

Multiple myeloma (MM) is a heterogeneous disease originating from the transformation of a post-germinal center B cell. MM pathogenesis is a multistep process with chromosomal changes, genetic and epigenetic events occurring at different stages during the course of the disease.1
MicroRNAs (miRNAs) are well known to be transcriptional repressors, possessing tumor suppressor (TS) or oncogenic function. They are involved in several signaling pathways affecting cell differentiation, proliferation, and development. miRNAs involvement in solid tumors and hematological malignancies has been well documented.2 Deregulated miRNAs and their interaction with factors with an established role in MM biology, such as p53, VEGF-A, NFκB, cyclins, and several other factors has been demonstrated in different stages of the disease.3,4 miRNAs expression can be epigenetically regulated by promoter hypermethylation and histone modifications, whereas they might target directly or indirectly the epigenetic machinery (epi-miRNAs). The miR-29 family members belong to the epi-miRNAs, as they directly affect DNA methyltransferase 3a/b (DNMT3a/b) expression.5 It has been previously demonstrated in xenograft models that the TS miR-29b has in vivo anti-MM activity. Most interestingly, miR-29b was attributed with demethylating activity by direct negative regulation of DNMT3a/b in MM cells. Thus, that particular miRNA might promote epigenetic changes in malignant plasma cells by reducing global DNA methylation levels.6
The most recent data published by Prof. Tassone’s collaborative group7 describe the effect of miR-29b on reverting the aberrantly hypermethylated profile of SOCS-1 gene promoter in MM and the biological significance of such interplay. Authors showed that SOCS-1 mRNA was downregulated in MM primary samples and positively correlated with miR-29b levels. Using Sequenom MassArray EpiTYPER, a high-throughput quantitative locus-specific analysis method, they identified differentially methylated CpG sites within the amplicons of SOCS-1 promoter that were fully demethylated after transfection with miR-29b synthetic mimics. In fact, enhanced expression of miR-29b resulted in increased SOCS-1 protein levels in MM cell lines. Demethylation was probably achieved through inhibition of DNMT3 by the re-expressed miR-29b, as previously reported.6 Moreover, it was demonstrated that bortezomib enhanced the expression of miR-29b and indirectly induced demethylation and consequent upregulation of SOCS-1. SOCS-1-restored expression was miR-29b-dependent and proteasome-independent. These findings highlight another putative mechanism of bortezomib action, as it seems that indirectly affects epigenetic modifications through modulation of miR-29b expression. SOCS-1 is known to be involved in the regulation of cell proliferation, migration, and adhesion of cancer cells through inhibition of the JAK/STAT signaling pathway. miR-29b mimics transfection resulted in reduced STAT3 (frequently constitutively active in MM) phosphorylation and subsequent downregulation of STAT3 downstream targets such as VEGF-A, IL-8, MMP2, and NFκB. These factors are well known to be deregulated in MM, contributing to disease pathogenesis and propagation. The final result of miR-29b-enhanced expression was the decreased angiogenesis, motility, and migration of plasma cells. According to the authors, this could be considered a paracrine effect of miR-29b on bone marrow microenvironment. Overall, in light of these recent results, a miR29b─DNMT3─SOCS1─STAT3 downstream effectors axis can be proposed (Fig. 1).
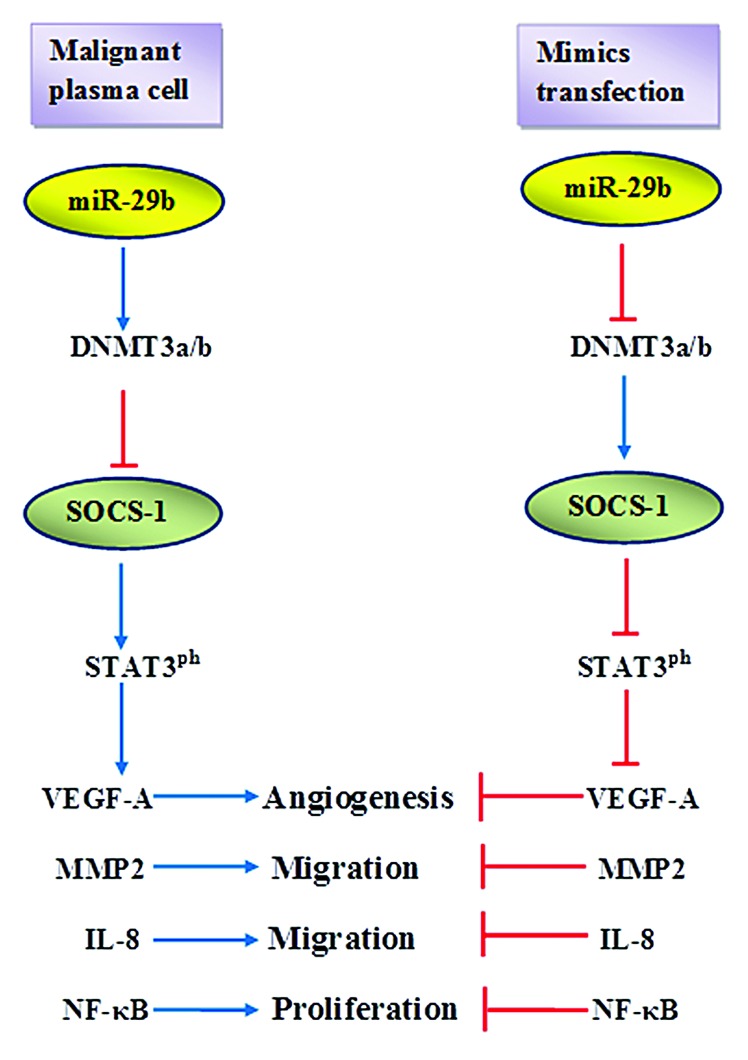
Figure 1. In malignant plasma cells, the initial downregution of miR-29b results in increased levels of DNMT3a/b which in turn causes epigenetic silencing of SOCS-1, increases STAT3 phosphorylation, and upregulation of downstream factors promoting angiogenesis, proliferation, and migration of plasma cells. miR-29b mimics transfection downregulates DNMT3a/b, leading to enhanced SOCS-1 expression which subsequently inhibits STAT3 phosphorylation and indirectly blocks downstream effectors.
The findings reported in this paper have further implications. First, synthesis of oligomiRNAs (miR-29b mimics) could have clinical application by promoting re-expression of miR-29b, demethylation of target TS genes, and inactivation of pathogenetic factors in a cascade of events. Second, bortezomib was found to exhibit an indirect effect on DNA methylation via modulation of miR-29b expression through a still-unidentified mechanism. However, further investigation is needed, because bortezomib could similarly upregulate oncogenic miRNAs. Furthermore, it would be interesting to study whether miR-29b is involved in active DNA demethylation, which results from the removal of methyl group from the 5-methylcytosine (5meC). The ten-11 translocation 1–3 (TET1–3) family of proteins, which play a role in several developmental processes including hematopoiesis, have been established as capable of oxidizing 5meC to 5-hydroxymethylcytosine, thus promoting active DNA demethylation.8 According to miRanda and RNA22 prediction tools, TET2 is a miR-29b putative target, and therefore, a possible functional interaction between miR-29b and TET2 should be evaluated in active DNA demethylation in the context of MM.
In conclusion, pharmacological re-expression of miR-29b in MM should be explored given that it affects epigenetic modifications and indirectly regulates several fundamental pathogenetic factors.
Amodio N, et al. Cell Cycle. 2013;12:3650–62. doi: 10.4161/cc.26585.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26975
References
- 1.Palumbo A, et al. N Engl J Med. 2011;364:1046–60. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 2.Zhao H, et al. Crit Rev Oncol Hematol. 2010;74:149–55. doi: 10.1016/j.critrevonc.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Benetatos L, et al. Cancer. 2012;118:878–87. doi: 10.1002/cncr.26297. [DOI] [PubMed] [Google Scholar]
- 4.Tagliaferri P, et al. Curr Cancer Drug Targets. 2012;12:838–46. doi: 10.2174/156800912802429355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, et al. Eur J Cell Biol. 2013;92:123–8. doi: 10.1016/j.ejcb.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Amodio N, et al. Oncotarget. 2012;3:1246–58. doi: 10.18632/oncotarget.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amodio N, et al. Cell Cycle. 2013;12:3650–62. doi: 10.4161/cc.26585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen L, et al. Curr Opin Cell Biol. 2013:25289–96. [Google Scholar]