Khodr et al., 2011; Martin et al., 2011).
Several microarray expression studies have shown that siRNAs can alter the expression of large numbers of “off-target” genes (Jackson et al., 2003; Semizarov et al., 2003; Persengiev et al., 2004; Bilanges and Stokoe, 2005). A study involving siRNA-mediated silencing of insulin-like growth factor receptor (IGF1R) and mitogen-activated protein kinase-1 (MAPK14/p38α) revealed both dose-dependent and sequence-dependent off-target effects (Jackson et al., 2003). Similar observations were reported in a separate study involving siRNA-mediated silencing of the retinoblastoma (RB1), protein-kinase B (Akt1/Pkb), and polo-like kinase (Plk1) genes (Semizarov et al., 2003). In both of these studies the siRNAs silenced the expression of the target gene(s) but also produced significant changes in nontargeted genes. The gene expression signatures were specific for individual siRNA sequences, indicating that the off-target effects were sequence related (Jackson et al., 2003; Semizarov et al., 2003). Temporal grouping of the affected genes revealed that the off-target effects were in place before silencing of the target gene (Jackson et al., 2003), suggesting that the off-target changes in gene expression are not the consequence of target gene silencing. Sequence analysis of the effected off-target genes revealed partial complementarity with each of the siRNAs used (Jackson et al., 2003).
The observation that siRNAs and miRNAs could silence gene expression by similar mechanisms has led to a better understanding of off-target effects (Doench et al., 2003). Several parallel studies have shown that siRNAs mediate their off-target effects through miRNA-like translational repression (Saxena et al., 2003; Scacheri et al., 2004; Jackson et al., 2006b). Target recognition in the miRNA pathway depends on complementary base pairing between the seed site and the target site on the mRNA. Complementary base pairing between siRNA seed sites and off-target transcripts has been shown to increase nonspecific effects (Birmingham et al., 2006; Jackson et al., 2006b), and siRNAs with low complementary seed frequencies have been demonstrated to generate fewer off-target effects (Anderson et al., 2008). The realization that significant numbers of human genes bear seed site matches for miRNAs suggests that the potential for off-target gene silencing by siRNAs may be high (Lewis et al., 2005). It is therefore important to attempt to predict potential mRNAs that may be unintentionally silenced when designing siRNAs, and algorithms are becoming available to do this (Yamada and Morishita, 2005; Anderson et al., 2008). Pooling multiple siRNAs targeting a specific gene (each at a lower dose) can help to mitigate off-target silencing (Kittler et al., 2007). This is thought to be due to competition between the different siRNAs within the pool, which each have a unique off-target signature. The lower concentration of each siRNA within the pool may also contribute to the reduced off-target effects. It has also been reported that chemical modification to the seed region, such as a 2′-O-methyl ribosyl substitution at position 2 of the guide strand can significantly reduce off-target effects (Jackson et al., 2006a). It is hypothesized that this modification causes conformational changes in the RISC and/or guide strand that disrupts RISC formation, so that weaker imperfect binding by siRNAs to off-target sequences dissociates before being cleaved. The 2′-O-methyl modification has the additional benefit that it can be easily amenable to both in vitro and in vivo applications.
Another concern is the activation of innate immune pathways by RNA-silencing techniques. The early development of RNAi in flies and nematodes was first explored with long dsRNA molecules, without significant negative side effects. However, transfection of exogenous dsRNA (>30 nucleotides) into mammalian cells has been shown to activate the IFN pathway (Katze et al., 2002; Sledz et al., 2003). A number of groups have now shown that siRNAs can increase the expression of three key ISGs: OAS1 (Bridge et al., 2003; Sledz et al., 2003; Fish and Kruithof, 2004; Pebernard and Iggo, 2004; Bauer et al., 2008), protein kinase R (PKR) (Sledz et al., 2003; Read et al., 2009), and myxovirus resistance-1 (Mx1) (Read et al., 2009; Suggate et al., 2009). Binding of dsRNA causes PKR to phosphorylate the eukaryotic translation initiation factor-2α (eIF-2α), resulting in a global disruption in translation, whereas OAS1 triggers nonspecific degradation of cellular mRNA by stimulating RNase-L and Mx1 inhibits RNA synthesis. In an early elegant study by Sledz and colleagues (2003), siRNA-mediated knockdown of both glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and laminin A/C resulted in dose-dependent upregulation of Stat1, a transcriptional activator involved in the expression of PKR and OAS1 in response to IFNs. This response was both siRNA and IFN dependent. In addition, microarray analysis indicated that multiple ISGs were upregulated. The mechanism of this response appeared to involve activation of PKR. Increasing autophosphorylation of PKR correlated with increasing siRNA concentration. In addition, PKR-null cell lines did not show an IFN response after transfection of siRNAs (Sledz et al., 2003). In a study from our laboratory we observed upregulated expression of PKR and OAS1 but not Mx1 in cultured CNS neurons after lentiviral vector-mediated shRNA expression. This was associated with reduced expression of off-target genes and reduced cell viability (Hutson et al., 2012a). The capacity of siRNAs to induce an immune response presents a major obstacle for the interpretation of therapeutic effects and the future clinical application of RNA silencing. The identification of an appropriate nontargeting control siRNA is complicated by the fact that the immunostimulatory response of each siRNA sequence is unique and difficult to predict. This was highlighted in a set of studies demonstrating that an siRNA thought to induce sequence-specific silencing of vascular endothelial growth factor (VEGF) produced a therapeutic effect, which was not observed when a commonly used negative control siRNA targeting eGFP was used. However, further studies showed that the control eGFP siRNA elicited a particularly low immunostimulatory response and that the observed therapeutic effect of the siRNA targeting VEGF was actually due to the induction of an IFN response by the VEGF siRNA (Michels et al., 2006; Kleinman et al., 2008; Rossi, 2009). This raises concerns that other reported RNAi-based therapies may not be the result of sequence-specific silencing but instead of an unwanted nonspecific IFN response and highlights the importance of checking for an IFN response.
The nonspecific effects of siRNAs have also been linked to a separate branch of the immune system after the discovery that siRNAs can activate Toll-like receptors (TLRs) in immune cells. TLRs are activated in response to microbial by-products such as lipopolysaccharide (Katze et al., 2002); however, a number of subtypes, including TLR3, TLR7, and TRL8, have also been implicated in recognizing bacterial and viral nucleic acids (Alexopoulou et al., 2001; Hornung et al., 2005). After activation, TLRs can induce IFNs and proinflammatory cytokines such as TNF-α and IL-6, resulting in a global downregulation of gene expression. siRNAs have been shown to activate TLR3 in keratinocytes in a sequence-independent manner and result in the production of IFNs (Karikó et al., 2004). Furthermore siRNAs have been implicated in the activation of TLR7 and the production of IFNs in plasmacytoid dendritic cells (Alexopoulou et al., 2001; Hornung et al., 2005) and in the activation of TLR8 and cytokine production in myeloid dendritic cells (Judge et al., 2005; Sioud, 2005). Unlike TLR3, the activation of TLR7 and TLR8 is thought to be sequence dependent, with GU-rich and 5′-UGU-3′ motifs being particularity immunostimulatory (Judge et al., 2005). However, these are not the only immunostimulatory motifs, as others have demonstrated that AU-rich sequences can also activate TRL8 and the presence of multiple uridine ribonucleotide residues is sufficient to stimulate the activation of TLR7 (Diebold et al., 2006; Forbach et al., 2008).
To mitigate siRNA-mediated immune stimulation, chemical modifications to the 2′-OH group in the ribose backbone of siRNAs, including 2′-O-methyl, 2′-fluoro, and 2′-deoxy modifications, and locked nucleic acids (LNAs), have been shown to be effective. Substituting more than 90% of the siRNA nucleotides with a combination of 2′-O-methyl-, 2′-fluoro-, and 2′-deoxy-modified nucleotides can prevent the immunostimulatory response (Morrissey et al., 2005). However, caution must be taken if undertaking extensive chemical modifications to an siRNA as this can significantly impair its silencing activity (Judge and MacLaclan, 2008). The use of just 2′-fluoro or 2′-deoxy modifications has also been shown to reduce immune stimulation; however, compared with 2′-O-methyl modifications their efficacy is more difficult to predict (Cekaite et al., 2007; Judge and MacLaclan, 2008). Incorporating LNAs that contain a methylene bridge between the 2′-O and the 4′-C of the ribose at the 3′ end of the passenger strand has been shown to attenuate the immunostimulatory response, although this can also affect silencing efficiency (Braasch et al., 2001; Hornung et al., 2005). In a key study, Judge and colleagues (2006) demonstrated that by selectively substituting only ∼5% of the ribonucleotides on the passenger strand with 2′-O-methyl-uridine or -guanosine nucleotides, the siRNA-induced immune response could be abrogated without affecting silencing efficacy (Judge et al., 2006). The results from this study indicate that incorporation of only a modest number of 2′-O-methyl modifications into an siRNA may be enough to abolish the unwanted immunostimulatory and off-target effects (Jackson et al., 2006a; Judge et al., 2006). The mechanism by which incorporation of 2′-O-methyl nucleotides can abolish the immunostimulatory activity of siRNAs is not well understood. However, it has been suggested that the modifications may disrupt recognition and binding of the siRNA duplex to immune receptors or proteins. It has also been proposed that the addition of natural nucleotide modifications to an siRNA may reduce the activation of TLRs that preferentially recognize pathogen-derived RNA, which contain fewer of these modifications (Karikó et al., 2005; Judge et al., 2006; Jackson and Linsley, 2010). Indeed, it has been demonstrated that the 2′-O-methyl modification can antagonize TLR7 (Robbins et al., 2007), providing support for this proposed mechanism. Therefore the addition of 2′-O-methyl modifications to siRNAs appears to inhibit both TLR-dependent and -independent IFN responses and prevents the recognition of siRNAs by the immune system. However, care must be exercised as extensive chemical modification to siRNAs can significantly reduce their silencing efficiency (Hornung et al., 2006; Judge et al., 2006; Judge and MacLaclan, 2008). As an alternative to 2′-O-methyl modifications, making minor structural changes to siRNAs, such as substituting uridine residues for thymidine or 2′-deoxyuridine, has also been shown to diminish the IFN response without affecting silencing activity (Eberle et al., 2008).
Dysregulation of gene expression and cytotoxicity can also occur as a result of saturating the endogenous RNA-silencing machinery. High-level expression of exogenous shRNAs can outcompete the endogenous miRNAs for cellular miRNA machinery such as exportin-5 and Ago1–Ago4, thereby disrupting miRNA biogenesis and endogenous RNA silencing (Yi et al., 2005; Davidson and Boudreau, 2007; Grimm et al., 2010). This can cause a global disruption in gene expression, which may contribute to the cytotoxicity observed in vitro and lethality in vivo after high-level shRNA expression in the CNS or liver (Grimm et al., 2006, 2010; McBride et al., 2008; Boudreau et al., 2009; Ehlert et al., 2010; Khodr et al., 2011; Martin et al., 2011). Therefore when using viral vector-mediated shRNA expression it is important to determine the lowest viral vector dose that provides efficient gene silencing while avoiding any nonspecific cellular toxicity, and it has been demonstrated that effective dose windows can be determined (Ulusoy et al., 2009; Grimm, 2011). In addition, the use of shRNA-miRs that are produced at lower levels than shRNAs and undergo full processing through the miRNA pathway, which may slow the rate at which they are loaded into the RISC, has been shown to mediate reduced competition with endogenous miRNAs and therefore reduced toxicity (Rao and Wilkinson, 2006; Castanotto et al., 2007; Boudreau et al., 2008, 2009; Maczuga et al., 2013). In addition to shRNAs, it has been reported that siRNA duplexes can also interrupt endogenous RNA silencing by competing with endogenous miRNAs for factors downstream of exportin-5 such as components of the RISC (Khan et al., 2009). Another strategy to reduce saturation of the endogenous miRNA machinery is to use weaker promoters, such as the H1 and 7SK promoters, which produce lower levels of shRNA expression and thereby mitigate the shRNA-meditated toxic affects without significantly reducing silencing efficiency (An et al., 2006; Grimm et al., 2010). Overexpressing specific components of the endogenous miRNA pathway, such as exportin-5 and Ago-2 that become saturated by high-level shRNA expression, can also alleviate the shRNA-mediated toxic effects; however, although effective in vitro, this would be impractical for long-term in vivo applications (Grimm et al., 2010).
These drawbacks have not significantly reduced the use of RNA silencing and studies have demonstrated that careful designing of siRNA sequences, the use of appropriate modifications and controls, and titrating viral vectors to a effective dose window can significantly reduce off-target effects (Jackson et al., 2003; Semizarov et al., 2003; Judge et al., 2006; Anderson et al., 2008; Grimm, 2011). Preclinical studies using viral vectors and/or RNAi strategies should also measure the regulation of key ISGs as an integral part of the experimental design. If this is not done, then one cannot determine whether or not any effects are the result of the desired (e.g., therapeutic) intervention rather than an ISG side effect.
Conclusions
Although there have been steady advancements in our knowledge and understanding of RNA silencing, issues still remain regarding the design, use, and side effects of shRNA-miRs, shRNAs, and siRNAs. However, the potential RNA silencing offers for studying gene function and as a novel therapeutic strategy for CNS disorders is undeniable, and as long as the appropriate design criteria, modifications, and controls are used, it can provide effective gene silencing and become a successful future therapeutic option.
Third-generation, integration-deficient lentiviral vectors are one of the leading gene therapy delivery systems for the CNS. Although some concerns regarding their immunogenicity remain, their large coding capacity and ability to efficiently and stably transduce postmitotic cells mean they are ideal mediators of RNA silencing for the CNS. Lentiviral vector-mediated RNA silencing therefore shows great promise as a future therapeutic strategy for CNS disorders.
Acknowledgments
The authors acknowledge support from a Research Councils U.K. Academic Fellowship (L.M.), the British Pharmacological Society's Integrative Pharmacology Fund (L.M.), Friends of Guy's Hospital Research Grants (L.M., R.J.Y.-M.), Biotechnology and Biological Sciences Research Council's Doctoral Training Grants (T.H. and E.F.), a grant from the Henry Smith Charity (L.M. and T.H.), financial support from the 7th EU Framework Programme (PERSIST Project, grant agreement no. 222878, R.J.Y.-M.), and a grant from Genoma España (R.J.Y.-M.).
Author Disclosure Statement
The authors declare that there are no conflicts of interest.
References
- Abordo-Adesida E., Follenzi A., Barcia C., et al. (2005). Stability of lentiviral vector-mediated transgene expression in the brain in the presence of systemic antivector immune responses. Hum. Gene Ther. 16, 741–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed Z., Dent R.G., Suggate E.L., et al. (2005). Disinhibition of neurotrophin-induced dorsal root ganglion cell neurite outgrowth on CNS myelin by siRNA-mediated knockdown of NgR, p75NTR and Rho-A. Mol. Cell Neurosci. 28, 509–523 [DOI] [PubMed] [Google Scholar]
- Alexopoulou L., Holt A.C., Medzhitov R., et al. (2001). Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413, 732–738 [DOI] [PubMed] [Google Scholar]
- Alves S., Nascimento-Ferreira I., Auregan G., et al. (2008). Allele-specific RNA silencing of mutant ataxin-3 mediates neuroprotection in a rat model of Machado-Joseph disease. PLoS One 3, e3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarzguioui M., and Prydz H. (2004). An algorithm for selection of functional siRNA sequences. Biochem. Biophys. Res. Commun. 316, 1050–1058 [DOI] [PubMed] [Google Scholar]
- An D.S., Qin F.X., Auyeung V.C., et al. (2006). Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors. Mol. Ther. 14, 494–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E.M., Birmingham A., Baskerville S., et al. (2008). Experimental validation of the importance of seed complement frequency to siRNA specificity. RNA 704–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle P.A., and Henkel T. (1994). Function and activation of NF-κB in the immune system. Annu. Rev. Immunol. 12, 141–179 [DOI] [PubMed] [Google Scholar]
- Bao L., Guo H., Huang X., et al. (2009). High-titer lentiviral vectors stimulate fetal calf serum-specific human CD4 T-cell responses: Implications in human gene therapy. Gene Ther. 16, 788–795 [DOI] [PubMed] [Google Scholar]
- Barks J.D., Nair M.P., Schwartz S.A., et al. (1993). Potentiation of N-methyl-d-aspartate-mediated brain injury by a human immunodeficiency virus-1-derived peptide in perinatal rodents. Pediatr. Res. 34, 192–198 [DOI] [PubMed] [Google Scholar]
- Bauer M., Kinkl N., Meixner A., et al. (2008). Prevention of interferon-stimulated gene expression using microRNA-designed hairpins. Gene Ther. 16, 142–147 [DOI] [PubMed] [Google Scholar]
- Baum C., and Fehse B. (2003). Mutagenesis by retroviral transgene insertion: Risk assessment and potential alternatives. Curr. Opin. Mol. Ther. 5, 458–462 [PubMed] [Google Scholar]
- Baum C., Kustikova O., Modlich U., et al. (2006). Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum. Gene Ther. 17, 253–263 [DOI] [PubMed] [Google Scholar]
- Behm-Ansmant I., Rehwinkel J., Doerks T., et al. (2006). mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 20, 1885–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentwich I., Avniel A., Karov Y., et al. (2005). Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 37, 766–770 [DOI] [PubMed] [Google Scholar]
- Bernstein E., Caudy A.A., Hammond S.M., et al. (2001). Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409, 363–366 [DOI] [PubMed] [Google Scholar]
- Bilanges B., and Stokoe D. (2005). Direct comparison of the specificity of gene silencing using antisense oligonucleotides and RNAi. Biochem. J. 388, 573–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birmingham A., Anderson E.M., Reynolds A., et al. (2006). 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat. Methods 3, 199–204 [DOI] [PubMed] [Google Scholar]
- Blomer U., Naldini L., Kafri T., et al. (1997). Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. J. Virol. 71, 6641–6649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau R.L., Monteys A.M., and Davidson B.L. (2008). Minimizing variables among hairpin-based RNAi vectors reveals the potency of shRNAs. RNA 14, 1834–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau R.L., Martins I., and Davidson B.L. (2009). Artificial microRNAs as siRNA shuttles: Improved safety as compared with shRNAs in vitro and in vivo. Mol. Ther. 17, 169–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braasch D.A., and Corey D.R. (2001). Locked nucleic acid (LNA): Fine-tuning the recognition of DNA and RNA. Chem. Biol. 8, 1–7 [DOI] [PubMed] [Google Scholar]
- Braun J.E., Huntzinger E., Fauser M., et al. (2011). GW182 proteins directly recruit cytoplasmic deadenylase complexes to miRNA targets. Mol. Cell 44, 120–133 [DOI] [PubMed] [Google Scholar]
- Bridge A.J., Pebernard S., Ducraux A., et al. (2003). Induction of an interferon response by RNAi vectors in mammalian cells. Nat. Genet. 34, 263–264 [DOI] [PubMed] [Google Scholar]
- Brown H.E., Chen H., and Engelman A. (1999). Structure based mutagenesis of the human immunodeficiency virus type 1 DNA attachment site: Effects on integration and cDNA synthesis. J. Virol. 73, 9011–9020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp T.R., Bernards R., and Agami R. (2002). A system for stable expression of short interfering RNAs in mammalian cells. Science 296, 550–553 [DOI] [PubMed] [Google Scholar]
- Burns J.C., Friedmann T., Driever W., et al. (1993). Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: Concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. U.S.A. 90, 8033–8037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cara A., Cereseto A., Lori F., et al. (1996). HIV-1 protein expression from synthetic circles of DNA mimicking the extrachromosomal forms of viral DNA. J. Biol. Chem. 271, 5393–5397 [DOI] [PubMed] [Google Scholar]
- Castanotto D., Sakurai K., Lingeman R., et al. (2007). Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC. Nucleic Acids Res. 35, 5154–5164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cekaite L., Furset G., Hovig E., et al. (2007). Gene expression analysis in blood cells in response to unmodified and 2′-modified siRNAs reveals TLR-dependent and independent effects. J. Mol. Biol. 365, 90–108 [DOI] [PubMed] [Google Scholar]
- Chalk A.M., Wahlestedt C., and Sonnhammer E.L. (2004). Improved and automated prediction of effective siRNA. Biochem. Biophys. Res. Commun. 319, 264–274 [DOI] [PubMed] [Google Scholar]
- Charneau P., Alizon M., and Clavel F. (1992). A second origin of DNA plus-strand synthesis is required for optimal human immunodeficiency virus replication. J. Virol. 66, 2814–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekulaeva M., Mathys H., Zipprich J.T., et al. (2011). miRNA repression involves GW182-mediated recruitment of CCR4-NOT through conserved W-containing motifs. Nat. Struct. Mol. Biol. 18, 1218–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chendrimada T.P., Gregory R.I., Kumaraswamy E., et al. (2005). TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436, 740–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen M., and Schratt G.M. (2009). microRNA involvement in developmental and functional aspects of the nervous system and in neurological diseases. Neurosci. Lett. 466, 55–62 [DOI] [PubMed] [Google Scholar]
- Craigie R. (2001). HIV integrase, a brief overview from chemistry to therapeutics. J. Biol. Chem. 276, 23213–23216 [DOI] [PubMed] [Google Scholar]
- Cullen B.R. (1991). Regulation of HIV-1 gene expression. FASEB J. 5, 2361–2368 [DOI] [PubMed] [Google Scholar]
- Cullen B.R. (2005). RNAi the natural way. Nat. Genet. 37, 1163–1165 [DOI] [PubMed] [Google Scholar]
- Cullen B.R. (2006). Induction of stable RNA interference in mammalian cells. Gene Ther. 13, 503–508 [DOI] [PubMed] [Google Scholar]
- Daly T.J., Cook K.S., Gray G.S., et al. (1989). Specific binding of HIV-1 recombinant Rev protein to the Rev-responsive element in vitro. Nature 342, 816–819 [DOI] [PubMed] [Google Scholar]
- Das A.T., Koken S.E., Essink B.B., et al. (1994). Human immunodeficiency virus uses tRNALys,3 as primer for reverse transcription in HeLa-CD4+ cells. FEBS Lett. 341, 49–53 [DOI] [PubMed] [Google Scholar]
- Davidson B.L., and Boudreau R.L. (2007). RNA interference: A tool for querying nervous system function and an emerging therapy. Neuron 53, 781–788 [DOI] [PubMed] [Google Scholar]
- Debouck C. (1991). Substrate specificity of the human (type 1) and simian immunodeficiency virus proteases. Adv. Exp. Med. Biol. 306, 407–415 [DOI] [PubMed] [Google Scholar]
- Delhaye S., Paul S., Blakqori G., et al. (2006). Neurons produce type I interferon during viral encephalitis. Proc. Natl. Acad. Sci. U.S.A. 103, 7835–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denli A.M., Tops B.B., Plasterk R.H., et al. (2004). Processing of primary microRNAs by the Microprocessor complex. Nature 432, 231–235 [DOI] [PubMed] [Google Scholar]
- Dickins R.A., Hemann M.T., Zilfou J.T., et al. (2005). Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat. Genet. 37, 1289–1295 [DOI] [PubMed] [Google Scholar]
- Diebold S.S., Massacrier C., Akira S., et al. (2006). Nucleic acid agonists for Toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur. J. Immunol. 36, 3256–3267 [DOI] [PubMed] [Google Scholar]
- Doench J.G., and Sharp P.A. (2004). Specificity of microRNA target selection in translational repression. Genes Dev. 18, 504–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G., Petersen C.P., and Sharp P.A. (2003). siRNAs can function as miRNAs. Genes Dev. 17, 438–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doss-Pepe E.W., Stenroos E.S., Johnson W.G., et al. (2003). Ataxin-3 interactions with rad23 and valosin-containing protein and its associations with ubiquitin chains and the proteasome are consistent with a role in ubiquitin-mediated proteolysis. Mol. Cell. Biol. 23, 6469–6483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T., Zufferey R., Kelly M., et al. (1998). A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72, 8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eacker S.M., Dawson T.M., and Dawson V.L. (2009). Understanding microRNAs in neurodegeneration. Nat. Rev. Neurosci. 10, 837–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle F., Giessler K., Deck C., et al. (2008). Modifications in small interfering RNA that separate immunostimulation from RNA interference. J. Immunol. 180, 3229–3237 [DOI] [PubMed] [Google Scholar]
- Ehlert E.M., Eggers R., Niclou S.P., et al. (2010). Cellular toxicity following application of adeno-associated viral vector-mediated RNA interference in the nervous system. BMC Neurosci. 18, 11–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir S.M., Harborth J., Lendeckel W., et al. (2001a). Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494.–498 [DOI] [PubMed] [Google Scholar]
- Elbashir S.M., Lendeckel W., and Tuschl T. (2001b). RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 15, 188.–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir S.M., Martinez J., Patkaniowska A., et al. (2001c). Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 20, 6877.–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir S.M., Harborth J., Weber K., et al. (2002). Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods 26, 199–213 [DOI] [PubMed] [Google Scholar]
- Engelman A. (1999). In vivo analysis of retroviral integrase structure and function. Adv. Virus Res. 52, 411–426 [DOI] [PubMed] [Google Scholar]
- Engels B.M., and Hutvagner G. (2006). Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene 25, 6163–6169 [DOI] [PubMed] [Google Scholar]
- Eulalio A., Huntzinger E., Nishihara T., et al. (2009a). Deadenylation is a widespread effect of miRNA regulation. RNA 15, 21.–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A., Tritschler F., and Izaurralde E. (2009b). The GW182 protein family in animal cells: New insights into domains required for miRNA-mediated gene silencing. RNA 15, 1433.–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian M.R., Cieplak M.K., Frank F., et al. (2011). miRNA-mediated deadenylation is orchestrated by GW182 through two conserved motifs that interact with CCR4-NOT. Nat. Struct. Mol. Biol. 18, 1211–1217 [DOI] [PubMed] [Google Scholar]
- Farnet C.M., and Haseltine W.A. (1991). Determination of viral proteins present in the human immunodeficiency virus type 1 preintegration complex. J. Virol. 65, 1910–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov Y., Anderson E.M., Birmingham A., et al. (2006). Off-target effects by siRNA can induce toxic phenotype. RNA 12, 1188–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipowicz W., Bhattacharyya S.N., and Sonenberg N. (2008). Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 9, 102–114 [DOI] [PubMed] [Google Scholar]
- Fischer U., Huber J., Boelens W.C., et al. (1995). The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell 82, 475–483 [DOI] [PubMed] [Google Scholar]
- Fish R.J., and Kruithof E.K. (2004). Short-term cytotoxic effects and long-term instability of RNAi delivered using lentiviral vectors. BMC Mol. Biol. 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiszer A., Mykowska A., Krzyzosiak W. J. (2011). Inhibition of mutant huntingtin expression by RNA duplex targeting expanded CAG repeats. Nucleic Acids Res. 39, 5578–5585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbach A., Nemorin J.G., Montino C., et al. (2008). Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. J. Immunol. 180, 3729–3738 [DOI] [PubMed] [Google Scholar]
- Ge Q., Dallas A., Ilves H., et al. (2010a). Effects of chemical modification on the potency, serum stability, and immunostimulatory properties of short shRNAs. RNA 16, 118.–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Q., Ilves H., Dallas A., et al. (2010b). Minimal-length short hairpin RNAs: The relationship of structure and RNAi activity. RNA 16, 106.–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez A.J., Mishima Y., Rihel J., et al. (2006). Zebrafish miR-430 promotes deadenylation and clearance of maternal mRNAs. Science 312, 75–79 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Alegre P., Bode N., Davidson B.L., et al. (2005). Silencing primary dystonia: Lentiviral-mediated RNA interference therapy for DYT1 dystonia. J. Neurosci. 25, 10502–10509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D. (2011). The dose can make the poison: Lessons learned from adverse in vivo toxicities caused by RNAi overexpression. Silence 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D., Streetz K.L., Jopling C.L., et al. (2006). Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441, 537–541 [DOI] [PubMed] [Google Scholar]
- Grimm D., Wang L., Lee J.S., et al. (2010). Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. J. Clin. Invest. 120, 3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S., Von Kalle C., Schmidt M., et al. (2003). LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302, 415–419 [DOI] [PubMed] [Google Scholar]
- Hammond S.M., Boettcher S., Caudy A.A., et al. (2001). Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 293, 1146–1150 [DOI] [PubMed] [Google Scholar]
- Han J., Lee Y., Yeom K.-H., et al. (2006). Molecular basis for the recognition of primary microRNAs by the Drosha–DGCR8 complex. Cell 125, 887–901 [DOI] [PubMed] [Google Scholar]
- Hornung V., Guenthner-Biller M., Bourquin C., et al. (2005). Sequence-specific potent induction of IFN-α by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 11, 263–270 [DOI] [PubMed] [Google Scholar]
- Hornung V., Ellegast J., Kim S., et al. (2006). 5′-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 [DOI] [PubMed] [Google Scholar]
- Horvath L., Van Marion, I., Tai K., et al. (2011). Knockdown of GAD67 protein levels normalizes neuronal activity in a rat model of Parkinson's disease. J. Gene Med. 13, 188–197 [DOI] [PubMed] [Google Scholar]
- Hu J., Matsui M., Gagnon K.T., et al. (2009). Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat. Biotechnol. 27, 478–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Liu J., and Corey D.R. (2010). Allele-selective inhibition of huntingtin expression by switching to an miRNA-like RNAi mechanism. Chem. Biol. 17, 1183–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson T.H., Foster E., Dawes J.M., et al. (2012a). Lentiviral vectors encoding short hairpin RNAs efficiently transduce and knockdown LINGO-1 but induce an interferon response and cytotoxicity in central nervous system neurones. J. Gene Med. 14, 299.–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson T.H., Verhaagen J., Yáñez-Muñoz R.J., et al. (2012b). Corticospinal tract transduction: A comparison of seven adeno-associated viral vector serotypes and a non-integrating lentiviral vector. Gene Ther. 19, 49.–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvagner G., and Simard M.J. (2008). Argonaute proteins: Key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 9, 22–32 [DOI] [PubMed] [Google Scholar]
- Jackson A.L., and Linsley P.S. (2010). Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 9, 57–67 [DOI] [PubMed] [Google Scholar]
- Jackson A.L., Bartz S.R., Schelter J., et al. (2003). Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 21, 635–637 [DOI] [PubMed] [Google Scholar]
- Jackson A.L., Burchard J., Leake D., et al. (2006a). Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA 12, 1197.–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson A.L., Burchard J., Schelter J., et al. (2006b). Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 12, 1179–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakymiw A., Lian S., Eystathioy T., et al. (2005). Disruption of GW bodies impairs mammalian RNA interference. Nat. Cell Biol. 7, 1267–1274 [DOI] [PubMed] [Google Scholar]
- Jankowski M.P., Cornuet P.K., Mcilwrath S., et al. (2006). SRY-box containing gene 11 (Sox11) transcription factor is required for neuron survival and neurite growth. Neuroscience 143, 501–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski M.P., Mcilwrath S.L., Jing X., et al. (2009). Sox11 transcription factor modulates peripheral nerve regeneration in adult mice. Brain Res. 1256, 43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge A., and MacLachlan I. (2008). Overcoming the innate immune response to small interfering RNA. Hum. Gene Ther. 19, 111–124 [DOI] [PubMed] [Google Scholar]
- Judge A.D., Sood V., Shaw J.R., et al. (2005). Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 23, 457–462 [DOI] [PubMed] [Google Scholar]
- Judge A.D., Bola G., Lee A.C., et al. (2006). Design of noninflammatory synthetic siRNA mediating potent gene silencing. Mol. Ther. 13, 494–505 [DOI] [PubMed] [Google Scholar]
- Julius D., and Basbaum A.I. (2001). Molecular mechanisms of nociception. Nature 413, 203–210 [DOI] [PubMed] [Google Scholar]
- Karikó K., Bhuyan P., Capodici J., et al. (2004). Exogenous siRNA mediates sequence-independent gene suppression by signaling through Toll-like receptor 3. Cells Tissues Organs 177, 132–138 [DOI] [PubMed] [Google Scholar]
- Karikó K., Buckstein M., Ni H., et al. (2005). Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 [DOI] [PubMed] [Google Scholar]
- Katze M.G., He Y., and Gale M. (2002). Viruses and interferon: A fight for supremacy. Nat. Rev. Immunol. 2, 675–687 [DOI] [PubMed] [Google Scholar]
- Kay M.A., Glorioso J.C., and Naldini L. (2001). Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 7, 33–40 [DOI] [PubMed] [Google Scholar]
- Kaye J.F., Richardson J.H., and Lever A.M. (1995). Cis-acting sequences involved in human immunodeficiency virus type 1 RNA packaging. J. Virol. 69, 6588–6592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen N.J., Gait M.J., and Karn J. (1996). Human immunodeficiency virus type-1 Tat is an integral component of the activated transcription–elongation complex. Proc. Natl. Acad. Sci. U.S.A. 93, 2505–2510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A.A., Betel D., Miller M.L., et al. (2009). Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat. Biotechnol. 27, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodr C.E., Sapru M.K., Pedapati J., et al. (2011). An α-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson's disease, but displays toxicity in dopamine neurons. Brain Res. 1395, 94–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khvorova A., Reynolds A., and Jayasena S.D. (2003). Functional siRNAs and miRNAs exhibit strand bias. Cell 115, 209–216 [DOI] [PubMed] [Google Scholar]
- Kim A.H., and Bonni A. (2007). Thinking within the D box: Initial identification of Cdh1-APC substrates in the nervous system. Mol. Cell. Neurosci. 34, 281–287 [DOI] [PubMed] [Google Scholar]
- Kim D.-H., Behlke M.A., Rose S.D., et al. (2005). Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat. Biotechnol. 23, 222–226 [DOI] [PubMed] [Google Scholar]
- Kim V.N., Mitrophanous K., Kingsman S.M., et al. (1998). Minimal requirement for a lentivirus vector based on human immunodeficiency virus type 1. J. Virol. 72, 811–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchner R., Vogtherr M., Limmer S., et al. (1998). Secondary structure dimorphism and interconversion between hairpin and duplex form of oligoribonucleotides. Antisense Nucleic Acid Drug Dev. 8, 507–516 [DOI] [PubMed] [Google Scholar]
- Kittler R., Surendranath V., Heninger A.K., et al. (2007). Genome-wide resources of endoribonuclease-prepared short interfering RNAs for specific loss-of-function studies. Nat. Methods 4, 337–344 [DOI] [PubMed] [Google Scholar]
- Kleinman M.E., Yamada K., Takeda A., et al. (2008). Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature 452, 591–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y., Stegmuller J., Matsuda T., et al. (2004). Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 303, 1026–1030 [DOI] [PubMed] [Google Scholar]
- Kosik K.S. (2006). The neuronal microRNA system. Nat. Rev. Neurosci. 7, 911–920 [DOI] [PubMed] [Google Scholar]
- Krichevsky A.M., King K.S., Donahue C.P., et al. (2003). A microRNA array reveals extensive regulation of microRNAs during brain development. RNA 9, 1274–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J., Loedige I., and Filipowicz W. (2010). The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 11, 597–610 [DOI] [PubMed] [Google Scholar]
- Krueger U., Bergauer T., Kaufmann B., et al. (2007). Insights into effective RNAi gained from large-scale siRNA validation screening. Oligonucleotides 17, 237–250 [DOI] [PubMed] [Google Scholar]
- Kulkarni M., Ozgur S., and Stoecklin G. (2010). On track with P-bodies. Biochem. Soc. Trans. 38, 242–251 [DOI] [PubMed] [Google Scholar]
- Kwon B.K., Liu J., Lam C., et al. (2007). Brain-derived neurotrophic factor gene transfer with adeno-associated viral and lentiviral vectors prevents rubrospinal neuronal atrophy and stimulates regeneration-associated gene expression after acute cervical spinal cord injury. Spine 32, 1164–1173 [DOI] [PubMed] [Google Scholar]
- Leavitt A.D., Robles G., Alesandro N., et al. (1996). Human immunodeficiency virus type 1 integrase mutants retain in vitro integrase activity yet fail to integrate viral DNA efficiently during infection. J. Virol. 70, 721–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y., Ahn C., Han J., et al. (2003). The nuclear RNase III Drosha initiates microRNA processing. Nature 425, 415–419 [DOI] [PubMed] [Google Scholar]
- Lee Y., Kim M., Han J., et al. (2004). MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23, 4051–4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y., Hur I., Park S.-Y., et al. (2006). The role of PACT in the RNA silencing pathway. EMBO J. 25, 522–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch J.K., Kuo F., Motti D., et al. (2012). Isoform diversity and regulation in peripheral and central neurons revealed through RNA-Seq. PLoS One 7, e30417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever A., Gottlinger H., Haseltine W., et al. (1989). Identification of a sequence required for efficient packaging of human immunodeficiency virus type 1 RNA into virions. J. Virol. 63, 4085–4087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis B.P., Shih I.H., Jones-Rhoades M.W., et al. (2003). Prediction of mammalian microRNA targets. Cell 115, 787–798 [DOI] [PubMed] [Google Scholar]
- Lewis B.P., Burge C.B., and Bartel D.P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20 [DOI] [PubMed] [Google Scholar]
- Li L., Olvera J.M., Yoder K.E., et al. (2001). Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. EMBO J. 20, 3272–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Lin X., Khvorova A., et al. (2007). Defining the optimal parameters for hairpin-based knockdown constructs. RNA 13, 1765–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Dullmann J., Schiedlmeier B., et al. (2002). Murine leukemia induced by retroviral gene marking. Science 296, 497. [DOI] [PubMed] [Google Scholar]
- Lingel A., Simon B., Izaurralde E., et al. (2003). Structure and nucleic-acid binding of the Drosophila Argonaute 2 PAZ domain. Nature 426, 465–469 [DOI] [PubMed] [Google Scholar]
- Liu J., Carmell M.A., Rivas F.V., et al. (2004). Argonaute2 is the catalytic engine of mammalian RNAi. Science 305, 1437–1441 [DOI] [PubMed] [Google Scholar]
- Liu J., Valencia-Sanchez M.A., Hannon G.J., et al. (2005). MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat. Cell Biol. 7, 719–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewen N., Leske D.A., Chen Y., et al. (2003). Comparison of wild-type and class I integrase mutant-FIV vectors in retina demonstrates sustained expression of integrated transgenes in retinal pigment epithelium. J. Gene Med. 5, 1009–1017 [DOI] [PubMed] [Google Scholar]
- Lombardi M.S., Jaspers L., Spronkmans C., et al. (2009). A majority of Huntington's disease patients may be treatable by individualized allele-specific RNA interference. Exp. Neurol. 217, 312–319 [DOI] [PubMed] [Google Scholar]
- Lv B., Yuan W., Xu S., et al. (2012). Lentivirus-siNgR199 promotes axonal regeneration and functional recovery in rats. Int. J. Neurosci. 122, 133–139 [DOI] [PubMed] [Google Scholar]
- Ma W., and Bisby M.A. (1998). Increased activation of nuclear factor κB in rat lumbar dorsal root ganglion neurons following partial sciatic nerve injuries. Brain Res. 797, 243–254 [DOI] [PubMed] [Google Scholar]
- Maczuga P., Lubelski J., van Logtenstein R., et al. (2013) Embedding siRNA sequences targeting Apolipoprotein B100 in shRNA and miRNA scaffolds results in differential processing and in vivo efficacy. Mol. Ther. 21, 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov S.S. (2000). NF-κB as a therapeutic target in chronic inflammation: Recent advances. Mol. Med Today 6, 441–448 [DOI] [PubMed] [Google Scholar]
- Malmberg A.B., Chen C., Tonegawa S., et al. (1997). Preserved acute pain and reduced neuropathic pain in mice lacking PKCγ. Science 278, 279–283 [DOI] [PubMed] [Google Scholar]
- Martin J.N., Wolken N., Brown T., et al. (2011). Lethal toxicity caused by expression of shRNA in the mouse striatum: Implications for therapeutic design. Gene Ther. 18, 666–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin W.J., Malmberg A.B., and Basbaum A.I. (2001). PKCγ contributes to a subset of the NMDA-dependent spinal circuits that underlie injury-induced persistent pain. J. Neurosci. 21, 5321–5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T., Planelles V., Krogstad P., et al. (1995). Genetic analysis of human immunodeficiency virus type 1 integrase and the U3 att site: Unusual phenotype of mutants in the zinc finger-like domain. J. Virol. 69, 6687–6696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T., Kuroda M.J., and Harada S. (1998). Specific and independent recognition of U3 and U5 att sites by human immunodeficiency virus type 1 integrase in vivo. J. Virol. 72, 8396–8402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride J.L., Boudreau R.L., Harper S.Q., et al. (2008). Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: Implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. U.S.A. 105, 5868–5873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels S., Schmidt-Erfurth U., and Rosenfeld P.J. (2006). Promising new treatments for neovascular age-related macular degeneration. Expert Opin. Investig. Drugs 15, 779–793 [DOI] [PubMed] [Google Scholar]
- Miyoshi H., Blomer U., Takahashi M., et al. (1998). Development of a self-inactivating lentivirus vector. J. Virol. 72, 8150–8157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore D.L., Blackmore M.G., Hu Y., et al. (2009). KLF family members regulate intrinsic axon regeneration ability. Science 326, 298–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey D.V., Lockridge J.A., Shaw L., et al. (2005). Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 23, 1002–1007 [DOI] [PubMed] [Google Scholar]
- Naldini L. (2011). Ex vivo gene transfer and correction for cell based therapies. Nat. Rev. Genet. 12, 301–315 [DOI] [PubMed] [Google Scholar]
- Naldini L., Blomer U., Gage F.H., et al. (1996a). Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. U.S.A. 93, 11382.–11388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L., Blomer U., Gallay P., et al. (1996b). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263.–267 [DOI] [PubMed] [Google Scholar]
- Nightingale S.J., Hollis R.P., Pepper K.A., et al. (2006). Transient gene expression by nonintegrating lentiviral vectors. Mol. Ther. 13, 1121–1132 [DOI] [PubMed] [Google Scholar]
- Paddison P.J., Caudy A.A., Bernstein E., et al. (2002). Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 16, 948–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pebernard S., and Iggo R.D. (2004). Determinants of interferon-stimulated gene induction by RNAi vectors. Differentiation 72, 103–111 [DOI] [PubMed] [Google Scholar]
- Peluffo H., Foster E., Ahmed S.G., et al. (2013). Efficient gene expression from integration-deficient lentiviral vectors in the spinal cord. Gene Ther. 20, 645–657 [DOI] [PubMed] [Google Scholar]
- Peng C., Ho B.K., Chang T.W., et al. (1989). Role of human immunodeficiency virus type 1-specific protease in core protein maturation and viral infectivity. J. Virol. 63, 2550–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persengiev S.P., Zhu X., and Green M.R. (2004). Non-specific, concentration-dependent stimulation and repression of mammalian gene expression by small interfering RNAs (siRNAs). RNA 10, 12–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister E.L., Kennington L., Staubhaar J., et al. (2009). Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington's disease patients. Curr. Biol. 19, 774–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe S., Sarkis C., Barkats M., et al. (2006). Lentiviral vectors with a defective integrase allow efficient and sustained transgene expression in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 103, 17684–17689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott N.J., and Thrasher A.J. (2007) Use of nonintegrating lentiviral vectors for gene therapy. Hum. Gene Ther. 18, 483–489 [DOI] [PubMed] [Google Scholar]
- Piedrahita D., Hernandez I., Lopez-Tobon A., et al. (2010). Silencing of CDK5 reduces neurofibrillary tangles in transgenic Alzheimer's mice. J. Neurosci. 30, 13966–13976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahim A.A., Wong A.M.S., Howe S.J., et al. (2009). Efficient gene delivery to the adult and fetal CNS using pseudotyped non-integrating lentiviral vectors. Gene Ther. 16, 509–520 [DOI] [PubMed] [Google Scholar]
- Ralph G.S., Radcliffe P.A., Day D.M., et al. (2005). Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med. 11, 429–433 [DOI] [PubMed] [Google Scholar]
- Ramezani A., and Hawley R.G. (2002). Overview of the HIV-1 lentiviral vector system. Curr. Protoc. Mol. Biol. Chapter 16, Unit 16.21 [DOI] [PubMed] [Google Scholar]
- Rao M.K., and Wilkinson M.F. (2006). Tissue-specific and cell type-specific RNA interference in vivo. Nat. Protoc. 1, 1494–1501 [DOI] [PubMed] [Google Scholar]
- Raoul C., Abbas-Terki T., Bensadoun J.C., et al. (2005). Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat. Med. 11, 423–428 [DOI] [PubMed] [Google Scholar]
- Read M.L., Mir S., Spice R., et al. (2009). Profiling RNA interference (RNAi)-mediated toxicity in neural cultures for effective short interfering RNA design. J. Gene Med. 11, 523–534 [DOI] [PubMed] [Google Scholar]
- Reynolds A., Leake D., Boese Q., et al. (2004). Rational siRNA design for RNA interference. Nat. Biotechnol. 22, 326–330 [DOI] [PubMed] [Google Scholar]
- Reynolds A., Anderson E.M., Vermeulen A., et al. (2006). Induction of the interferon response by siRNA is cell type- and duplex length-dependent. RNA 12, 988–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins M., Judge A., Liang L., et al. (2007). 2′-O-Methyl-modified RNAs act as TLR7 antagonists. Mol. Ther. 15, 1663–1669 [DOI] [PubMed] [Google Scholar]
- Rossi J.J. (2009). Innate Immunity confounds the clinical efficacy of small interfering RNAs (siRNAs). Gene Ther. 16, 579–580 [DOI] [PubMed] [Google Scholar]
- Saenz D.T., Loewen N., Peretz M., et al. (2004). Unintegrated lentivirus DNA persistence and accessibility to expression in nondividing cells: Analysis with class I integrase mutants. J. Virol. 78, 2906–2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel C.E. (2001). Antiviral actions of interferons. Clin. Microbiol. Rev. 14, 778–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapru M.K., Yates J.W., Hogan S., et al. (2006). Silencing of human α-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. Exp. Neurol. 198, 382–390 [DOI] [PubMed] [Google Scholar]
- Saxena S., Jonsson Z.O., and Dutta A. (2003). Small RNAs with imperfect match to endogenous mRNA repress translation: Implications for off-target activity of small inhibitory RNA in mammalian cells. J. Biol. Chem. 278, 44312–44319 [DOI] [PubMed] [Google Scholar]
- Scacheri P.C., Rozenblatt-Rosen O., Caplen N.J., et al. (2004). Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 101, 1892–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schambach A., Zychlinski D., Ehrnstroem B., et al. (2013). Biosafety features of lentiviral vectors. Hum. Gene Ther. 24, 132–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder A.R., Shinn P., Chen H., et al. (2002). HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110, 521–529 [DOI] [PubMed] [Google Scholar]
- Schwarz D.S., Hutvagner G., Du T., et al. (2003). Asymmetry in the assembly of the RNAi enzyme complex. Cell 115, 199–208 [DOI] [PubMed] [Google Scholar]
- Semizarov D., Frost L., Sarthy A., et al. (2003). Specificity of short interfering RNA determined through gene expression signatures. Proc. Natl. Acad. Sci. U.S.A. 100, 6347–6352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen G.L., and Blau H.M. (2005). Argonaute 2/RISC resides in sites of mammalian mRNA decay known as cytoplasmic bodies. Nat. Cell Biol. 7, 633–636 [DOI] [PubMed] [Google Scholar]
- Seredenina T., Gokce O., and Luthi-Carter R. (2011). Decreased striatal RGS2 expression is neuroprotective in Huntington's disease (HD) and exemplifies a compensatory aspect of HD-induced gene regulation. PLoS One 6, e22231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J.M., Li M.Z., Chang K., et al. (2005). Second-generation shRNA libraries covering the mouse and human genomes. Nat. Genet. 37, 1281–1288 [DOI] [PubMed] [Google Scholar]
- Singer O., Marr R.A., Rockenstein E., et al. (2005). Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat. Neurosci. 8, 1343–1349 [DOI] [PubMed] [Google Scholar]
- Siolas D., Lerner C., Burchard J., et al. (2005). Synthetic shRNAs as potent RNAi triggers. Nat. Biotechnol. 23, 227–231 [DOI] [PubMed] [Google Scholar]
- Sioud M. (2005). Induction of inflammatory cytokines and interferon responses by double-stranded and single-stranded siRNAs is sequence-dependent and requires endosomal localization. J. Mol. Biol. 348, 1079–1090 [DOI] [PubMed] [Google Scholar]
- Sledz C.A., Holko M., De Veer M.J., et al. (2003). Activation of the interferon system by short-interfering RNAs. Nat. Cell Biol. 5, 834–839 [DOI] [PubMed] [Google Scholar]
- Sledz C.A., and Williams B.R. (2004). RNA interference and double-stranded-RNA-activated pathways. Biochem. Soc. Trans. 32, 952–956 [DOI] [PubMed] [Google Scholar]
- Song J.J., Smith S.K., Hannon G.J., et al. (2004). Crystal structure of Argonaute and its implications for RISC slicer activity. Science 305, 1434–1437 [DOI] [PubMed] [Google Scholar]
- Stegmuller J., Konishi Y., Huynh M.A., et al. (2006). Cell-intrinsic regulation of axonal morphogenesis by the Cdh1-APC Target SnoN. Neuron 50, 389–400 [DOI] [PubMed] [Google Scholar]
- Stein B.S., and Engleman E.G. (1990). Intracellular processing of the gp160 HIV-1 envelope precursor: Endoproteolytic cleavage occurs in a cis or medial compartment of the Golgi complex. J. Biol. Chem. 265, 2640–2649 [PubMed] [Google Scholar]
- Stevenson M., Haggerty S., Lamonica C.A., et al. (1990). Integration is not necessary for expression of human immunodeficiency virus type 1 protein products. J. Virol. 64, 2421–2425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland I.T., Richards L., Holmes F.E., et al. (2011). Axotomy-induced miR-21 promotes axon growth in adult dorsal root ganglion neurons. PLoS One 6, e23423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudarsky L., and Coutinho P. (1995). Machado-Joseph disease. Clin. Neurosci. 3, 17–22 [PubMed] [Google Scholar]
- Suggate E.L., Ahmed Z., Read M.L., et al. (2009). Optimisation of siRNA-mediated RhoA silencing in neuronal cultures. Mol. Cell. Neurosci. 40, 451–462 [DOI] [PubMed] [Google Scholar]
- Sun T., Luo J., Jia M., et al. (2012). Small interfering RNA-mediated knockdown of NF-κBp65 attenuates neuropathic pain following peripheral nerve injury in rats. Eur. J. Pharmacol. 682, 79–85 [DOI] [PubMed] [Google Scholar]
- Tegeder I., Costigan M., Griffin R.S., et al. (2006). GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat. Med. 12, 1269–1277 [DOI] [PubMed] [Google Scholar]
- Themis M., Waddington S.N., Schmidt M., et al. (2005). Oncogenesis following delivery of a nonprimate lentiviral gene therapy vector to fetal and neonatal mice. Mol. Ther. 12, 763–771 [DOI] [PubMed] [Google Scholar]
- Thomas C.E., Ehrhardt A., and Kay M.A. (2003). Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 4, 346–358 [DOI] [PubMed] [Google Scholar]
- Tomari Y., Matranga C., Haley B., et al. (2004). A protein sensor for siRNA asymmetry. Science 306, 1377–1380 [DOI] [PubMed] [Google Scholar]
- Tuschl T., Zamore P.D., Lehmann R., et al. (1999). Targeted mRNA degradation by double-stranded RNA in vitro. Genes Dev. 13, 3191–3197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulusoy A., Sahin G., Björklund T., et al. (2009). Dose optimization for long-term rAAV-mediated RNA interference in the nigrostriatal projection neurons. Mol. Ther. 17, 1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valsamakis A., Zeichner S., Carswell S., et al. (1991). The human immunodeficiency virus type 1 polyadenylylation signal: A 3′ long terminal repeat element upstream of the AAUAAA necessary for efficient polyadenylylation. Proc. Natl. Acad. Sci. U.S.A. 88, 2108–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bilsen P.H., Jaspers L., Lombardi M.S., et al. (2008). Identification and allele-specific silencing of the mutant huntingtin allele in Huntington's disease patient-derived fibroblasts. Hum. Gene Ther. 19, 710–719 [DOI] [PubMed] [Google Scholar]
- Wang H., Sun H., Della Penna K., et al. (2002). Chronic neuropathic pain is accompanied by global changes in gene expression and shares pathobiology with neurodegenerative diseases. Neuroscience 114, 529–546 [DOI] [PubMed] [Google Scholar]
- Wang Y., Sheng G., Juranek S., et al. (2008). Structure of the guide-strand-containing argonaute silencing complex. Nature 456, 209–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanisch K., and Yáñez-Muñoz R.J. (2009). Integration-deficient lentiviral vectors: A slow coming of age. Mol. Ther. 17, 1316–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson D.J., Kobinger G.P., Passini M.A., et al. (2002). Targeted transduction patterns in the mouse brain by lentivirus vectors pseudotyped with VSV, Ebola, Mokola, LCMV, or MuLV envelope proteins. Mol. Ther. 5, 528–537 [DOI] [PubMed] [Google Scholar]
- Wong L.F., Azzouz M., Walmsley L.E., et al. (2004). Transduction patterns of pseudotyped lentiviral vectors in the nervous system. Mol. Ther. 9, 101–111 [DOI] [PubMed] [Google Scholar]
- Wu L., Fan J., and Belasco J.G. (2006). MicroRNAs direct rapid deadenylation of mRNA. Proc. Natl. Acad. Sci. U.S.A. 103, 4034–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. (2004). HIV-1 gene expression: Lessons from provirus and non-integrated DNA. Retrovirology 1, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. (2008). The second chance story of HIV-1 DNA: Unintegrated? Not a problem! Retrovirology 5, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H.S., Huang Q.H., Zhang F.X., et al. (2002). Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proc. Natl. Acad. Sci. U.S.A. 99, 8360–8365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T., and Morishita S. (2005). Accelerated off-target search algorithm for siRNA. Bioinformatics 21, 1316–1324 [DOI] [PubMed] [Google Scholar]
- Yáñez-Muñoz R.J., Balaggan K.S., MacNeil A., et al. (2006). Effective gene therapy with nonintegrating lentiviral vectors. Nat. Med. 12, 348–353 [DOI] [PubMed] [Google Scholar]
- Yi R., Qin Y., Macara I.G., et al. (2003). Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 17, 3011–3016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi R., Doehle B.P., Qin Y., et al. (2005). Overexpression of exportin 5 enhances RNA interference mediated by short hairpin RNAs and microRNAs. RNA 11, 220–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore P.D. (2001). RNA interference: Listening to the sound of silence. Nat. Struct. Biol. 8, 746–750 [DOI] [PubMed] [Google Scholar]
- Zeng Y., and Cullen B.R. (2004). Structural requirements for pre-microRNA binding and nuclear export by Exportin 5. Nucleic Acids Res. 32, 4776–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y., and Cullen B.R. (2005). Efficient processing of primary microRNA hairpins by Drosha requires flanking nonstructured RNA sequences. J. Biol. Chem. 280, 27595–27603 [DOI] [PubMed] [Google Scholar]
- Zeng Y., Wagner E.J., and Cullen B.R. (2002). Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol. Cell 9, 1327–1333 [DOI] [PubMed] [Google Scholar]
- Zeng Y., Yi R., and Cullen B.R. (2003). MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc. Natl. Acad. Sci. U.S.A. 100, 9779–9784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y., Cai X., and Cullen B.R. (2005). Use of RNA polymerase II to transcribe artificial microRNAs. Methods Enzymol. 392, 371–380 [DOI] [PubMed] [Google Scholar]
- Zennou V., Petit C., Guetard D., et al. (2000). HIV-1 genome nuclear import is mediated by a central DNA flap. Cell 101, 173–185 [DOI] [PubMed] [Google Scholar]
- Zou H., Ho C., Wong K., et al. (2009). Axotomy-induced Smad1 activation promotes axonal growth in adult sensory neurons. J. Neurosci. 29, 7116–7123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W., Song Z., Guo Q., et al. (2011). Intrathecal lentiviral-mediated RNA interference targeting PKCγ attenuates chronic constriction injury-induced neuropathic pain in rats. Hum. Gene Ther. 22, 465–475 [DOI] [PubMed] [Google Scholar]
- Zufferey R., Dull T., Mandel R.J., et al. (1998). Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 72, 9873–9880 [DOI] [PMC free article] [PubMed] [Google Scholar]