

Abstract
Germline mutations of the breast cancer 1 (BRCA1) gene are a major cause of familial breast and ovarian cancer. The BRCA1 protein displays E3 ubiquitin ligase activity, and this enzymatic function is thought to be required for tumor suppression. To test this hypothesis, we generated mice that express an enzymatically defective Brca1. We found that this mutant Brca1 prevents tumor formation to the same degree as does wild-type Brca1 in three different genetically engineered mouse (GEM) models of cancer. In contrast, a mutation that ablates phosphoprotein recognition by the BRCA C terminus (BRCT) domains of BRCA1 elicits tumors in each of the three GEM models. Thus, BRCT phosphoprotein recognition, but not the E3 ligase activity, is required for BRCA1 tumor suppression.
Germline mutations of the breast cancer 1 (BRCA1) tumor suppressor are a common cause of hereditary breast and ovarian cancer. The BRCA1 protein harbors an N-terminal RING motif characteristic of many ubiquitin E3 ligases and two BRCA C terminus (BRCT) motifs that form a phosphoprotein recognition domain (1-4). BRCA1 interacts with BRCA1-associated RING domain protein 1 (BARD1) to form a potent E3 ligase (5, 6) that is thought to regulate multiple pathways, including those responsible for tumor suppression (1-4). To test whether the E3 ligase activity of BRCA1 is essential for tumor suppression, we examined mice that express Brca1FH-I26A, an enzymatically defective protein with a missense mutation (I26A) in the RING domain that allows assembly of the BRCA1/BARD1 heterodimer but abrogates its E3 ligase activity (7, 8). (Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; and Y, Tyr. In the mutants, other amino acids were substituted at certain locations; for example, I26A indicates that isoleucine at position 26 was replaced by alanine.) Brca1FH-I26A/+ and Brca1FH-WT/+ mice were generated from isogenic embryonic stem (ES) cells that express either Brca1FH-I26A or the corresponding wild-type Brca1FH-WT protein (9) and bred to homozygosity. Brca1FH-I26A/FH-I26A pups were born at the expected Mendelian ratio and survived to adulthood. Because Brca1-null mice invariably undergo embryonic lethality (10, 11), the viability of Brca1FH-I26A/FH-I26A mice indicates that E3 ligase activity is not essential for all BRCA1 functions. Nonetheless, Brca1FH-I26A/FH-I26A, but not Brca1FH-WT/FH-WT, males are sterile and have smaller testes, and their seminiferous tubules lack elongated spermatids and spermatozoa (fig. S1), which suggests a block in spermatogenesis. Apart from male sterility and a modest decrease in adult body weight (5 to 10%), Brca1FH-I26A/FH-I26A mice appear indistinguishable from their Brca1FH-WT/FH-WT littermates. Moreover, analyses of Brca1FH-I26A/FH-I26A mouse embryonic fibroblasts (MEFs) revealed that the I26A mutation had no measurable effect on cellular proliferation, chromosomal stability, senescence induction, centrosome number, spindle formation, resistance to genotoxic stress, or ubiquitin foci formation at sites of DNA damage—unlike the hypomorphic Brca1 lesions in MEFs of Brca1tr/tr (12) and Brca1Δ11/Δ11 mice (13), which are known to disrupt tumor suppression (fig. S2).
To evaluate whether the E3 ligase activity affects tumor suppression, we initially used a mouse model of pancreatic cancer in which the Pdx1-cre transgene triggers KrasG12D and p53R172H expression in pancreatic progenitor cells (14). To test whether Brca1 suppresses formation of these tumors, we generated Pdx1-cre animals carrying KrasLSL-G12D (14) together with conditional-null Brca1flex2 (15) and/or p53flex7 (16) alleles. Although double-mutant KrasLSL-G12D/p53flex7/flex7/Pdx1-cre mice succumbed to pancreatic tumors with an average latency (T50) of 68 days, tumor latency was dramatically reduced in triple-mutant animals (KrasLSL-G12D/p53flex7/flex7/Brca1flex2/flex2/Pdx1-cre) with conditional-null Brca1flex2 (T50 = 40 days; P < 0.0001), indicating that wild-type Brca1 suppresses pancreatic tumor development (Fig. 1A). In contrast, triple-mutant animals (KrasLSL-G12D/p53flex7/flex7/Brca1flex2/FH-I26A/Pdx1-cre) expressing Brca1FH-I26A developed pancreatic tumors with a similar latency (T50 = 65 days) as that of double-mutant KrasLSL-G12D/p53flex7/flex7/Pdx1-cre mice (T50 = 68 days; P = 0.2595) (Fig. 1A). Thus, the tumor suppression potential of enzymatically inactive Brca1 in the pancreas is indistinguishable from that of wild-type Brca1.
Fig. 1.
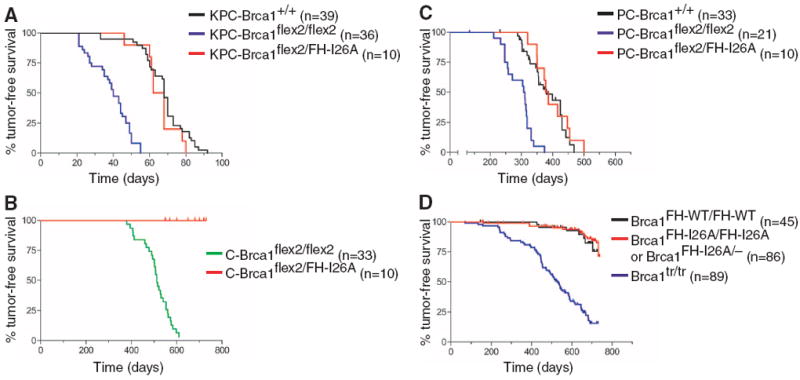
The enzymatic activity of Brca1 is dispensable for tumor suppression. (A) Kaplan-Meier tumor-free survival curves of KrasLSL-G12D/p53flex7/flex7/Pdx1-cre (KPC-Brca1+/+; black curve; T50 = 68 days) mice compared with KrasLSL-G12D/p53flex7/flex7/Pdx1-cre/Brca1flex2/flex2 (KPC-Brca1flex2/flex2; blue curve; T50 = 40 days; P < 0.0001) and KrasLSL-G12D/p53flex7/flex7/Pdx1-cre/Brca1flex2/FH-I26A (KPC-Brca1flex2/FH-I26A; red curve; T50 = 65 days; P = 0.2595) mice. (B) Kaplan-Meier survival curves of Brca1flex2/flex2/Wapcre/+ (C-Brca1flex2/flex2; green curve; T50 = 512 days) females compared with Brca1flex2/FH-I26A/Wapcre/+ (C-Brca1flex2/FH-I26A; red curve; P < 0.0001) females. (C) Kaplan-Meier survival curves of p53LSL-R270H/+/Wapcre/+ (PC-Brca1+/+; black curve; T50 = 380 days) females compared with Brca1flex2/flex2/p53LSL-R270H/+/Wapcre/+ (PC-Brca1flex2/flex2; blue curve; T50 = 309 days; P < 0.0001) and Brca1flex2/FH-I26A/p53LSL-R270H/+/Wapcre/+ (PC-Brca1flex2/FH-I26A; red curve; T50 = 382 days; P = 0.5354) females. (D) Kaplan-Meier survival curves of control Brca1FH-WT/FH-WT (black curve) mice compared with Brca1tr/tr (blue curve; T50 = 529 days; P < 0.0001) and Brca1FH-I26A/FH-I26A and Brca1FH-I26A/– (red curve; P = 0.5197) mice.
We next applied a mouse model of familial breast cancer in which the Wapcre gene elicits mammary-specific inactivation of the conditional-null Brca1flex2 allele (15). However, unlike Brca1flex2/flex2/Wapcre/+ females, which form tumors resembling the basal-like breast carcinomas of human BRCA1 mutation carriers (15), all mice expressing enzymatically inactive Brca1 (Brca1flex2/FH-I26A/Wapcre/+) remained tumor-free over the 24-month observation period (Fig. 1B).
We also monitored three cohorts of Wapcre/+ mice sensitized for tumor development by a conditional p53 mutation (p53LSL-R270H). Whereas mammary tumors developed in control p53LSL-R270H/+/Wapcre/+ females with an average latency of 380 days, which is consistent with previous studies (17), tumor formation was accelerated (T50 = 308 days) by conditional Brca1 inactivation in Brca1flex2/flex2/p53LSL-R270H/+/Wapcre/+ females (P < 0.0001) (Fig. 1C). The kinetics of tumor development in Brca1flex2/FH-I26A/p53LSL-R270H/+/Wapcre/+ females was indistinguishable from that of control p53LSL-R270H/+/Wapcre/+ females (P = 0.7502) and significantly slower than that of Brca1flex2/flex2/p53LSL-R270H/+/Wapcre/+ females (P < 0.0001) (Fig. 1C). Moreover, representative oligonucleotide microarray analysis (18) revealed a “simplex” pattern of genomic copy number variation in Brca1flex2/FH-I26A/p53flex7/flex7/Wapcre/+ tumors, similar to that of p53flex7/flex7/Wapcre/+ tumors but distinct from the “complex sawtooth” pattern of Brca1flex2/flex2/p53flex7/flex7/Wapcre/+ tumors (fig. S3). Thus, mammary-specific loss of Brca1 enzymatic activity does not promote basal-like breast carcinoma in a manner analogous to complete Brca1 inactivation.
Although mice lacking Brca1 enzymatic activity (Brca1FH-I26A/FH-I26A and Brca1FH-I26A/−) are viable, some (18.6%) died of tumors at a very advanced age (Fig. 1D). However, the frequency and kinetics of spontaneous tumor formation in the mutant mice were comparable with those of Brca1FH-WT/FH-WT control animals (P = 0.5197) and significantly lower than those of Brca1tr/tr mice (P < 0.0001) (12). Thus, the E3 ligase activity of BRCA1 is dispensable for tumor suppression in each of the three GEM cancer models.
The BRCT motifs of BRCA1 form a phospho-recognition domain that preferentially binds the phosphorylated isoforms of repair proteins Abraxas/CCDC98, BACH1/FancJ, and CtIP (1, 2). Because most tumor-associated BRCA1 alleles have frameshift/nonsense mutations that eliminate one or both BRCT motifs, BRCT phospho-recognition may be critical for tumor suppression. Indeed, in some families breast cancer susceptibility can be ascribed to missense mutations that cause a single amino acid substitution (for example, S1655F) that disrupts the interaction between the BRCT domain and its cognate phospho-ligands. Structural studies show that BRCA1 residue S1655 donates a hydrogen bond to the phosphate group of these phospho-ligands, and that mutation of this residue disrupts their interaction with BRCA1 (19-23). To determine whether BRCT phospho-recognition is required for genome stability and tumor suppression, we mutated the corresponding mouse residue (S1598F) to generate heterozygous (Brca1S1598F/+) and homozygous (Brca1S1598F/S1598F) ES clones (fig. S4). The Brca1S1598F/+ ES cells were injected into blastocysts to derive germline chimeras, and heterozygous animals were then intercrossed to produce homozygous Brca1S1598F/S1598F offspring, which appeared at the expected Mendelian ratio. Apart from male sterility, these mice developed normally and provided a source of Brca1S1598F/S1598F MEFs.
The mutant Brca1 protein of Brca1S1598F/S1598F MEFs is expressed at normal levels and fails to bind Bach1/FancJ (Fig. 2A). Brca1S1598F/S1598F ES cells are hypersensitive to genotoxic stress (Fig. 2B) and defective for homology-directed DNA repair (Fig. 2C). In addition, Brca1S1598F/S1598F MEFs display proliferation defects, chromosomal instability, centrosome amplification, and diminished recruitment of repair proteins to sites of DNA damage (figs. S5 and S6). Thus, the S1598F mutation disrupts BRCA1 function in the DNA damage response. To evaluate its effect on tumor suppression, we monitored an experimental cohort of Brca1flex2/S1598F/p53LSL-R270H/+/Wapcre/+ females and control cohorts of the p53LSL-R270H/+/Wapcre/+, Brca1flex2/flex2/Wapcre/+, and Brca1flex2/flex2/p53LSL-R270H/+/Wapcre/+ females described above (Fig. 1, B and C). As shown in Fig. 3A, mammary tumorigenesis was accelerated in experimental Brca1flex2/S1598F/p53LSL-R270H/+/Wapcre/+ mice (T50 = 308 days) relative to p53LSL-R270H/+/Wapcre/+ animals (T50 = 380 days; P < 0.0001), and the shortened latency was indistinguishable from that of Brca1flex2/flex2/p53LSL-R270H/+/Wapcre/+ animals (T50 = 309 days; P = 0.4543). Thus, the S1598F mutation impairs mammary tumor suppression to a comparable degree as does complete Brca1 inactivation. Pancreatic tumor formation in mice expressing Brca1S1598F (KrasLSL-G12D/p53flex7/flex7/Brca1flex2/S1598F/Pdx1-cre; T50 = 45 days) was also accelerated relative to those expressing wild-type Brca1 (KrasLSL-G12D/p53flex7/flex7/Pdx1-cre mice; T50 = 68 days; P < 0.0001) (Fig. 3B) and comparable with conditional-null Brca1 mice (KrasLSL-G12D/p53flex7/flex7/Brca1flex2/flex2/Pdx1-cre) (T50 = 40 days; P = 0.2632). Furthermore, homozygous Brca1S1598F/S1598F mice were highly tumor-prone (49 out of 72; 68.1%) relative to control animals (P < 0.0001) (Fig. 3C). Thus, BRCT phospho-recognition is critical for BRCA1-mediated tumor suppression in all three GEM cancer models.
Fig. 2.
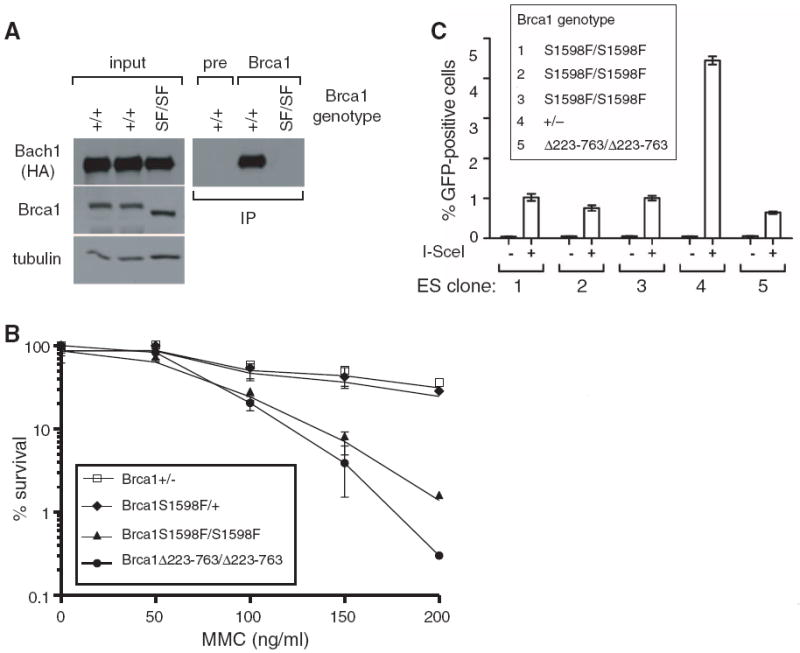
The BRCT phospho-recognition property of Brca1 is critical for the DNA damage response. (A) S1598F ablates the interaction between Bach1/FancJ and endogenous Brca1. MEFs were prepared from mice with a knock-in allele (Bach1WT-FH) encoding wild-type Bach1 with C-terminal Flag-hemagglutinin (HA) epitopes. Bach1WT-FH and Brca1 levels were examined by immunoblotting lysates of Brca1+/+/Bach1WT-FH/+ and Brca1S1598F/S1598F/Bach1WT-FH/+ MEFs (left). To evaluate the Brca1/Bach1WT-FH interaction, lysates were immunoprecipitated with a Brca1-specific antiserum or corresponding preimmune serum (pre) and immunoblotted with HA-specific antibodies (right). The input (left) represents 2.5% of the protein amount used for immunoprecipitation (right). (B) Brca1S1598F/S1598F cells are sensitive to genotoxic stress. ES cells proficient (Brca1+/− and Brca1S1598F/+) or deficient (Brca1S1598F/S1598F) for BRCT phospho-recognition were examined for mitomycin C (MMC) sensitivity in clonogenic survival assays, together with ES cells homozygous for the hypomorphic Brca1Δ223-763 mutation. (C) BRCT phospho-recognition by Brca1 is essential for homology-directed DNA repair. Brca1+/−, Brca1S1598F/S1598F, and Brca1Δ223-763/Δ223-763 ES subclones containing the direct repeat–green fluorescent protein (DR-GFP) substrate integrated into the Pim1 locus were transfected with either an I-SceI expression vector or empty vector. I-SceI strongly induced the number of GFP-positive cells in Brca1+/− cells (clone 4), indicating efficient homology-directed repair, but not in Brca1S1598F/S1598F cells (clones 1 to 3). The reduction in GFP-positive cells in Brca1S1598F/S1598F cells was similar to that of Brca1Δ223-763/Δ223-763 ES cells (clone 5), which are known to be deficient in homology-directed repair (28).
Fig. 3.
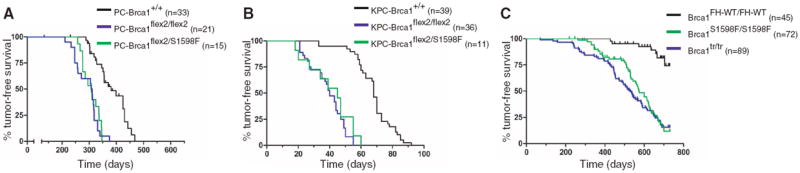
BRCT phospho-recognition is essential for Brca1 tumor suppression. (A) Kaplan-Meier tumor-free survival curves of p53LSL-R270H/+/Wapcre/+ (PC-Brca1+/+; black curve; T50 = 380 days) females compared with Brca1flex2/flex2/p53LSL-R270H/+/Wapcre/+ (PC-Brca1flex2/flex2; blue curve; T50 = 309 days; P < 0.0001) and Brca1flex2/S1598F/p53LSL-R270H/+/Wapcre/+ (PC-Brca1flex2/S1598F; green curve; T50 = 308 days; P < 0.0001) females. (B) Kaplan-Meier survival curves of KrasLSL-G12D/p53flex7/flex7/Pdx1-cre (KPC-Brca1+/+; black curve; T50 = 68 days) mice compared with KrasLSL-G12D/p53flex7/flex7/Pdx1-cre/Brca1flex2/flex2 (KPC-Brca1flex2/flex2; blue curve; T50 = 40 days; P < 0.0001) and KrasLSL-G12D/p53flex7/flex7/Pdx1-cre/Brca1flex2/S1598F (KPC-Brca1flex2/S1598F; green curve; T50 = 45 days; P < 0.0001) mice. (C) Kaplan-Meier survival curves of Brca1FH-WT/FH-WT (black curve) mice compared with Brca1tr/tr (blue curve; P < 0.0001) and Brca1S1598F/S1598F (green curve; P < 0.0001) mice.
BRCA1 is thought to regulate diverse cellular processes by ubiquitinating multiple protein substrates (1-4). Indeed, on the basis of the same mutation (I26A) used here the E3 ligase activity has been implicated in BRCA1 control of centrosome duplication (24), checkpoint activation (25), mitotic spindle assembly (26), and tumor cell motility (27). In light of these observations, the viability of mice lacking BRCA1 enzymatic activity was unexpected. Because mice homozygous for null Brca1 mutations undergo embryonic lethality (10, 11), our results suggest that the E3 ligase activity of BRCA1 is dispensable for much of normal development. Thus, many of the known and unknown functions of BRCA1 may be mediated independent of its ability to catalyze ubiquitination.
From a medical standpoint, the key function of BRCA1 is its ability to suppress tumor formation in breast and ovarian tissues. Like BRCA1, many other oncoproteins and tumor suppressors possess enzymatic activity. In most cases, the enzymatic function has proven to be integral to the oncogenic process, and in some cases it can be targeted for therapeutic gain, as illustrated by the clinical success of certain tyrosine kinase inhibitors. However, because enzymatically inert Brca1 is sufficient to suppress tumor formation in several settings (Fig. 1), including a model of human basal-like breast cancer, the E3 ligase activity of BRCA1 appears to be dispensable for tumor suppression.
A role for the E3 ligase activity in BRCA1 tumor suppression was predicated partly on the fact that this activity is ablated by certain tumor-associated lesions of the RING domain, such as the C61G and C64G missense mutations (6). However, these mutations also disrupt the interaction between BRCA1 and its partner protein BARD1 (5). Because mammary-specific inactivation of either Brca1 or Bard1 elicits breast tumors with the same basal-like phenotype (15), the tumor suppression activity of BRCA1 is probably mediated by the BRCA1/BARD1 heterodimer. Unlike the tumorigenic C61G and C64G mutations, the synthetic I26A mutation used in this study specifically ablates the enzymatic activity of BRCA1 but allows proper assembly of the BRCA1/BARD1 heterodimer (7). Thus, the tumorigenic RING mutations are likely to compromise BRCA1-mediated tumor suppression primarily by impairing BRCA1/BARD1 heterodimerization.
The mouse model of hereditary breast cancer has provided meaningful insights into the molecular mechanisms of BRCA1-mediated tumor suppression. First, the identical pattern of breast carcinogenesis in conditional Brca1-, Bard1-, and Brca1/Bard1-null mice implies that tumor suppression is dependent on the BRCA1/BARD1 heterodimer (15). Second, as shown here tumor suppression does not require the E3 ligase activity of BRCA1/BARD1. Third, our data also suggest that the ability of the BRCT domain to bind its phospho-ligands is critical for BRCA1 tumor suppression. In light of this result, it is noteworthy that BRCA1/BARD1 forms distinct protein complexes (A, B, and C) based on BRCT-mediated interactions with three different repair proteins (Abraxas/CCDC98, BACH1/FancJ, and CtIP) (1, 2). Because tumor suppression appears to be dependent on BRCA1 association with one or more of these BRCT phospho-ligands (and/or others yet to be discovered), these interactions may provide valuable targets for therapeutic intervention.
Supplementary Material
Acknowledgments
We thank X. Sun for technical assistance, V. Murty for advice, M. Wigler for discussions and encouragement, and NYSCF for access to the confocal microscope. This work was supported by NIH grants R01-CA137023 (R.B. and T.L.), P01-CA97403 (R.B. and T.L.), and R01-HD40916 (M.J.). R.S. was supported by a Susan G. Komen Breast Cancer fellowship, L.J.R. by a Kirschstein National Research Service Award fellowship (F31-CA132626), C.R.R. by fellowships from the National Cancer Institute (T32-CA09503) and U.S. Department of Defense (DOD) (BC083089), and F.C. by a Kirschstein National Research Service Award fellowship (F32-HD51392). K.R. and J.B.H. were supported by grants to M. Wigler and J.B.H. from DOD (W81XWH04-1-0477) and the Breast Cancer Research Foundation. Microarray data have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus (GEO) with GEO Series accession number GSE31673.
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/334/6055/525/DC1
Materials and Methods
Figs. S1 to S6
References
References and Notes
- 1.Moynahan ME, Jasin M. Nat Rev Mol Cell Biol. 2010;11:196. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huen MS, Sy SM, Chen J. Nat Rev Mol Cell Biol. 2010;11:138. doi: 10.1038/nrm2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu W, Koike A, Takeshita T, Ohta T. Cell Div. 2008;3:1. doi: 10.1186/1747-1028-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baer R, Ludwig T. Curr Opin Genet Dev. 2002;12:86. doi: 10.1016/s0959-437x(01)00269-6. [DOI] [PubMed] [Google Scholar]
- 5.Wu LC, et al. Nat Genet. 1996;14:430. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- 6.Hashizume R, et al. J Biol Chem. 2001;276:14537. doi: 10.1074/jbc.C000881200. [DOI] [PubMed] [Google Scholar]
- 7.Brzovic PS, et al. Proc Natl Acad Sci U S A. 2003;100:5646. doi: 10.1073/pnas.0836054100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christensen DE, Brzovic PS, Klevit RE. Nat Struct Mol Biol. 2007;14:941. doi: 10.1038/nsmb1295. [DOI] [PubMed] [Google Scholar]
- 9.Reid LJ, et al. Proc Natl Acad Sci U S A. 2008;105:20876. doi: 10.1073/pnas.0811203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hakem R, et al. Cell. 1996;85:1009. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- 11.Ludwig T, Chapman DL, Papaioannou VE, Efstratiadis A. Genes Dev. 1997;11:1226. doi: 10.1101/gad.11.10.1226. [DOI] [PubMed] [Google Scholar]
- 12.Ludwig T, Fisher P, Ganesan S, Efstratiadis A. Genes Dev. 2001;15:1188. doi: 10.1101/gad.879201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu X, et al. Mol Cell. 1999;3:389. doi: 10.1016/s1097-2765(00)80466-9. [DOI] [PubMed] [Google Scholar]
- 14.Hingorani SR, et al. Cancer Cell. 2005;7:469. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 15.Shakya R, et al. Proc Natl Acad Sci U S A. 2008;105:7040. doi: 10.1073/pnas.0711032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z, et al. Nature. 2005;436:725. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wijnhoven SW, et al. Cancer Res. 2005;65:8166. doi: 10.1158/0008-5472.CAN-05-1650. [DOI] [PubMed] [Google Scholar]
- 18.Hicks J, et al. Genome Res. 2006;16:1465. doi: 10.1101/gr.5460106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Botuyan MV, et al. Structure. 2004;12:1137. doi: 10.1016/j.str.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clapperton JA, et al. Nat Struct Mol Biol. 2004;11:512. doi: 10.1038/nsmb775. [DOI] [PubMed] [Google Scholar]
- 21.Shiozaki EN, Gu L, Yan N, Shi Y. Mol Cell. 2004;14:405. doi: 10.1016/s1097-2765(04)00238-2. [DOI] [PubMed] [Google Scholar]
- 22.Varma AK, Brown RS, Birrane G, Ladias JA. Biochemistry. 2005;44:10941. doi: 10.1021/bi0509651. [DOI] [PubMed] [Google Scholar]
- 23.Williams RS, Lee MS, Hau DD, Glover JN. Nat Struct Mol Biol. 2004;11:519. doi: 10.1038/nsmb776. [DOI] [PubMed] [Google Scholar]
- 24.Sankaran S, Starita LM, Simons AM, Parvin JD. Cancer Res. 2006;66:4100. doi: 10.1158/0008-5472.CAN-05-4430. [DOI] [PubMed] [Google Scholar]
- 25.Yu X, Fu S, Lai M, Baer R, Chen J. Genes Dev. 2006;20:1721. doi: 10.1101/gad.1431006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joukov V, et al. Cell. 2006;127:539. doi: 10.1016/j.cell.2006.08.053. [DOI] [PubMed] [Google Scholar]
- 27.Coene ED, et al. J Cell Biol. 2011;192:497. doi: 10.1083/jcb.201004136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moynahan ME, Chiu JW, Koller BH, Jasin M. Mol Cell. 1999;4:511. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.