

The importance of incorporating fluorinated subunits into organic molecules is well established in the pharmaceutical, agrochemical, and materials industries.1,2 Developing methods of incorporating such fluorinated moieties into organic substrates in a safe, selective, and facile manner, therefore, has been an area of great interest.3 Among the various fluorinated moieties, the strongly electron-withdrawing trifluoromethyl group (CF3) has garnered much attention because of the profound properties it can effect upon being implanted within a molecule.4 However, most of the synthetic efforts have been focused on introducing CF3 in functionalized arenes.5
By contrast, general methods for the incorporation of CF3 into alkyl systems is less well developed,6–8 yet increasingly more important in terms of structural diversity and expanding chemical space.9 β-Trifluoroethyl carbanion synthons represent an attractive set of reagents for the installation of fluorinated substructures within the realm of alkyl building blocks. However, owing to a strong driving force for the formation of metal-fluorine (M–F) bonds, the propensity for β-elimination of M–F moieties is high (eq 1), and thus the chemistry of β-trifluoroethyl carbanion synthons is astonishingly limited and underdeveloped.10
 |
(1) |
Although Brown and Ramachandran have previously generated α-trifluoromethylated organoboranes via a hydroboration route,11 these intermediates were never isolated but rather simply oxidized in situ.12 With the boron oxidized to the corresponding alcohol, the considerable synthetic value of the trifluoroethyl subunit in subsequent transformations is diminished. Unlike the organoborons, the alcohol cannot function as a bench-stable trifluoroethyl anion or trifluoroethyl radical precursor, and the versatility characteristic of the organoborons in further functionalization (e.g., C–C bond formation via metal-catalyzed cross-coupling13 or radical reactions,14 Rh-catalyzed 1,2-additions,15 amination,16 etc.) is lost.
Herein, we report the synthesis of diverse libraries of indefinitely bench-stable trifluoromethylated building blocks, which can be further diversified through versatile organoboron transformations. This has the potential to improve the existing paradigm for the introduction of CF3 at sp3 centers, which is limited mostly to CF3-based reagents such as Me3SiCF3 that minimally enhance molecular complexity. Our strategy was to utilize trifluoroethylidene in conjunction with tricoordinate organoborons to generate unprecedented α-trifluoromethylated organoborons through an established α-transfer mechanism (Scheme 1).
Scheme 1.
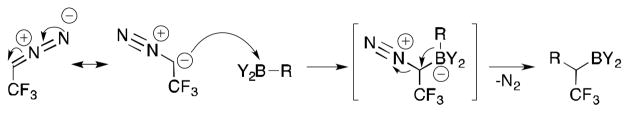
α-Transfer Mechanism
The synthesis of 2,2,2-trifluorodiazoethane (CF3CHN2) from the corresponding ammonium salt was first described in the 1940s and was reported on a scale as large as 100–200 mmols.17 However, CF3CHN2 was not used extensively in organic synthesis until the Carreira group developed a method to generate the material and react it in situ with other organic compounds. As demonstrated by the Carreira group, CF3CHN2 has a similar reactivity profile to that of ethyl diazoacetate.18 The reactivity of CF3CHN2 toward organoboron compounds, however, has never been explored, although several organoborons have been shown to react with ethyl diazoacetate and other α-diazocarbonyl compounds to give α-arylated, -vinylated, or -alkylated carbonyl compounds after protodeboronation.19 Unlike the reactions of organoborons with diazo compounds such as ethyl diazoacetate, where an enol boronate is formed,19 the B-C bond in the present process was expected to remain intact after reaction with CF3CHN2, giving rise to unprecedented organoboron compounds bearing an α-trifluoromethyl substituent.
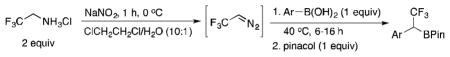
Following Carreira’s method,18 stock solutions of CF3CHN2 in several organic solvents (heptanes, toluene, dichloromethane and chlorobenzene) at varying concentrations (0.1–1 M) were prepared in 75–90% yield. With these stock solutions of CF3CHN2 in hand, their reactivity with various boron species was investigated. Initial attempts were made with commercially available aryl pinacol boronates. No reactivity was observed with these substrates, and the starting materials were fully recovered. The lack of reactivity may be explained by the low Lewis acidity of boronate esters.20 With boronic acids, reactivity was observed in various solvents and at different temperatures. After optimization of the reaction conditions, the desired α-trifluoromethylated organoborons were detected in good yields by 1H NMR after quenching the reaction mixtures with pinacol (Table 1).
Table 1.
Reactions of 2,2,2-Trifluorodiazoethane with Boronic Acids
 1a
1a


Yields based on 1H NMR of the crude mixture.
Isolated yield
Although the crude 1H NMR yields of the desired products were good, in most cases (especially when using electron poor boronic acids) the α-trifluoromethylated pinacol boronates were prone to oxidation during purification using silica gel chromatography. In certain cases, simple exposure to air at room temperature led to the corresponding alcohols, and the isolated yields suffered drastically. Conversion of the trifluoromethylated, tricoordinate boronic acids to the more stable tetracoordinate potassium organotrifluoroborates by quenching the crude mixture with KHF2 led to mixtures, and the desired products could not be isolated in high yields after successive recrystallizations.
Further to the aforementioned purification problems, the use of boronic acids as limiting reagents in reaction with 2,2,2-trifluorodiazoethane became rapidly unappealing for other reasons. Along with the well-known instability of some classes of boronic acids when exposed to air even at low temperatures,21 their equilibrium with cyclic boroxines also leads to an uncertain stoichiometry. Further, boronic acids and boroxines were reported to have different Lewis acidities and consequently different reactivity rates toward the diazo compounds.19b
The use of potassium organotrifluoroborates (RBF3K) as starting materials was envisioned as a more favorable alternative to boronic acids, because of their precise stoichiometry and excellent stability across all classes of substrates (alkyl, alkenyl, alkynyl, aryl and heteroaryls). Vedejs22a and Matteson22b have shown that potassium organotrifluoroborates can be converted to dihaloboranes (RBX2, X = F or Cl) upon treatment with TMSCl or SiCl4, respectively. After screening a variety of silicon sources, solvents, and temperatures, the use of CH2Cl2 at room temperature or toluene at 40 °C, with TMSCl or p-tolylSiCl3 as a fluorophile proved optimal (Table 2). CH2Cl2 and toluene often provided similar results, but the conversion in toluene was slower than in CH2Cl2 at room temperature. Therefore, a slight increase in temperature for the toluene reactions (40 °C) was necessary to achieve high conversions. During the optimization, it was observed that all reagents could be added at once, which obviated the step of preforming RBF2 prior to the addition of the diazo solutions and facilitated the experimental setup.
Table 2.
Substrate Scope of Potassium Organotrifluoroborates.

| ||||
|---|---|---|---|---|
| substrate | product | solvent | [Si] | isolated yield (%) |

|
 2a
2a
|
CH2Cl2 | TMSCl | 71 |
|
|
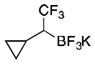 2b
2b
|
toluene | TMSCl | 80 |
|
|
 2c
2c
|
toluene | TMSCl | 78 |
|
|
|
CH2Cl2 | p-tolylSiCl3 | 78 |
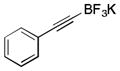
|
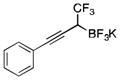 2e
2e
|
CH2Cl2 | TMSCl | 92 |

|
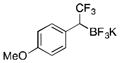 2f
2f
|
CH2Cl2 | p-tolylSiCl3 | 71(67)a |
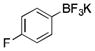
|
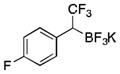 2g
2g
|
CH2Cl2 | TMSCl | 72 |
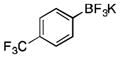
|
 2h
2h
|
toluene | p-tolylSiCl3 | 83 |

|
 2i
2i
|
CH2Cl2 | TMSCl | 70 |
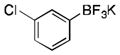
|
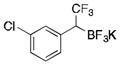 2j
2j
|
CH2Cl2 | p-tolylSiCl3 | 71 |
|
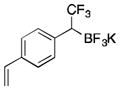 2k
2k
|
CH2Cl2 | TMSCl | 89 |

|
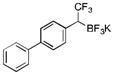 2l
2l
|
CH2Cl2 | p-tolylSiCl3 | 65 |
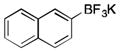
|
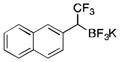 2m
2m
|
CH2Cl2 | TMSCl | 88 |
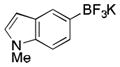
|
 2n
2n
|
CH2Cl2 | TMSCl | 90 |

|
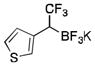 2o
2o
|
CH2Cl2 | TMSCl | 72 |

|
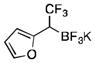 2p
2p
|
toluene | TMSCl | 86 |
Reaction performed on a 10 mmol scale
To avoid the oxidation problem mentioned in reference to Table 1, the crude mixture was quenched with KHF2, and the desired products were purified in good to excellent yields as tetracoordinate potassium trifluoroborates. The substrate scope of the transformation is extremely broad, and the purification only requires hot acetone extraction and recrystallization, avoiding the use of column chromatography. Primary and secondary alkyl trifluoroborates reacted in high yields (2a–2c). Because of the importance of allylic and propargylic organoborons in organic synthesis,23 our attention then focused toward alkenyl and alkynyl potassium trifluoroborates as starting materials. These two classes of substrates smoothly underwent the reaction with 2,2,2-trifluorodiazoethane, leading to novel allylic and propargylic α-trifluoromethylated trifluoroborates that are indefinitely stable on the bench top (2d, 2e). No borotropic shift was observed when an alkenyltrifluoroborate was used, and no allenylboron was detected when starting with alkynyltrifluoroborate. The reaction of various aryltrifluoroborates was then investigated, and the desired α-trifluoromethylated benzylic products were obtained in good to excellent yields in all cases. Various functional groups such as ethers (2f), nitriles (2i), halides (2j), and olefins (2k) were tolerated under the reaction conditions, and both electron-donating and electron-withdrawing groups could be present on the aryltrifluoroborates without affecting the yields. Important heterocyclic systems such as indole (2n), thiophene (2o), and furan (2p) were also successfully converted.
All these classes of α-trifluoromethylated products isolated as trifluoroborates have been kept on the bench for over 6 months without any sign of decomposition. These trifluoroborates represent the first indefinitely stable α-trifluoromethylated alkylborons.
To demonstrate the potential value of the α-trifluoromethylated organoborons in synthesis, preliminary studies for the functionalization of the carbon-boron bond were performed. Reactions were carried out in situ on the α-trifluoromethylated, tricoordinate organoboron species. Oxidation was performed by quenching the crude reaction mixture with pinacol, followed by treatment with NaOH/H2O2 (eq 2). This method is complementary to the synthesis of α-trifluoromethyl alcohols from aldehydes and the expensive Ruppert-Prakash reagent24 or the reduction of trifluoromethyl ketones, which have limited commercial availability.25
 |
(2) |
Methods to prepare benzylic α-trifluoromethyl bromides involve multistep syntheses, and require the isolation of the α-trifluoromethylated alcohol intermediates, followed by treatment with stoichiometric amounts of NBS and triphenylphosphite, leading to low to moderate overall yields.26 Using a one pot procedure, starting from the commercially available potassium p-methoxyphenyltrifluoroborate, the α–trifluoromethyl benzyl bromide was prepared in 51% yield without any further optimization (Table 3, 3b). Interestingly, when alkenyltrifluoroborates were used as starting materials, rearrangement occurred to provide the secondary allylic bromide and chloride in a regio- and stereodefined manner (Table 3, 3c and 3d). To our best knowledge, this class of trifluoromethylated secondary allyl halides has never reported. Previous literature reports only describe isolated multistep syntheses of a few primary trifluoromethylated allyl bromides and chlorides, along with their potential applications in organic transformations (SN2, addition to carbonyls).27
Table 3.
Halogenation of α-Trifluoromethyl Organoborons.
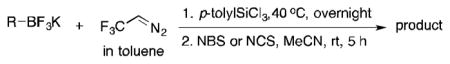
| ||
|---|---|---|
| substrate | product | isolated yield (%) |

|
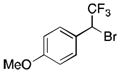 3b
3b
|
51 |
|
|
|
60 |
|
|
|
61 |
Protodeboronation of the crude α-trifluoromethylated pinacol boronate was smoothly achieved using Aggarwal’s conditions (eq 3).28 Unlike any existing procedures,29 this transformation provides an approach to metal-free trifluoroethylation of a variety of organic substructures. Thus, in addition to avoiding the transition-metal waste, the substrate scope of the trifluoroborate starting materials is very broad (as shown in Table 2), potentially allowing the introduction of CF3CH2 on primary and secondary alkyls, alkenyls, alkynyls, aryls and heteroryls via the same general experimental procedure.
 |
(3) |
β-Fluoride elimination is a major unresolved problem when a trifluoromethyl group and a metal are situated on the same C(sp3). For an α-trifluoromethylated organolithium or Grignard reagent, β-fluoride elimination occurs instantaneously unless strongly electron-withdrawing groups are introduced to stabilize the trifluoroethyl anions at very low temperatures.10 The α-trifluoromethylated dihaloboranes reported herein could be heated to 40 °C under Ar without any sign of β-F elimination. However, it was observed that a controlled elimination could be induced by increasing the temperature to 75 °C, wherein β-F elimination occurred to form substituted 1,1-difluoro-1,3-butadienes30 (3f). Although this transformation can be applied to aryl trifluoroborates to synthesize difluorostyrenes, higher yield and selectivity were obtained with an alkenyl trifluoroborate under these conditions (eq 4).
 |
(4) |
Finally, to prove another advantage of retaining the boron intact after the trifluorodiazoethane insertion, a reaction with a second diazo compound was performed. When ethyl diazoacetate (EDA) was added in situ to the α-trifluoromethylated dihaloborane, the “double diazo” insertion product was obtained in an overall (unoptimized) yield of 43%. After the insertion of EDA, the resulting boron enolate is rapidly protonated, leading directly to the observed ester.19 It is important to note that this approach represents a complementary method to access the same class of molecules that are obtained by the addition of the Ruppert-Prakash reagent to α,β-unsaturated esters (eq 5),31 with the potential to provide access to greater structural diversity as well: the range of organoboron precursors available far exceeds that of the requisite α,β-unsaturated carbonyl substrates required for the conjugate addition reaction of the Ruppert reagent.
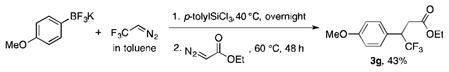 |
(5) |
In conclusion, a family of shelf-stable α-trifluoromethylated organoborons has been generated for the first time. Not only is the method for generating these materials operationally simple and effective, but it can also be carried out without resorting to metal mediation. Numerous carbon-carbon and carbon-heteroatom bond-forming transformations appear to be feasible with these reagents, employing well-known reactivity patterns of organoborons. These reagents thus represent potentially powerful building blocks for selective incorporation of the CF3 unit at sp3 carbon centers in complex molecules.
Experimental Section
Potassium organotrifluoroborate (1 mmol) was added to a 20 mL Biotage microwave vial equipped with a stir bar. The vial was sealed and purged with argon three times. The solution of 2,2,2-trifluorodiazoethane in CH2Cl2 (~0.5 M, 4 mL) or toluene (~0.5 M, 4 mL) was added under Ar, then freshly distilled Me3SiCl (120 mg, 1.1 mmol) or p-tolylSiCl3 (248 mg, 1.1 mmol) was added, and the reaction was stirred at room temperature (in CH2Cl2) or 40 °C (in toluene) overnight. The pressure was vented under Ar pressure. The reaction was cooled to 0 °C, then a saturated solution of KHF2 (1 mL, 4.5 M in H2O) was added dropwise under Ar. Acetone (3 mL) was added to increase the solubility and improve the stirring of the reaction. The reaction was allowed to warm to rt and stirred for an additional 30 min under argon. The solvent was evaporated from the crude reaction mixture, and the product was extracted into dry acetone to eliminate the inorganic salts. The acetone was evaporated and the desired product was recrystallized from the crude mixture.
Supplementary Material
Acknowledgments
We thank Frontier Scientific for boronic acids and potassium organotrifluoroborates. Financial support has been provided by NIH (NIGMS R01 GM035249). Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining HRMS data. J.-B. Coty (University of Pennsylvania) is acknowledged for repeating the experiment using p-methoxyphenyltrifluoroborate (2f) and running the experiment using p-trifluoromethylphenyl-trifluoroborate (2h).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c.30–40% of agrochemicals and 20–25% of pharmaceuticals on the market are estimated to contain fluorine: Thayer AM. Chem Eng News. 2006;84:15–24.
- 2.Scheirs J. Modern Fluoropolymers: High Performance Polymers for Diverse Applications. Wiley; Chichester: 1997. [Google Scholar]
- 3.Gouverneur V, Lozano O. In: Science of Synthesis, Stereoselective Syntheses. De Vries JG, Molander GA, Evans PA, editors. Vol. 3. Thieme; Stuttgart: 2011. pp. 851–930. [Google Scholar]
- 4.Yamazaki T, Taguchi T, Ojima I. In: Fluorine in Medicinal Chemistry and Chemical Biology. Ojima I, editor. Wiley-Blackwell; Chichester: 2009. pp. 1–46. [Google Scholar]
- 5.a) Tomashenko OA, Grushin V. Chem Rev. 2011;111:4475–4521. doi: 10.1021/cr1004293. [DOI] [PubMed] [Google Scholar]; b) Ye Y, Sanford M. Synlett. 2012;23:2005–2013. doi: 10.1055/s-0032-1316988. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Besset T, Schneider C, Cahard D. Angew Chem. 2012;124:5134–5136. doi: 10.1002/anie.201201012. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2012;51:5048–5050. doi: 10.1002/anie.201201012. [DOI] [PubMed] [Google Scholar]
- 6.Allylic and benzylic trifluoromethylations to afford C(sp3)–CF3 bonds: Parsons AT, Buchwald SL. Angew Chem. 2011;123:9286–9289.Angew Chem Int Ed. 2011;50:9120–9123. doi: 10.1002/anie.201104053.Xu J, Fu Y, Luo DF, Jiang YY, Xiao B, Liu ZJ, Gong TJ, Liu L. J Am Chem Soc. 2011;133:15300–15303. doi: 10.1021/ja206330m.Wang X, Ye Y, Zhang S, Feng J, Xu Y, Zhang Y, Wang J. J Am Chem Soc. 2011;133:16410–16413. doi: 10.1021/ja207775a.Kawai H, Furukawa T, Nomura Y, Tokunga E, Shibata N. Org Lett. 2011;13:3596–3599. doi: 10.1021/ol201205t.Shimizu R, Egami H, Hamashima Y, Sodeoka M. Angew Chem. 2012;124:4655–4658. doi: 10.1002/anie.201201095.Angew Chem Int Ed. 2012;51:4577–4580. doi: 10.1002/anie.201201095.Mizuta S, Galicia-Lopez O, Engle KM, Verhoog S, Wheelhouse K, Rassias G, Gouverneur V. Chem Eur J. 2012;18:8583–8587. doi: 10.1002/chem.201201707.Zhu L, Liu S, Douglas JT, Altman RA. Chem Eur J. 2013 doi: 10.1002/chem.201302328.Chu L, Qing FL. Org Lett. 2012;14:2106–2109. doi: 10.1021/ol300639a.
- 7.α- or β-Trifluoromethylated carbonyl compounds containing C(sp3)–CF3 bonds: Pham PV, Nagib DA, MacMillan DWC. Angew Chem. 2011;123:6243–6246. doi: 10.1002/anie.201101861.Angew Chem Int Ed. 2011;50:6119–6122. doi: 10.1002/anie.201101861.Herrmann AT, Smith LL, Zakarian A. J Am Chem Soc. 2012;134:6976–6979. doi: 10.1021/ja302552e.Deng QH, Wadepohl H, Gade LH. J Am Chem Soc. 2012;134:10769–10772. doi: 10.1021/ja3039773.Matousek V, Togni A, Bizet V, Cahard D. Org Lett. 2011;13:5762–5765. doi: 10.1021/ol2023328.Liu X, Xiong F, Huang X, Xu L, Li P, Wu X. Angew Chem. 2013;125:7100–7104. doi: 10.1002/anie.201302673.Angew Chem Int Ed. 2013;52:6962–6966. doi: 10.1002/anie.201302673.Chen ZM, Bai W, Wang SH, Yang BM, Tu YQ, Zhang FM. Angew Chem. 2013;125:9963–9967. doi: 10.1002/anie.201304557.Angew Chem Int Ed. 2013;52:9781–9785. doi: 10.1002/anie.201304557.
- 8.General methods for the formation of C(sp3)–CF3 bonds: Xu J, Xiao B, Xie CQ, Luo DF, Liu L, Fu Y. Angew Chem. 2012;124:12719–12722. doi: 10.1002/anie.201206681.Angew Chem Int Ed. 2012;51:12551–12554. doi: 10.1002/anie.201206681.Mizuta S, Verhoog S, Engle KM, Khotavivattana T, O’Duill M, Wheelhouse K, Rassias G, Medebielle M, Gouverneur V. J Am Chem Soc. 2013;135:2505–2508. doi: 10.1021/ja401022x.Wilger DJ, Gesmundo NJ, Nicewicz DA. Chem Sci. 2013;4:3160–3165.Wu X, Chu L, Qing FL. Angew Chem Int Ed. 2013;52:2198–2202. doi: 10.1002/anie.201208971.
- 9.Lovering F, Bikker J, Humblet C. J Med Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 10.Uneyama K, Katagiri T, Amii H. Acc Chem Res. 2008;41:817–829. doi: 10.1021/ar7002573. [DOI] [PubMed] [Google Scholar]
- 11.a) Brown HC, Chen GM, Jenning MP, Ramachandran PV. Angew Chem. 1999;111:2088–2090. [Google Scholar]; Angew Chem Int Ed. 1999;38:2052–2054. [Google Scholar]; b) Ramachandran PV, Jenning MP. Org Lett. 2001;3:3789–3790. doi: 10.1021/ol016779e. [DOI] [PubMed] [Google Scholar]
- 12.To our best knowledge, 1-methyl-2,2,2-trifluoroethyl pinacolborane is the only α-CF3 alkylboron reported and fully characterized. However, the compound was obtained in low yeids (4–11%), in an inseparable mixture with other products: Braun T, Solomon MA, Altenhöner K, Teltevskoi M, Hinze S. Angew Chem. 2009;121:1850–1854. doi: 10.1002/anie.200805041.Angew Chem Int Ed. 2009;48:1818–1822. doi: 10.1002/anie.200805041.
- 13.a) Sandrock DL. In: Science of Synthesis, Cross Coupling and Heck-Type Reactions. Molander GA, Wolfe JP, Larhed M, editors. Vol. 1. Thieme; Stuttgart: 2013. pp. 323–358. [Google Scholar]; b) Imao D, Glasspoole BW, Laberge VS, Crudden CM. J Am Chem Soc. 2009;131:5024–5025. doi: 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]; c) Yang C-T, Zhang Z-Q, Tajuddin H, Wu C-C, Liang J, Liu J-H, Fu Y, Czyzewska M, Steel PG, Marder TB, Liu L. Angew Chem Int Ed. 2012;51:528–532. doi: 10.1002/anie.201106299. [DOI] [PubMed] [Google Scholar]
- 14.a) Molander GA, Colombel V, Braz V. Org Lett. 2011;13:1852–1855. doi: 10.1021/ol2003572. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sorin G, Martinez MR, Contie Y, Baralle A, Malacria AM, Goddard JP, Fensterbank L. Angew Chem. 2010;122:8903–8905. doi: 10.1002/anie.201004513. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:8721–8723. doi: 10.1002/anie.201004513. [DOI] [PubMed] [Google Scholar]
- 15.a) Ros A, Aggarwal VK. Angew Chem. 2009;121:6407–6410. [Google Scholar]; Angew Chem, Int Ed. 2009;48:6289–6292. doi: 10.1002/anie.200901900. [DOI] [PubMed] [Google Scholar]; b) Zhang C, Yun J. Org Lett. 2013;15:3416–3419. doi: 10.1021/ol401468v. [DOI] [PubMed] [Google Scholar]
- 16.a) Mlynarski SN, Karns AS, Morken JP. J Am Chem Soc. 2012;134:16449–16451. doi: 10.1021/ja305448w. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Matteson DS, Kim GY. Org Lett. 2002;4:2153–2155. doi: 10.1021/ol025973d. [DOI] [PubMed] [Google Scholar]; b) Bagutski V, Elford TG, Aggarwal VK. Angew Chem. 2011;123:1112–1115. doi: 10.1002/anie.201006037. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:1080–1083. doi: 10.1002/anie.201006037. [DOI] [PubMed] [Google Scholar]; c) Brown HC, Kim KW, Cole TE, Singaram B. J Am Chem Soc. 1986;108:6761–6764. [Google Scholar]
- 17.Gilman H, Jones RG. J Am Chem Soc. 1943;65:1458–1460. [Google Scholar]
- 18.a) Morandi B, Carreira EM. Angew Chem. 2010;122:950–953. [Google Scholar]; Angew Chem Int Ed. 2010;49:938–941. [Google Scholar]; b) Morandi B, Carreira EM. Angew Chem. 2010;122:4390–4392. [Google Scholar]; Angew Chem Int Ed. 2010;49:4294–4296. doi: 10.1002/anie.201000787. [DOI] [PubMed] [Google Scholar]; c) Morandi B, Carreira EM. Angew Chem. 2011;123:9251–9254. [Google Scholar]; Angew Chem Int Ed. 2011;50:9085–9088. doi: 10.1002/anie.201103526. [DOI] [PubMed] [Google Scholar]; d) Morandi B, Mariampillai B, Carreira EM. Angew Chem. 2011;123:1133–1136. doi: 10.1002/anie.201004269. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:1101–1104. doi: 10.1002/anie.201004269. [DOI] [PubMed] [Google Scholar]
- 19.a) Hooz J, Bridson JN, Calzada JG, Brown HC, Midland MM, Levy AB. J Org Chem. 1973;38:2574–2576. [Google Scholar]; b) Peng C, Zhang W, Yan G, Wang J. Org Lett. 2009;11:1667–1670. doi: 10.1021/ol900362d. [DOI] [PubMed] [Google Scholar]
- 20.Bawn CEH, Ledwith A, Matthies P. J Polym Sci. 1959;34:93–106. [Google Scholar]
- 21.Knapp DM, Gillis EP, Burke MD. J Am Chem Soc. 2009;131:6961–6963. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Vedejs E, Chapman RW, Fields SC, Lin S, Schrimpf MR. J Org Chem. 1995;60:3020–3027. [Google Scholar]; b) Kim BJ, Matteson DS. Angew Chem. 2004;116:3118–3120. [Google Scholar]; Angew Chem Int Ed. 2004;43:3056–3058. doi: 10.1002/anie.200453690. [DOI] [PubMed] [Google Scholar]
- 23.a) Lachance H, Hall DG. In: Organic Reactions. Denmark SE, editor. Vol. 73. John Wiley & Sons; Hoboken: 2008. [Google Scholar]; b) Hesse MJ, Butts CP, Willis CL, Aggarwal VK. Angew Chem. 2012;124:12612–12616. doi: 10.1002/anie.201207312. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2012;51:12444–12448. doi: 10.1002/anie.201207312. [DOI] [PubMed] [Google Scholar]; b) Partridge BM, Chausset-Boissarie L, Burns M, Pulis AP, Aggarwal VK. Angew Chem. 2012;124:11965–11969. doi: 10.1002/anie.201203198. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2012;51:11795–11799. doi: 10.1002/anie.201203198. [DOI] [PubMed] [Google Scholar]
- 24.Prakash GKS, Krishnamurti R, Olah GA. J Am Chem Soc. 1989;111:393–395. [Google Scholar]
- 25.a) Yamauchi Y, Hara S, Senboku H. Tetrahedron. 2010;66:473–479. [Google Scholar]; b) Rudzinski DM, Kelly CB, Leadbeater NE. Chem Commun. 2012;48:9610–9612. doi: 10.1039/c2cc35037h. [DOI] [PubMed] [Google Scholar]
- 26.a) Okana T, Sugiura H, Fumoto M, Matsubara H, Kusukawa T, Fujita M. J Fluorine Chem. 2002;114:91–98. [Google Scholar]; b) Yamauchi Y, Hara S, Senboku H. Tetrahedron. 2010;66:473–479. [Google Scholar]
- 27.a) Martin V, Molines H, Wekselman C. J Fluorine Chem. 1993;62:63–68. [Google Scholar]; b) Loh TP, Li XR. Eur J Org Chem. 1999;8:1893–1899. [Google Scholar]; c) Chen Q, Qiu XL, Qing FL. J Org Chem. 2006;71:3762–3777. doi: 10.1021/jo0601157. [DOI] [PubMed] [Google Scholar]; d) Pinna GA, Cignarella G, Ruiu S, Loriga G, Murineddu G, Villa S, Grella GE, Cossu G, Fratta W. Bioorg Med Chem. 2003;11:4015–4026. doi: 10.1016/s0968-0896(03)00373-0. [DOI] [PubMed] [Google Scholar]
- 28.Nave S, Sonawane RP, Elford TG, Aggarwal VK. J Am Chem Soc. 2010;132:17096–17098. doi: 10.1021/ja1084207. [DOI] [PubMed] [Google Scholar]
- 29.a) Zhao Y, Hu J. Angew Chem. 2012;124:1057–1060. [Google Scholar]; Angew Chem Int Ed. 2012;51:1033–1036. doi: 10.1002/anie.201106742. [DOI] [PubMed] [Google Scholar]; b) Liang A, Li X, Liu D, Li J, Zou D, Wu Y, Wu Y. Chem Commun. 2012;48:8273–8275. doi: 10.1039/c2cc31651j. [DOI] [PubMed] [Google Scholar]; c) Matsubara S, Mitani M, Utimoto K. Tetrahedron Lett. 1987;28:5857–5860. [Google Scholar]; d) Feng YS, Xie CQ, Qiao WL, Xu HJ. Org Lett. 2013;15:936–939. doi: 10.1021/ol400099h. [DOI] [PubMed] [Google Scholar]; (e) Kreis LM, Krautwald S, Pfeiffer N, Martin RE, Carreira EM. Org Lett. 2013;15:1634–1637. doi: 10.1021/ol400410m. [DOI] [PubMed] [Google Scholar]
- 30.a) Ichikawa J, Ikeura C, Minami T. Synlett. 1992;9:739–740. [Google Scholar]; b) Ichikawa J. J Fluorine Chem. 2000;105:257–263. [Google Scholar]
- 31.Zemtsov AA, Levin VV, Dilman AD, Struchkova MI, Belyakov PA, Tartakovsky VA. Tetrahedron Lett. 2009;50:2998–3000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.