

Abstract
Therapeutics which reduce the pathology in sickle cell syndromes are needed, particularly non-cytotoxic therapeutics. Fetal hemoglobin (HbF, α2γ2) is established as a major regulator of disease severity; increased HbF levels correlate with milder clinical courses and improved survival. Accordingly, sodium dimethylbutyrate (HQK-1001), an orally-bioavailable, promoter-targeted fetal globin gene-inducing agent, was evaluated in a randomized, blinded, dose-ranging Phase I/II trial in 24 adult patients with HbSS or S/β thalassemia, to determine safety and tolerability of three escalating dose levels. The study therapeutic was administered once daily for two 6-week cycles, with a 2-week interim dose holiday. Twenty-one patients completed the study. Five patients received study drug at 10 or 20 mg/kg doses, seven patients received study drug at 30 mg/kg/dose, and 4 patients received placebo. HQK-1001 was well-tolerated with no unexpected drug-related adverse events; a dose-limiting toxicity was not identified. Plasma drug levels were sustained above targeted levels for 24 hours. Increases in HbF above baseline were observed particularly with 30 mg/kg/day doses; in five of seven treated patients, a mean absolute increase in HbF of 0.2 g/dl and a mean increase in total hemoglobin (Hgb) of 0.83 g/dl above baseline were observed, whereas no increases occurred in placebo-treated controls. These findings of favorable PK profiles, tolerability, early rises in HbF and total Hgb indicate that trials of longer duration appear warranted to more definitively evaluate the therapeutic potential of HQK-1001 in sickle cell disease.
INTRODUCTION
Sickle cell disease, characterized by HbS (α2βS2), is a genetic blood disorder which is recognized by WHO as a global health burden.1 Sickle cell disease causes hemolytic anemia, vaso-occlusion, and cell adhesion, producing widespread organ damage and early mortality.2-3 Fetal globin, or HbF (α2γ2), has been established as the major modulator of disease severity in biochemical, epidemiologic, and clinical trials.4-22 While HbF levels >8.6% correlate with improved survival, levels >20% are associated with amelioration of most complications.8-14,17-18,21-23 One therapeutic, hydroxyurea, is approved for treatment of sickle cell disease and confers considerable benefit in some patients; reduced frequency of clinical events and improved survival in adult patients are associated with rises in absolute HbF levels > 0.5 g/dl.17-18,20 Additional therapeutics that induce HbF expression by different mechanisms are needed for many patients to achieve levels of HbF which are disease-modifying.4,23
Previously, two other classes of therapeutics have produced high HbF levels in adult sickle cell patients: demethylating agents (5-azacytidine and decitabine) and short chain fatty acids, of which some are histone deacetylase inhibitors, such as arginine butyrate. 24-26 Clinical trials of these agents resulted in a mean 3-fold increase in HbF, to levels of 18-30%, in adult patients with an associated rise in total hemoglobin of 1 gm/dl above baseline. 24-28 However, these agents require parenteral administration, while oral therapeutics are more feasible and preferable for long-term use. Sodium 2,2 dimethylbutyrate (SDMB, HQK-1001) is a short-chain fatty acid derivative which induces expression of the γ–globin gene promoter in reporter gene assays, γ–globin mRNA and HbF in cultured erythroid progenitors, and of HbF in anemic baboons.29-32 SDMB is orally bio-available with a half-life of several hours in non-human primates and in normal human volunteers.30,33 A dose-ranging trial to evaluate safety, tolerability, and pharmacokinetics in normal human volunteers demonstrated no significant drug-related adverse events, and produced pharmacokinetic profiles which are favorable for once-daily dosing, with a t1/2 of 9-11 hours at doses projected to be required for therapeutic induction of fetal globin.33 Accordingly, a blinded, sequential dose-escalating safety and tolerability study of sodium 2,2 dimethylbutyrate (HQK-1001) was performed in patients with sickle cell disease. The drug was well-tolerated, and early signals of induced fetal globin expression were observed in some subjects during this short-term drug administration.
MATERIALS AND METHODS
The primary objective of this trial (NCT00842088) was to determine the safety and tolerability of HQK-1001 administered for 12 weeks at three dose levels in subjects with sickle cell disease. Secondary objectives were to evaluate the pharmacokinetic (PK) profiles of the drug with repeated doses, to assess potential pharmacodynamic effects on fetal globin expression within the relatively brief dosing period, and to identify dose regimens and schedules suitable for further evaluation in follow-on trials. The design was a randomized, blinded, placebo-controlled, sequential dose-escalating trial and was conducted in seven clinical sites, six in the United States and one in Jamaica, with approval of their Investigational Review Boards. Eligibility criteria required that subjects have an established diagnosis of Hb SS or sickle/β-thalassemia, were ages 12-60 years inclusive, and had a history of an average of one significant sickle cell-related medical event requiring medical attention per year for in the preceding three years, or at least one hospitalization for acute chest syndrome in the previous five years. Concomitant hydroxyurea therapy was allowed, if a stable dose was established in the subject for at least six months. A baseline screening HbF level >2% was required, as this threshold was necessary for responses to arginine butyrate in previous trials.26 Exclusion criteria included a red blood cell transfusion within 3 months before beginning drug or participation in a regular transfusion program, acute vaso-occlusive events within 3 weeks, a history of >4 hospitalizations for sickle cell related events in the preceding 12 months, renal or hepatic compromise, or pulmonary hypertension requiring oxygen therapy. Randomization to active treatment or placebo was assigned according to a table generated by random numbers prepared by biostatisticians in an independent contract research organization, Quintiles, Inc., which monitored the study.
The study drug was administered once-daily for 2 cycles of 6 weeks each. Because arginine butyrate was more effective when administered in an intermittent or pulsed fashion, a 2-week dose holiday was provided between the two treatment cycles.26 Initial doses were administered with observation in a clinical research unit. Cohorts of 10, 20, and 30 mg/kg/doses included at least one placebo subject and six or seven active treatment subjects, and dose escalation between cohorts was allowed sequentially after review of the first four subjects’ courses by an independent Safety Monitoring Committee. Patients received supplements with folic acid, 1 mg/day orally, and ferrous gluconate if their ferritin was <1000ng/ml, to support erythropoiesis, as previously shown to be necessary for effective HbF induction.34 Safety monitoring by blinded investigators included physical examinations, laboratory studies of hematologic, renal, and hepatic function every two weeks, periodic coagulation analyses, electrocardiograms, monthly pregnancy tests, and adverse event monitoring every two weeks during the dosing period and for four weeks after dosing was completed.
HbF was assayed by HPLC at a central reference laboratory by blinded personnel, and F-cells, proportions of red blood cells containing HbF, was assessed by flow cytometry also by blinded personnel. Samples were collected for PK profiles on day 6; drug levels were assayed by previously established methods.33 Pharmacokinetic parameters which were analyzed included area under the curve from time 0 to last quantifiable concentration (AUC(0-lqc)), AUC from time 0 to infinity (AUC(0-∞)), maximum concentration of drug (Cmax), minimum concentration of drug (Cmin), average concentration of drug (Cavg), time of maximum concentration (Tmax), elimination half-life (T1/2), total body clearance (CL/F), and elimination rate constant (λz). Pharmacokinetic analyses were performed using WinNonLinR Professional version 5.2 (Parsight Corp., Mountain View, CA.), as previously described.33 Descriptive statistics were prepared and statistical analyses were performed by J.M. White Inc., using Statistical Analysis Software (SAS) version 9 or higher. Adverse events were characterized by dose level or placebo using MedDRA version 9 preferred term coding, and laboratory abnormality severity was graded using NCI CTC version 3. All 24 subjects who enrolled received at least one dose of the study drug, and all were included in the safety analysis. The blinded principle investigator (PI) at each site determined adverse event (AE) relationship to study drug.
RESULTS
Thirty patients were screened for eligibility criteria; twenty-four subjects met the required criteria, signed informed consent, and were enrolled. Demographic characteristics and baseline hematologic features of the study participants are shown in Table I. Patients ranged in age from 14 to 57 years; the mean ratio of males to females was 0.6 in the four cohorts. The subjects’ genotypes were homozygous Hb SS except for one patient who had HbS-β+ thalassemia. No apparent differences in demographic characteristics of age, ethnicity, or baseline height and weight were observed across treatment groups. The patients were of Jamaican or African American ethnicity. All subjects had significant past medical histories characteristic of sickle cell disease, including vaso-occlusive crises, osteonecrosis, pulmonary hypertension, cholelithiasis, priapism, proliferative retinopathy, and joint replacement for avascular necrosis. Twelve subjects had concomitant hydroxyurea (HU) use, including 75% of placebo subjects, and in 57, 50, and 29% of the 10, 20, and 30 mg/kg dose cohorts, respectively. Baseline median HbF levels were 14.5, 7.7, 11.2, and 10.8% in the placebo-treated, 10, 20, and 30 mg/kg dose cohorts, respectively. Mean baseline HbF was in those subjects receiving treatment with HU was 14.4% and in subjects not receiving HU was 7.6%. Baseline total hemoglobin (Hgb) levels ranged from 5.9 to 11.8 g/dl; the means at baseline were 8.8, 8.4, and 9.7 g/dl in the 10, 20, and 30 mg/kg dose 1001-treated cohorts respectively and 9.3 g/dl in the placebo treated group. Baseline total Hb and % HbF in individual subjects by assigned treatment or placebo group and HU use are shown in Table Ib.
Table IA. Basic characteristics.
| Characteristics | HQK-1001 (n=20) | Placebo (n = 4) |
|---|---|---|
| Male Gender | 11 (60 %) | 3 (75%) |
| Age in years, median (Range) |
29 (13-56) | 34 (20-48) |
| Subjects taking Hydroxyurea | 9 (45 %) | 3 (75 %) |
| Weight in kg, median (Range) | 63 (53 – 94) | 59 (54 – 74) |
Table IB. Baseline total hemoglobin and % HbF in individual subjects. Hydroxyurea treatment status is designated in the far right column.
| ID | Group | Hb (g/dL) | HbF (%) | HU |
|---|---|---|---|---|
| 90301 | Placebo | 10.0 | 3.0 | Yes |
| 90803 | Placebo | 8.7 | 18.3 | Yes |
| 91101 | Placebo | 7.4 | 2.5 | |
| 91103 | Placebo | 9.9 | 10.8 | Yes |
|
| ||||
| 90101 | 10 mg/kg | 9.2 | 7.7 | Yes |
| 90102 | 10 mg/kg | 8.8 | 5.8 | Yes |
| 90103 | 10 mg/kg | 11.9 | 14.7 | Yes |
| 90202 | 10 mg/kg | 9.3 | 8.4 | |
| 90203 | 10 mg/kg | 8.0 | 2.9 | |
| 91102 | 10 mg/kg | 6.1 | 1.7 | |
| 91104 | 10 mg/kg | 5.9 | 12.2 | Yes |
|
| ||||
| 90105 | 20 mg/kg | 9.5 | 32.8 | Yes |
| 90204 | 20 mg/kg | 8.5 | 8.3 | |
| 90205 | 20 mg/kg | 9.2 | 7.0 | |
| 90801 | 20 mg/kg | 11.8 | 10.2 | Yes |
| 90802 | 20 mg/kg | 9.9 | 28.2 | Yes |
| 90901 | 20 mg/kg | 11.4 | 12.2 | |
|
| ||||
| 90106 | 30 mg/kg | 10.3 | 13.0 | |
| 90206 | 30 mg/kg | 6.3 | 6.4 | |
| 90804 | 30 mg/kg | 9.5 | 19.8 | Yes |
| 90805 | 30 mg/kg | 8.4 | 2.9 | |
| 90806 | 30 mg/kg | 10.2 | 8.7 | Yes |
| 90902 | 30 mg/kg | 7.5 | 3.7 | |
| 91301 | 30 mg/kg | 7.6 | 22.2 | |
Twenty-one patients completed the study and are included in the pharmacodynamic data analysis, all patients were included in the safety analysis, and the drug was well-tolerated. Reasons for early termination included development of more severe anemia (in association with a port infection) which required transfusion within one week after starting the study, and two study discontinuations unrelated to the study drug (inability to continue clinic visits due to work obligations in one subject and hypersensitivity to a concomitant medication in another). There were no unexpected, drug-related serious adverse events (SAEs). However, nine subjects experienced 19 SAEs, summarized in Table 2A. These included 6 subjects receiving active treatment who had 13 SAEs, and 3 placebo-treated subjects who had 6 SAEs. All the SAEs were considered to be expected sickle-related events, including vaso-occlusive crises, an indwelling port infection, worsening anemia (associated with the port infection), arthralgia, and extremity pain. One vaso-occlusive crisis in a placebo-treated subject was classified as possibly drug-related by the blinded investigators. No differences were detected in rates or severity of adverse events between patients who were or were not receiving concomitant HU. Adverse events which were graded as mild to moderate and reported in at least 10% of the subjects are shown in Table 2B; the most common events included vomiting, abdominal pain, arthralgia, bone pain. Other adverse events considered to be treatment-related included nausea and rash in two subjects. Minor transient laboratory abnormalities in liver function tests occurred in four subjects; increases in transaminases were graded as mild (Grade 1) or moderate (grade 2), and all returned to normal before the end of the dosing period. The highest elevation in ALT was 256 (range 68-256) and in AST was 169 U/L (range 73-169). Mild leukopenia occurred in one patient, in whom a decline in WBC from 4.4 to 3.1 × 109/L occurred transiently, with subsequent increase to 5.6 × 109/L without any change in study drug. There were no episodes of neutropenia or thrombocytopenia. No dose-dependent pattern was observed for incidence and severity of AEs, and there was no clear difference in the incidence and severity of AEs between the HQK-1001 groups compared to placebo treated subjects, or to HU use, in this small, dose-ranging study. Arthralgia was reported more frequently in the 30 mg/kg group, while bone pain, cough, and headache were more often reported in the 20 mg/kg group, and pain in an extremity was seen more frequently in the 10 mg/kg group.
Table IIA. Adverse events listed as serious.
| Serious Adverse Events | ||
|---|---|---|
| SAE Terms | HQK-1001 (n=20) N (%) |
Placebo (n=4) N (%) |
| Crisis | 5 (25) | 3 (75) |
| Anemia | 1 (5) | 0 |
| Catheter site infection | 1 (5) | 0 |
| Arthralgia | 1 (5) | 0 |
| Pain in extremity | 1 (5) | 0 |
Table IIB. Most common adverse events, occurring in 10% or more of subjects.
| Event | HQK-1001 (n = 20, all doses) % of subjects |
Placebo (n = 4) % of subjects |
|---|---|---|
| Sickle cell crisis | 45 | 75 |
| Nausea | 30 | 25 |
| Pain in extremity | 30 | 0 |
| Cough | 30 | 0 |
| Arthalgia | 25 | 25 |
| Headache | 25 | 25 |
| Chest Pain | 15 | 25 |
| URI | 15 | 25 |
| Back Pain | 15 | 25 |
| Vomiting | 15 | 0 |
Pharmacokinetic studies demonstrated mean PK profiles with dose-dependent increases in AUC and Cmax of HQK-1001 (Figure 1), with plasma concentrations sustained for >10 hours above concentrations (12 to 71 μg/mL) which induce HbF in erythroid progenitors in vitro.29 Mean maximum concentration increased as dose increased, ranging from 54.9 to 114.0 μg/mL over the 10 to 30 mg/kg dose range. The mean Tmax ranged from 2.0 to 2.4 hours. Mean minimum concentration at 24 hours was 15.5 μg/mL at 10 mg/kg, 17.8 μg/mL at 20 mg/kg, and 17.6 μg/mL at 30 mg/kg, which are HbF-inducing “therapeutic” levels in erythroid progenitors in vitro. The mean AUCinf ranged from 722 at 10 mg/kg to 1302 h*μg/mL at 30 mg/kg. Mean CL/F values increased from 0.015 at 10 mg/kg to 0.026 L/h/kg at 30 mg/kg. However, the mean terminal phase half-life for HQK-1001 decreased from 13.8 at 10 mg/kg to 8.4 hours at 30 mg/kg. The decrease in exposure with increase in dose could possibly be due to an increase in the intrinsic clearance. These studies confirm that this agent has more favorable PK profiles compared to prior-generation butyrates, which are rapidly metabolized and required significantly higher doses administered by infusions or multiple doses, three times/day, such as arginine butyrate and phenylbutyrate.26,35
FIGURE 1.
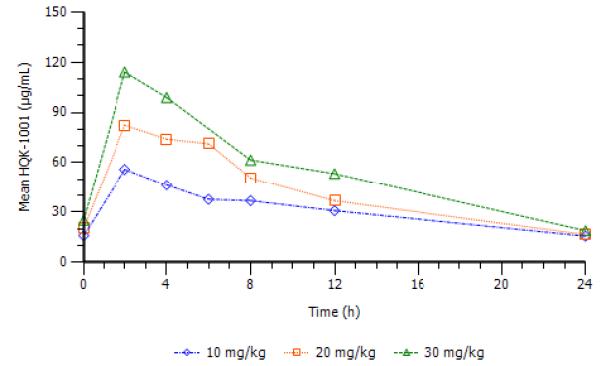
Mean plasma levels of HQK-1001 on day 6 are shown by dose cohort.
Increases in median percent HbF, absolute HbF, and total hemoglobin on the last observation at Day 97 were observed in at least half of the subjects in the 20 and 30 mg/kg dose cohorts, while no increases were detected in the placebo group. Increases in % HbF of > 1.1% above subjects’ baseline (which are reliably detected in the testing laboratory) were observed at the end of the treatment period in 2/5 subjects treated with 10 mg/kg/dose (by 2.6 and 3.1% above baseline), in 3/5 patients treated at 20 mg/kg doses (by 1.6 to 3% above baseline), and in 5/7 patients treated with 30 mg/kg doses, (by 1.1 to 3.7% above baseline). Individual responses are shown in Figure 2A. Absolute HbF also increased in the 30 mg/kg dose cohort in five of seven treated subjects; the median increase was 1.82 g/dl, compared to no increase in the 4 placebo-treated subjects. The per cent of F-cells increased from baseline to day 97 by a median of 4.7 %, 0.45%, and 6.1% in the 10,20,and 30 mg/kg 1001 dose cohorts respectively; in the placebo group, F-cells declined by −0.6%. A mean increase in total Hgb of 0.83 g/dl above averaged baseline levels was observed in the 30 mg/kg dose cohort (Figure 2B). Increases in HbF occurred in subjects both with and without concomitant hydroxyurea therapy.
FIGURE 2.
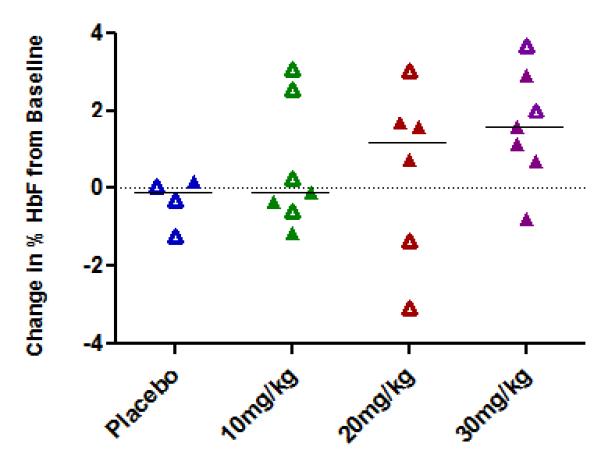
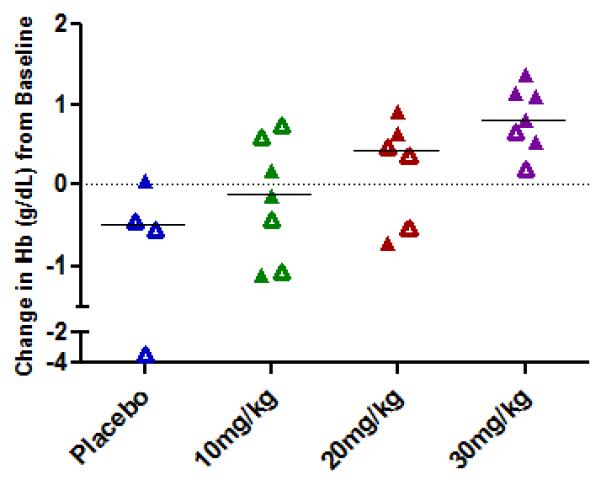
Changes from baseline in % HbF (top panel) and total Hgb (bottom panel) in the 3 dose cohorts.
A. Changes in % HbF are shown in individual subjects in each of the 4 treatment groups. Open symbols represent values in subjects on concomitant hydroxyurea treatment.
B. Changes from baseline in total Hgb are shown in individual subjects in each of the 4 treatment groups. Open symbols represent values in subjects on concomitant hydroxyurea treatment.
DISCUSSION
Sickle cell disease, a WHO-designated global health burden, still causes significant pain and disability in many patients, despite the beneficial effects of hydroxyurea in approximately half of treated adult patients, and higher responses in most children.15-21 Elevation of fetal hemoglobin in sickle cell disease is well-established as a modulator of disease severity in sickle cell disease, and agents which can be used without added toxicities with hydroxyurea are needed for many patients.4,12,23 Sodium dimethylbutyrate, an orally bioavailable, short chain fatty acid derivative fetal globin gene inducer was well-tolerated in this Phase I/II trial. Pharmacokinetic studies demonstrated profiles suitable for once daily dosing, which is preferable over multiple daily dosing in a treatment requiring long-term use.
Dose-limiting toxicity was not identified at the dose levels studied here. HU trials typically demonstrate increases in HbF after 6 months of treatment, and HbF responses in adult patients in the MSH trial averaged 3.5% above baseline.19 Achieving HbF levels > 0.5 g/dl is associated with increased survival in long (9 and 17 year) follow-up studies of HU in adult sickle cell patients.17-18
This dose-ranging safety trial was not designed, dosed for an adequate duration of time, or powered to detect changes in HbF. However, some increases in HbF were detected in subjects treated with the study drug and the findings suggest a trend of increasing response rates and a higher magnitude of effect on HbF and total Hb as the dose was increased. The early signals in HbF observed in this safety study, particularly at the 30 mg/kg/dose level, are therefore encouraging, and also suggest that higher HbF levels may be inducible with longer duration of administration.
Sickle cell disease is characterized by anemia related to short red cell lifespan, intravascular hemolysis, and inappropriately low levels of erythropoietin for the degree of anemia.2-3,11,35 The anemia typically becomes more severe with increasing age, perhaps related to renal compromise and reduced erythropoietin levels. This HbF-inducing therapeutic candidate, HQK-1001, was selected for clinical development for its improved PK profile in non-human primates compared to prior generation butyrates, and a second activity of increasing erythroid cell proliferation through STAT-5 signaling and enhanced Bcl-xL expression, in vitro and in vivo, in anemic baboons.29-31 Demonstration of any erythropoietic effect was not expected over the brief time-frame of this study in sickle cell disease, in the presence of markedly reduced red blood cell lifespan of 16 days, so it is encouraging that increases in total hemoglobin (mean rise of 0.8 g/dl) occurred in the 30 mg/kg dose cohort. It is also interesting that changes in total Hgb were observed to a lesser extent in subjects receiving concomitant hydroxyurea treatment, as a proliferative effect would be expected to be inhibited by the cytostatic effects of hydroxyurea.
Steinberg and others have strongly recommended that a combination therapeutic approach is needed to modify clinical disease in many patients with sickle cell disease, as multiple agents acting by different mechanisms are usually required for effective control of most systemic medical conditions, and differences in drug metabolism often render response rates to any drug 25-40% at best.10,23,35,38
Previous studies by Franco and colleagues documented that red cells which express HbF (F-cells), whether naturally-occurring or associated with a pharmacologic treatment, undergo selective survival and become enriched by 2-to 3-fold in sickle cell disease, resulting in a progressive increase in total HbF levels over time.10 These findings would predict that longer administration of HQK-1001 should result in higher levels of HbF than observed in this relatively brief trial in small cohorts.
The complex pathophysiology of sickle cell disease has been increasingly well-characterized, with multiple therapeutic targets identified, yet only one therapeutic to reduce the underlying pathology has been approved in over a decade. Stem cell transplant, the one curative approach, is available for a minority of patients for whom appropriate donors are found.37 Despite documented benefit from HbF induction >0.5 g/dl with hydroxyurea, many adults still suffer chronic morbidity, and there is still an early mortality rate > 40%.17-18 Several prior generation short chain fatty acids have HbF-inducing activity, and are appealing for their targeted effects in displacing repressor complexes, including BCL-11A and HDAC-3, and recruiting EKLF, but their feasibility for chronic use is problematic due to pharmacokinetic challenges.28,32,38-9 These early clinical findings now with sodium 2,2 dimethylbutyrate, in combination with prior studies in molecular, cellular, and nonhuman primate models, strongly suggest that HQK-1001 offers potential for increasing HbF in sickle cell disease without cytotoxicity, and its pharmacokinetic profile is feasible for long-term administration. Basal HbF levels vary widely in association with diverse genetic modifier profiles, which are likely to influence responses to HbF-inducing therapeutics. 40-48 It is therefore encouraging that five of seven subjects who received the highest dose level had a detectable rise in HbF above baseline in the presence of quite different basal HbF levels in this brief time-frame. Further trials with longer administration of HQK-1001 in larger numbers of sickle cell subjects appear warranted to more definitively evaluate its therapeutic potential.
Table IC. Baseline total hemoglobin and % HbF in individual subjects. Hydroxyurea treatment status is designated in the far right column.
| ID | Group | Hb (g/dL) | HbF (%) | HU |
|---|---|---|---|---|
| 90301 | Placebo | 10.0 | 3.0 | Yes |
| 90803 | Placebo | 8.7 | 18.3 | Yes |
| 91101 | Placebo | 7.4 | 2.5 | |
| 91103 | Placebo | 9.9 | 10.8 | Yes |
|
| ||||
| 90101 | 10 mg/kg | 9.2 | 7.7 | Yes |
| 90102 | 10 mg/kg | 8.8 | 5.8 | Yes |
| 90103 | 10 mg/kg | 11.9 | 14.7 | Yes |
| 90202 | 10 mg/kg | 9.3 | 8.4 | |
| 90203 | 10 mg/kg | 8.0 | 2.9 | |
| 91102 | 10 mg/kg | 6.1 | 1.7 | |
| 91104 | 10 mg/kg | 5.9 | 12.2 | Yes |
|
| ||||
| 90105 | 20 mg/kg | 9.5 | 32.8 | Yes |
| 90204 | 20 mg/kg | 8.5 | 8.3 | |
| 90205 | 20 mg/kg | 9.2 | 7.0 | |
| 90801 | 20 mg/kg | 11.8 | 10.2 | Yes |
| 90802 | 20 mg/kg | 9.9 | 28.2 | Yes |
| 90901 | 20 mg/kg | 11.4 | 12.2 | |
|
| ||||
| 90106 | 30 mg/kg | 10.3 | 13.0 | |
| 90206 | 30 mg/kg | 6.3 | 6.4 | |
| 90804 | 30 mg/kg | 9.5 | 19.8 | Yes |
| 90805 | 30 mg/kg | 8.4 | 2.9 | |
| 90806 | 30 mg/kg | 10.2 | 8.7 | Yes |
| 90902 | 30 mg/kg | 7.5 | 3.7 | |
| 91301 | 30 mg/kg | 7.6 | 22.2 | |
Acknowledgement
We thank Drs. Paul Swerdlow, George Atweh, Adam Lerner, David Nathan, Curt Scribner, and Wayne Wallis for helpful advice and the NIH RAID Pilot Program for making this trial possible.
REFERENCES
- 1.Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115:4331–4336. doi: 10.1182/blood-2010-01-251348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762–769. doi: 10.1056/NEJM199709113371107. [DOI] [PubMed] [Google Scholar]
- 3.Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical sub-phenotypes. Blood Rev. 2006;21:37–47. doi: 10.1016/j.blre.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinberg MH, Rodgers GP. Pharmacologic modulation of fetal hemoglobin. Medicine. 2001;80:328–44. doi: 10.1097/00005792-200109000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Watson J. The significance of the paucity of sickle cells in newborn Negro infants. Amer J Med Sci. 1948;215:419–423. doi: 10.1097/00000441-194804000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Goldberg MA, Husson MA, Bunn HF. Participation of hemoglobins A and F in polymerization of sickle hemoglobin. J Biol Chem. 1977;252:3414–3421. [PubMed] [Google Scholar]
- 7.Nagel RL, Bookchin RM, Johnson J, Labie D, et al. Structural bases of the inhibitory effects of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S. Proc Natl Acad Sci USA. 1979;76:670–2. doi: 10.1073/pnas.76.2.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perrine RP, Brown MJ, Clegg JB, Weatherall DJ, et al. Benign sickle-cell anaemia. Lancet. 1972;2:1163–7. doi: 10.1016/s0140-6736(72)92592-5. [DOI] [PubMed] [Google Scholar]
- 9.Perrine RP, Pembrey ME, John P, Perrine S, Shoup F. Natural history of sickle cell anemia in Saudi Arabs. A study of 270 subjects. Ann Intern Med. 1978;88:1–6. doi: 10.7326/0003-4819-88-1-1. [DOI] [PubMed] [Google Scholar]
- 10.Franco RS, Yasin Z, Palascak MB, Ciraolo P, et al. The effect of fetal hemoglobin on the survival characteristics of sickle cells. Blood. 2006;108:1073–6. doi: 10.1182/blood-2005-09-008318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perrine SP. Fetal globin stimulant therapies in the beta-hemoglobinopathies: principles and current potential. Pediatr Ann. 2008;37:339–46. doi: 10.3928/00904481-20080501-10. [DOI] [PubMed] [Google Scholar]
- 12.Steinberg MH. Predicting clinical severity in sickle cell anaemia. Br J Haematol. 2005;129:465–81. doi: 10.1111/j.1365-2141.2005.05411.x. [DOI] [PubMed] [Google Scholar]
- 13.Platt OS, Thorington BD, Brambilla DJ, Milner PF, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med. 1995;325:11–16. doi: 10.1056/NEJM199107043250103. [DOI] [PubMed] [Google Scholar]
- 14.Platt OS, Brambilla DJ, Rosse WF, Milner PF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–44. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 15.Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on frequency of painful crises in sickle cell anemia. Investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med. 1995;332(2):1317–1322. doi: 10.1056/NEJM199505183322001. RD, et al. [DOI] [PubMed] [Google Scholar]
- 16.Charache S, Dover GJ, Moyer MA, Moore JW. Hydroxyurea-induced augmentation of fetal hemoglobin production in patients with sickle cell anemia. Blood. 1987;69:109–16. [PubMed] [Google Scholar]
- 17.Steinberg MH, Barton F, Castro O, Pegelow CH, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289:1645–51. doi: 10.1001/jama.289.13.1645. [DOI] [PubMed] [Google Scholar]
- 18.Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol. 2010;85:403–408. doi: 10.1002/ajh.21699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinberg MH, Lu ZH, Barton FB, Terrin ML, et al. Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Multicenter study of hydroxyurea. Blood. 1997;89:1078–88. [PubMed] [Google Scholar]
- 20.Wang CW, Ware RE, Miller ST, Iyer RV, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomized, controlled trial (BABY HUG) Lancet. 2011;377:1663–1672. doi: 10.1016/S0140-6736(11)60355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inati A, Koussa S, Taher A, et al. Sickle cell disease; new insights into pathophysiology and treatment. Pediatr Ann. 2008;37:311–321. doi: 10.3928/00904481-20080501-07. [DOI] [PubMed] [Google Scholar]
- 22.Noguchi CT, Rodgers GP, Serjeant G, Schechter AN. Levels of fetal hemoglobin necessary for treatment of sickle cell disease. N Engl J Med. 1988;318:96–99. doi: 10.1056/NEJM198801143180207. [DOI] [PubMed] [Google Scholar]
- 23.Steinberg MH. Clinical trials in sickle cell disease: adopting the combination chemotherapy paradigm. Am J Hematol. 2008;83:1–3. doi: 10.1002/ajh.21033. [DOI] [PubMed] [Google Scholar]
- 24.Koshy M, Dorn L, Bressler L, Molokie R, et al. 2-Deoxy 5-azacytidine and fetal hemoglobin induction in sickle cell anemia. Blood. 2000;96:2379–2384. [PubMed] [Google Scholar]
- 25.Saunthararajah Y, Hillery CA, Lavelle D, et al. Effects of 5-aza-2′-deoxycytidine on fetal hemoglobin levels, red cell adhesion, and hematopoietic differentiation in patients with sickle cell disease. Blood. 2003;102:3865–3870. doi: 10.1182/blood-2003-05-1738. [DOI] [PubMed] [Google Scholar]
- 26.Atweh GF, Sutton M, Nassif I, Boosalis V, et al. Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. Blood. 1999;93:790–797. [PMC free article] [PubMed] [Google Scholar]
- 27.Perrine SP, Ginder GD, Faller DV, Dover GH, et al. A short-term trial of butyrate to stimulate fetal-globin gene expression in the beta-globin disorders. N Engl J Med. 1993;328:81–86. doi: 10.1056/NEJM199301143280202. [DOI] [PubMed] [Google Scholar]
- 28.Ikuta T, Kan Y, Swerdlow PS, Faller DV, et al. Alterations in protein-DNA interactions in the gamma-globin gene promoter in response to butyrate therapy. Blood. 1998;92:2924–33. [PubMed] [Google Scholar]
- 29.Boosalis MS, Bandyopadhyay R, Bresnick EH, Pace BS, et al. Short-chain fatty acid derivatives stimulate cell proliferation and induce STAT-5 activation. Blood. 2001;7:3259–67. doi: 10.1182/blood.v97.10.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pace BS, White GL, Dover GJ, Boosalis MS, et al. Short-chain fatty acid derivatives induce fetal globin expression and erythropoiesis in vivo. Blood. 2002;100:4640–8. doi: 10.1182/blood-2002-02-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castaneda S, Boosalis MS, Emery D, Thies A, et al. Enhancement of growth and survival and alterations in Bcl-family proteins in beta-thalassemic erythroid progenitors by novel short-chain fatty acid derivatives. Blood Cells, Mol, Dis. 2005;35:217–26. doi: 10.1016/j.bcmd.2005.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perrine SP, Mankidy R, Boosalis MS, Bieker JJ, et al. Erythroid Krüppel-like factor (EKLF) is recruited to the gamma-globin gene promoter as a co-activator and is required for gamma-globin gene induction by short-chain fatty acid derivatives. Eur J Haematol. 2009;82:466–76. doi: 10.1111/j.1600-0609.2009.01234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perrine SP, Wargin WA, Boosalis MS, Wallis WJ, Case S, Keefer JR, Faller DV, Welch WC, Berenson RJ. Evaluation of safety and pharmacokinetics of sodium 2, 2 dimethylbutyrate, a novel short chain fatty acid derivative, in a phase 1, double-blind, placebo-controlled, single- and repeat-dose studies in healthy volunteers. J Clin Pharm. 2011;51(8):1186–94. doi: 10.1177/0091270010379810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagel RL, Vichinsky EP, Shah M, et al. F-reticulocyte response in sickle cell anemia treated with recombinant human erythropoietin: a double blind study. Blood. 1992;81:9–14. [PubMed] [Google Scholar]
- 35.Sherwood JB, Goldwasser E, Chilcote R, et al. Sickle cell anemia patients have low erythropoietin levels for their degree of anemia. Blood. 1986;67:46–49. [PubMed] [Google Scholar]
- 36.Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352:2211–2221. doi: 10.1056/NEJMra032424. [DOI] [PubMed] [Google Scholar]
- 37.Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med. 2009;361:2309–2317. doi: 10.1056/NEJMoa0904971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Resar LM, Segal JB, Fitzpatric LK, Friedmann A, et al. Induction of fetal hemoglobin synthesis in children with sickle cell anemia on low-dose oral sodium phenylbutyrate therapy. J Pediatr Hematol Oncol. 2002;24:737–41. doi: 10.1097/00043426-200212000-00011. [DOI] [PubMed] [Google Scholar]
- 39.Mankidy R, Faller DV, Mabaera R, Lowrey CH, Perrine SP. Short-chain fatty acids induce gamma-globin gene expression by displacement of a HDAC3-NCoR repressor complex. Blood. 2006;108:3179–86. doi: 10.1182/blood-2005-12-010934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Z, Luo H, Steinberg MH, Chui DHK. BCL11A represses HBG transcription in K562 cells. Blood Cells Mols, Dis. 2009;41:144–149. doi: 10.1016/j.bcmd.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 41.Thein SL, Menzel S. Discovering the genetics underlying foetal haemoglobin production in adults. Br J Haematol. 2009;145:455–467. doi: 10.1111/j.1365-2141.2009.07650.x. [DOI] [PubMed] [Google Scholar]
- 42.Miller BA, Salameh M, Ahmed M, et al. Analysis of hemoglobin F production in Saudi Arabian families with sickle cell anemia. Blood. 1987;70:716–720. [PubMed] [Google Scholar]
- 43.Pembrey ME, Wood WG, Weatherall DJ, Perrine RP. Fetal haemoglobin production and the sickle gene in the oases of eastern Saudi Arabia. Br J Haematol. 1978;40:415–29. doi: 10.1111/j.1365-2141.1978.tb05813.x. [DOI] [PubMed] [Google Scholar]
- 44.Dover GJ, Smith KD, Chang YC, Purvis S, et al. Fetal hemoglobin levels in sickle cell disease and normal individuals are partially controlled by an X-linked gene located at Xp22.2. Blood. 1992;80:816–24. [PubMed] [Google Scholar]
- 45.Chang YC, Smith KD, Moore RD, Serjeant GR, et al. An analysis of fetal hemoglobin variation in sickle cell disease: the relative contributions of the X-linked factor, beta-globin haplotypes, alpha-globin gene number, gender and age. Blood. 1995;85:1111–7. [PubMed] [Google Scholar]
- 46.Ma Q, Wyszynski DF, Farrell JJ, Kutlar A, Farrer LA, Baldwin CT, Steinberg MH. Fetal hemoglobin in sickle cell anemia: genetic determinants of response to hydroxyurea SNPs, hydroxyurea and HbF in sickle cell anemia. Pharmacogenomics J. 2007;7:386–394. doi: 10.1038/sj.tpj.6500433. [DOI] [PubMed] [Google Scholar]
- 47.Akinsheye I, Alsultan A, Solovieff N, et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118:4495. doi: 10.1182/blood-2011-03-325258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perrine SP, Faller DV, Shen L, Boosalis MS, et al. HQK-1001 has additive HbF-inducing activity in combination with hydroxyurea and decitabine. Blood. 2009;114:977. [Google Scholar]
- 49.Sobash P, Kutlar F, Brown HC, Clair B, Kutlar A. BCL11A polymorphism and HbF response to hydroxyurea in adult patients with sickle cell disease. Blood. 2011;118:484a. [Google Scholar]
- 50.Lavelle D, Saunthararajah Y, Vaitkus K, Singh M, et al. S110, a novel decitabine dinucleotide, increases fetal hemoglobin levels in baboons (P. anubis) Journal of Translational Medicine. 2010;8:92. doi: 10.1186/1479-5876-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]