

Abstract
p31Comet is a well-known interacting partner of the spindle assembly checkpoint (SAC) effector molecule Mad2. At the molecular level it is well established that p31Comet promotes efficient mitotic exit, specifically the metaphase–anaphase transition, by antagonizing Mad2 function. However, there is little knowledge of how p31Comet is regulated or the physiological importance of controlling p31Comet. Here, we show that the Rb–E2F pathway regulates p31Comet expression. In multiple tumor types (including breast and lung) p31Comet expression is increased along with Mad2. Expression of this antagonist–target pair is coordinated in cells and correlated in cancer. Moreover, a narrow range of p31Comet:Mad2 ratios is compatible with cellular viability. Our data suggest that coordinate regulation is important for the outgrowth of oncogenic cell populations. Our findings suggest that altered p31Comet:Mad2 expression ratios may provide new insight into altered SAC function and the generation of chromosomal instability in tumors.
Keywords: spindle assembly checkpoint, Mad2, p31Comet, Rb, E2F, cell cycle, cancer
Introduction
The spindle assembly checkpoint (SAC) is an essential, evolutionarily conserved cellular surveillance mechanism, which ensures that chromosomes segregate with high fidelity during mitosis. In prometaphase (early mitosis), chromosomes lacking bipolar microtubule attachments activate the SAC. The Mad1–Mad2 heterodimer binds the kinetochores of unattached chromosomes and catalyzes the activation of free Mad2. Active Mad2, along with the BubR1-Bub3 heterodimer, bind and inhibit the ubiquitin ligase, APCCdc20, triggering mitotic arrest. Upon bipolar attachment of all chromosomes, the SAC is satisfied and APCCdc20 is released to drive chromosome segregation and mitotic exit, allowing continued cellular proliferation.
Chromosomal instability is widespread in cancer. Deficient SAC function has long been suspected to underlie this hallmark trait in tumors. Indeed, loss or mutation of SAC components clearly drives chromosomal instability in both animal and cell culture models.1-9 However, evidence for a cancer association of such defects in SAC function is lacking, likely due to the essential nature of the SAC.10-15 In contrast, many SAC components, including the effector molecules Mad2 and BubR1 are overexpressed in tumors.16,17 In particular, recent work in cell and animal models has demonstrated that hyperactivation of the SAC, caused by abnormally high levels of Mad2, drives chromosomal instability leading to tumorigenesis.9,16-18 However, Mad2-mediated hyperactivation of the SAC also leads to increased mitotic duration, reduced proliferation, and decreased viability.18 The mechanism that allows Mad2-overexpressing cells to escape the potentially tumor-suppressive characteristics of the hyperactive SAC remains to be determined.
The p31Comet protein was identified as a binding partner of Mad2.19 Overexpression of p31Comet bypasses hyperactivation of the SAC caused by microtubule poisons.20-23 Depletion of p31Comet delays the release of APCCdc20 from the Mad2–BubR1 complex after drug-induced activation of the SAC.22-25 p31Comet functions by antagonizing Mad2 and mutants that fail to bind Mad2 fail to silence SAC activity.20 The exact mechanism that p31Comet employs to silence SAC activity, e.g., preventing the generation of active Mad2 by the Mad1–Mad2 heterodimer, removing Mad2 from the inhibited Cdc20 complex, facilitating Mad2-mediated destruction of Cdc20, or all three, is still being delineated.20,22,23,26-28 Regardless of the mechanism, while many analyses of p31Comet function have been performed after treatment with microtubule poisons, which hyperactivate the SAC, we know relatively little about the biological importance of p31Comet in the absence of drug. In addition, regulation of p31Comet is essentially unknown at all levels. We therefore asked whether p31Comet is abnormally expressed in human cancers and whether this might be important for the growth of cells with intrinsically high SAC activity due to Mad2 overexpression. Here, we have analyzed the expression of p31Comet and examined the importance of coordinately increasing p31Comet expression along with Mad2 for cellular survival.
Results
p31Comet expression is upregulated in cancer
p31Comet is a well-known antagonist of the SAC effector Mad2, yet the biological importance of SAC silencing is largely uncharacterized.19-28 We queried the Oncomine database to examine the expression of p31Comet in human cancers (Fig. 1A). p31Comet expression is upregulated in tumors originating from multiple tissues. Notably, p31Comet expression is increased in tumors of breast and lung, where upregulation of Mad2 is known promote to tumorigenesis. To extend these analyses to changes in protein expression, we employed a highly characterized in vitro model of human mammary epithelial cell (HMEC) immortalization and transformation.29-38 HMECs undergo 2 major events, stasis and crisis, en route to full immortalization and susceptibility to transformation. Overcoming these hurdles is tightly associated with the silencing or functional loss of the tumor suppressors p16Ink4A and p53, respectively.34,39,40 We mimicked successful navigation of stasis and crisis by sequential introduction of p16Ink4A - and p53-targeting shRNAs (for full characterization of these cells, see ref. 29). p31Comet protein levels were barely detectable in primary HMECs (Fig. 1B and data not shown). In contrast, loss of p16Ink4A triggered a dramatic increase in p31Comet protein that was unaffected by the introduction of the oncogenes c-Myc and RasG12V. Mad2 protein levels exhibited a similar trend of increased expression, whereas Cyclin A levels exhibited a modest increase at this stage (Fig. 1B). These data suggest that increased p31Comet expression is not due to dramatic increase in proliferation, as Cyclin A is also required for proliferation. Thus, our data suggest that p31Comet increases along with its target Mad2, perhaps to attenuate the deleterious effects associated with Mad2 overexpression (see below).

Figure 1. Expression levels of p31Comet increase in cancer. (A) Comparison of p31Comet mRNA expression differences between normal and tumor samples for multiple tissues. The mean expression for each sample group is represented. The number of samples in each group are indicated in parentheses. Data were obtained from the Oncomine database. (B) p31Comet protein levels increase during the immortalization/transformation of HMECs. Cell lysates of primary HMECs and derivatives expressing the indicated shRNAs or oncogenes were probed for the indicated proteins by immunblot analysis.
p31Comet expression is regulated by Rb-E2F
p16Ink4A is a Cdk inhibitor and its loss leads to aberrant phosphorylation of the Rb tumor suppressor and subsequent activation of E2F family transcription factors. Consistent with its increased expression in this cell population, Mad2 is a direct target of the Rb–E2F pathway.9,17 We therefore investigated whether p31Comet might also be regulated by this pathway. To determine if p31Comet expression is directly regulated by E2F activity rather than indirectly via an E2F target, we sought to determine whether E2Fs act on the p31Comet promoter. We probed a repository of ChIP–chip data in the ENCODE database at UCSC (http://genome-preview.ucsc.edu/ENCODE/) to determine whether E2Fs bind to the p31Comet promoter. Indeed, all of the E2Fs tested in the ENCODE project, E2F1, E2F4, and E2F6, as well as exogenous HA-E2F1, bound to regions proximal to the transcription start sites of both p31Comet variants 1 and 2 (Fig. 2A and not shown).

Figure 2. p31Comet expression is controlled by the Rb-E2F pathway. (A) Schematic representation of the p31Comet genomic structure, indicating the position of exons and the transcription start sites for variants 1 and 2. The positions of E2F1 binding peaks for both variants as defined by ChIP–chip data form the ENCODE project are depicted for HeLa and MCF-7 cells. Similar peaks were identified for E2F4 and E2F6. (B) E2F1 drives p31Comet protein expression. HCT116 cells were transfected with control or HA-E2F1 constructs and harvested 24 h later for immunoblot analysis. (C) Loss of Rb family proteins results in increased p31Comet expression. Lysates of 3T3 cells derived from MEFs with the indicated genotypes were probed for protein expression. (D) Rb represses the expression of p31Comet. SAOS2 cells with tetracycline-inducible Rb construct were treated ± doxycycline to induce Rb expression for 24 h. Whole-cell lysates were probed with the indicated antibodies. (E and F) Expression data for the p31Comet and the indicated E2F family member were obtained and subject to correlation analysis.
The ENCODE data supported E2Fs as drivers of p31Comet expression. To test this possibility, we asked whether modulating E2F activity alters p31Comet expression. We expressed HA-E2F1 in HCT116 cells, where E2F1 has been shown to modulate Mad2 expression.17 p31Comet expression was increased by E2F1 (Fig. 2B). As controls, we monitored the protein levels of additional E2F1 targets, Mad2 and Cyclin A, which increased as expected (Fig. 2B). Because exogenous E2F activity drives p31Comet expression, we expected that cells with intrinsically higher endogenous E2F activity would also have higher p31Comet expression. We found that Rb−/− 3T3 cells express higher levels of p31Comet than wild-type 3T3 cells (Fig. 2C). Similar results were obtained in cells that have been deleted for the Rb family members p107 and p130 (Fig. 2C). These results are in good agreement with our initial observation that p31Comet is increased upon depletion of p16 in HMECs, which affects the activity of all 3 Rb family proteins (Fig. 1B). In contrast, reduction of E2F activity by expression of tetracycline-inducible Rb in the Rb−/− cell line SAOS-2, results in a loss of p31Comet as well as Cyclin A (Fig. 2D).41 Together with the ENCODE data, our results support p31Comet as an E2F target gene. Moreover, the ENCODE data represents E2F family binding to the p31Comet promoter in cells derived from non-transformed breast, breast cancer, cervical cancer, B lymphocytes, and testicular cancer, indicating that p31Comet is regulated by E2F in multiple tissues and tumor types. Indeed, correlation analyses of p31Comet expression with that of E2F family members indicate that multiple E2Fs target p31Comet (Fig. 2E and F). We analyzed the correlation between p31Comet expression and E2F family members in expression data for invasive ductal breast cancer and lung adenocarcinoma obtained from the Oncomine database. In breast cancer samples, p31Comet expression correlated most significantly with E2F5 (Fig. 2E). In contrast p31Comet expression correlated most highly with E2F4 in lung cancer (Fig. 2F). Similar results were obtained with Mad2 (not shown). Notably the regulation of p31Comet by multiple E2Fs is consistent with regulation by multiple Rb family members, which exhibit differential association with E2F proteins. These data indicate that regulation of p31Comet (as well as Mad2) by E2Fs may be stochastic, and that allowing multiple family members to drive expression would ensure coordinate regulation of protein levels.
p31Comet expression is cell cycle-regulated
Regulation by Rb-E2F suggests that p31Comet expression may be cell cycle-regulated. We examined p31Comet protein levels in HeLa cells that were synchronized at the G1/S by a double-thymidine block and followed through to the next S phase. Levels of p31Comet and Mad2 proteins did not oscillate dramatically during the cell cycle (Fig. 3A). In contrast, levels of the additional E2F targets and mitotic regulators Securin and Cyclin B dropped precipitously after mitosis and then re-accumulated as cells entered the next S phase (Fig. 3B). We reasoned that this discrepancy might reflect differences in the requirements to destroy these proteins in order to complete mitosis. Because rapid destruction of p31Comet and Mad2 is not required, residual protein in cycling cells may mask the appearance of newly synthesized proteins as cells progress toward the next mitosis. Moreover, as Rb function is compromised in HeLa cells, the window of p31Comet expression during the cell cycle may be less defined, contributing to the lack of oscillating protein levels. We therefore examined the expression of p31Comet in Rb-proficient T98G cells that had been blocked in G0 by serum starvation and stimulated to enter the cell cycle by addition of serum. Under these conditions we were able to observe an increase in p31Comet expression as cells progressed through the cell cycle. The increase in expression occurred at both the mRNA and protein levels. Moreover, p31Comet expression kinetics mirrored those of additional E2F targets, Mad2 and Cyclin A (Fig. 3B and C). Together these data support p31Comet as a cell cycle-regulated E2F target gene.
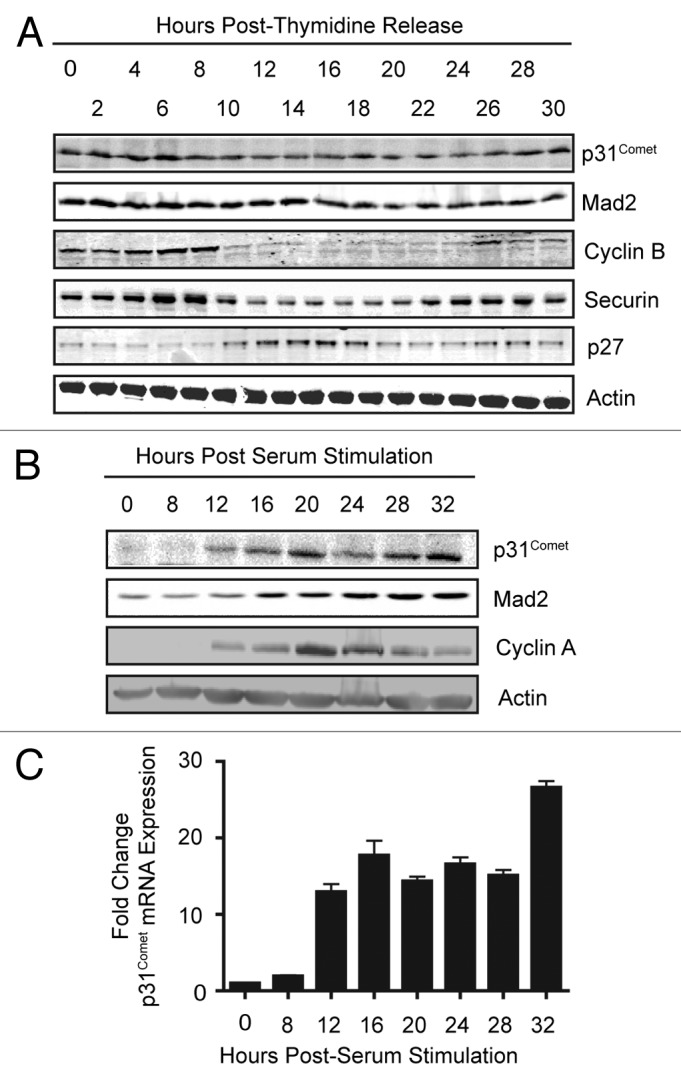
Figure 3. p31Comet expression is cell cycle regulated. (A) HeLa cells were synchronized at G1/S by a double thymidine block. After release into fresh media (0 h) cells were harvested at the indicated times. Whole-cell lysates were probed with antibodies against the proteins specified. (B and C) T98G cells were synchronized in G0 by incubation in serum-free media for 72 h. Cells were stimulated to reenter the cell cycle by addition of media containing serum, serum stimulation. Whole-cell lysates were probed with antibodies against the specified proteins (B), and mRNA collected in parallel samples was analyzed by qPCR (C).
Balanced expression of p31Comet and Mad2 is required for cellular proliferation
The results of our studies are consistent with the described role of p31Comet in promoting efficient mitotic exit.22-25 However, our data also has additional implications for the biological importance of p31Comet. Increased expression of Mad2 leads to hyperactivation of the SAC and diminished cellular viability and proliferation.18 Similarly, overexpression of p31Comet also results in decreased proliferative capacity.9,18 We reasoned that, in addition to being part of a cell cycle-regulated transcriptional module, coordinate regulation of both p31Comet and Mad2 by the Rb–E2F pathway ensures that the potential deleterious effects of each these proteins when overexpressed are buffered by the expression of the other. To test this hypothesis, we first determined that induction of p31Comet or ectopic expression of Mad2 in tetracycline-inducible p31Comet HeLa cells dramatically reduced proliferation (Fig. 4A). As expected, simultaneous induction of p31Comet with ectopic expression of Mad2 resulted in partial restoration of cellular proliferation. Examination of the total p31Comet and Mad2 levels in these cells revealed that the relative p31Comet:Mad2 ratio remained altered in our coexpressing population (Fig. 4B and C). The partial rescue we observed in these cells is likely due to the imbalanced ratio. Indeed, the level of p31Comet in this population is unable to rescue the increased mitotic index caused by Mad2 overexpression (Fig. 4D). To further examine the importance of the p31Comet:Mad2 ratio, we co-transfected p31Comet along with increasing amounts of Mad2 plasmid (Fig. 4E and F). Increasing Mad2 levels first rescued the proliferative defect resulting from exogenous p31Comet expression. However, further increases in Mad2 levels were detrimental, despite the presence of exogenous p31Comet. These results indicate that there is a specific range of p31Comet:Mad2 ratios that are compatible with cellular proliferation.

Figure 4. Coordinated expression of p31Comet and Mad2 is required for cell viability. (A) HeLa cells harboring a tetracycline-inducible p31Comet construct were transfected with control or Mad2 containing constructs along with a plasmid encoding puromycin resistance ± doxycycline, as indicated. After selection with puromycin, cells were allowed to grow for 10 d and stained with crystal violet. (B) Representative western blot of cells treated as in (A). (C) Quantitation of the ratio of total p31Comet:Mad2 ratios from (B). (D) Mitotic index of cells treated as in (A). Cells were analyzed visually for the presence of mitotic chromatin. (E) HeLa cells were transfected with vector or constructs encoding p31Comet and Mad2 as indicated, along with a puromycin resistance plasmid. Cells were selected as in (A). After 10 d the cells were stained with anti-β-actin antibodies and analyzed using the LI-COR Odyssey scanner. The mean relative cell number and standard deviation from 2 experiments performed in triplicate are shown. (F) Representative immunoblots of cells treated in (E). (G) Expression data for p31Comet and Mad2 was obtained for the indicated data sets and subject to correlation analysis. (H) The ratio of p31Comet:Mad2 for normal and cancer subsets of the Curtis Breast and Okayama Lung datasets in (G) were calculated and compared. Gray bars indicate the median for each group.
p31Comet and Mad2 are coordinately upregulated in cancer
The results of our in vitro studies support a model in which p31Comet expression must increase (or potentially decrease) upon deregulation of Mad2 expression during the transformation process. To test this model, we downloaded expression data for both p31Comet and Mad2 from the Oncomine database and performed correlation analyses. In data sets from breast cancer, lung cancer, and, directly in line with our analyses in HeLa cells, cervical cancer expression of p31Comet is significantly correlated with Mad2 expression (Fig. 4G). These data are consistent with the need for coordinate regulation. Recently it was reported that the p31Comet:Mad2 ratio varies among cell lines. Given that we observed increased expression of both proteins in the early steps of immortalization, we asked whether the distribution of the p31Comet:Mad2 ratio may be skewed in cancer. Indeed, we found that the median p31Comet:Mad2 ratio is substantially altered in tumor cells (Fig. 4H).
Discussion
Mad2 is a key effector molecule of the SAC and is required for inhibition of mitotic exit upon checkpoint activation. The role of p31Comet in promoting efficient mitotic exit by antagonizing Mad2 is well known.19-25,27,28 However, little is known about the regulation of p31Comet or the physiological importance of p31Comet function in the absence of microtubule poisons. Herein, we have provided novel insight into the regulation of p31Comet expression by the Rb-E2F pathway. We have shown that multiple means of altering the function of this pathway in a panel of different cell types results in modulation of p31Comet expression. We have also provided evidence that antagonism of Mad2 by p31Comet is required for the long-term proliferation of cells. In line with these observations, expression of p31Comet and Mad2 is coordinated in the cell cycle and during the immortalization of human mammary epithelial cells. Moreover, we found that increased expression of p31Comet in tumors correlates with increased Mad2 levels. Together these data provide new insight into the regulation of SAC activity by p31Comet and suggest that SAC silencing may play a role in tumorigenesis.
Deficient function of the Rb family of tumor suppressors is a frequent occurrence in cancer, including breast cancer. Silencing of p16Ink4A is a key step in circumventing the first barrier toward immortalization of HMECs, termed stasis.34,39,40 The induction of p31Comet in post-stasis HMECs expressing shRNA targeting p16Ink4A (shp16), which mimic selection, suggests that it may be an early event in cellular immortalization/transformation as well (Fig. 1B). Aberrant activation of E2F (e.g., by loss of p16Ink4A) leads to induction of the SAC effector Mad2.16,17 Indeed, we find that Mad2 is overexpressed in shp16 HMECs (Fig. 1B). Importantly, this change does not reflect altered proliferation, as no dramatic change in Cyclin A levels is observed (Fig. 1B). Furthermore, Mad2 expression does not correlate with proliferation.9,17 Mad2 is overexpressed in breast cancer, and E2F driven upregulation of Mad2 is a major driving force in the generation of aneuploidy upon loss of the Rb pathway and is required for mammary adenocarcinoma development in WAP-T121 (functionally deficient Rb family) mice.16 However, overexpression of Mad2 results in hyperactivation of the SAC, which leads to aneuploidy, reduced proliferation, and decreased viability.18,42 Nonetheless, elevated Mad2 levels are found in a number of tumor types (including breast, lung, cervix; Fig. 4G) and have been shown to promote chromosomal instability and tumor formation in pRb−/−/p107−/−/p130−/− (TKO) MEFs, p53 mutant mice, and mouse models of breast and lung cancer.9,16-18
The results above indicate that during selection/immortalization, cells must also compensate for the effects of increased Mad2 levels. Consistent with this idea, normalizing the levels of constitutively overexpressed Mad2 in TKO MEFs to that of wild-type MEFs results in a weakened SAC.16 These data suggest that these cells have an upregulated SAC silencing activity. Indeed, in pRb−/− or p107−/−/p130−/− cells we find that increased p31Comet levels accompany high levels of Mad2 expression (Fig. 2D). Notably, increased p31Comet expression, along with Mad2, is also found in precancerous lesions in humans; ductal carcinoma in situ (DCIS), a precursor to invasive ductal breast cancer; non-neoplastic nevi, precursor melanoma; and prostatic intraepithelial neoplasia (Fig. 1A and not shown). While increased expression of the Mad2 target Cdc20 in tumors could also counter elevated Mad2 in tumors, several observations argue against Cdc20 as a key buffer of Mad2. First, concomitant increase in BubR1 levels will inhibit Cdc20.9 Second, recent studies have shown that Mad2 activation triggers the destruction of Cdc20, which is potentiated by p31Comet.25,43-45 These data, in conjunction with our observation that increased p31Comet levels accompany elevated Mad2 levels in both HMECs and human tumors, indicate that induction of p31Comet buffers increased Mad2 activity to allow for outgrowth of the abnormal population. Our analysis of p31Comet:Mad2 ratio effects on the long-term cellular proliferation supports this indication as well (Fig. 4A and E). A range of p31Comet:Mad2 is compatible with viability, and deviations from this range in either direction result in a significant loss of proliferative capacity (Fig. 4E and F). A recent study found that the p31Comet:Mad2 ratio varies among cell lines and correlates with the degree of mitotic slippage during prolonged exposure to microtubule poisons.46 We have found that the distribution of the p31Comet:Mad2 ratio is significantly altered in cancer (Fig. 4H). Whether these altered ratios are associated with mitotic slippage in the absence of spindle poisons and may contribute to chromosomal instability will require future study.
The existence of the SAC recovery mechanism, mediated by p31Comet, is a relatively recent discovery, and its potential involvement in cancer is essentially unknown. Our data indicate that increased p31Comet expression contributes to cancer by promoting the survival of Mad2-overexpressing cells. Furthermore our findings suggest that altered p31Comet expression and regulation may contribute to cancer by promoting chromosomal instability. Moreover, the potential for alterations in post-transcriptional/translational regulation of p31Comet and Mad2 also exists and may further contribute to aberrant SAC function in cancer. Along this line, p31Comet undergoes multiple post-translational modifications, including phosphorylation, which regulates the p31Comet-Mad2 interaction (DAD, ACB, and MKS, manuscript in preparation). Mad2 is also regulated by phosphorylation. For example, phosphorylation of Mad2 by Chk1 (also an E2F target gene) stabilizes Mad2, resulting in increased protein levels.47 Deregulation of these mechanisms may further skew the balance of p31Comet and Mad2 functions, effectively altering the p31Comet:Mad2 ratio and further driving tumor development. Future studies examining these aspects of p31Comet and Mad2 regulation may help to reconcile in vivo function of the SAC with in silico modeling of SAC function and ultimately provide new insight into how defective SAC function drives chromosomal instability and tumor progression.48-51
Experimental Procedures
Cell lines and drug treatments
HeLa, HCT116 and T98G (obtained from ATCC) as well as 3T3 cells (provided by Dr William Taylor), were maintained in Dulbecco modified Eagle media (DMEM) supplemented with 10% FBS and penicillin/streptomycin in a humidified atmosphere of 10% CO2 and a temperature of 37 °C. Tetracycline-inducible SAOS2-Rb cells were obtained from Dr Michelle Longworth and maintained in DMEM supplemented with 15% FBS minus tetracycline.41 HMECs were cultured as described.29 Chemicals were obtained from Sigma Aldrich unless otherwise mentioned. Doxycycline was dissolved in water and used at a concentration of 0.5 μg/μl. Puromycin was dissolved in water and used at a concentration 0.5 μg/μl.
Transfections and plasmids
Transient transfections were performed using Transit-LT1 (Mirus). The transfection reagent and DNA were used at a ratio of 3:1 and suspended in antibiotic and serum-free DMEM to form DNA and polymer complexes. The mixture was incubated at room temperature for 20 min before being added to cells. The cells were incubated at 37 °C for 24–48 h prior to harvesting. HA-tagged full-length E2F was described.52 shRNAs and constructs used in the HMEC studies were described.29 The cells were incubated at 37 °C for 24 to 48 h prior to harvesting or drug selection. pENTR223 p31Comet variant 2 was obtained from Open Biosystems and subcloned into pGLAP2 using Gateway technology (Invitrogen).53
Cell synchronization, serum starvation, and colony formation
HeLa cells at 25–30% confluency were treated with 2 mM thymidine and incubated at 37 °C for 18 h. Cells were released from the block into fresh media and incubated for 9 h prior to second block in 2 mM thymidine for 18 h. Cells were released from the second block into fresh media and harvested as they progressed synchronously through G2–M phase. T98G cells at 30–40% confluency were starved in media without serum and incubated at 37 °C for 72 h. Cells were released into media with 20% serum for stimulation and incubated at 37 °C prior to being harvested at required time points. HeLa cells were transfected as above. Twenty-four hours after transfection, cells were split 1:3 or 1:6 and allowed to grow for an additional 24 h. Transfected cells were selected with 0.5 μg/ml puromycin for 48 h. Cells were released into fresh media and allowed to proliferate for 10 d.
Western blotting and antibodies
Cell extracts were generated in RIPA buffer composed of 10 mM Tris (pH 7.4), 150 mM NaCl, 1% NP-40, 1% DOC, 0.1% SDS, 1 mM DTT, protease inhibitors (Leupeptin, Pepstatin, and Chymotrypsin, each at 10 μg/ml) and phosphatase inhibitors (25 mM β-glycerophosphate, 5 mM NaF, 1 mM NaVO4) or EBC buffer 50 mM Tris (pH 8.0), 120 mM NaCl, 0.5% NP-40, 1 mM DTT, 25 mM β-glycerophosphate, 5 mM NaF, 1 mM NaVO4, and Leupeptin, Pepstatin, and Chymotrypsin, each at 10 μg/ml. Cell lysates were resolved by SDS-PAGE. The proteins were transferred to polyvinyldenefluoride (PVDF) membranes obtained from Millipore. Western blot blocking and primary antibody hybridization were performed in 5% (w/v) non-fat dry milk in phosphate-buffered saline and washed with Tris-buffered saline + 0.1% Tween-20 (TBST). Secondary antibodies (LI-COR) were incubated in TBST+ 0.02% SDS and visualized with the LI-COR Odyssey near infrared imaging system.
Commercial antibodies from Sigma were utilized for the following proteins p31comet (4G11), Mad1 (9B10), FLAG epitope (M2), and β-Actin (AC-15,). Antibodies to Cyclin B (GNS1), Cyclin A (H-432), RB (IF8), p31Comet (4RE23) were obtained from Santa Cruz Biotech. Polyclonal antibodies from Bethyl laboratories were used for Mad2 and Mad1. Antibodies against p27 and Securin were obtained from Invitrogen. Monoclonal antibody from Covance (HA.11) was used for HA tagged proteins. The anti-MYC antibody 9E10 was produced in house at the Lerner Research Institute.
Statistical methods
Correlation analyses, Mann–Whitney tests were performed using GraphPad Prism.
Acknowledgments
We thank William Flavahan for assistance with correlation analysis. We thank Peggy Farnham or assistance with ENCODE data analysis. We thank Carol Prives, Michelle Longworth, and William Taylor for reagents and cell lines. We thank M Longworth for helpful discussions. This work was supported by seed funds from the Lerner Research Institute and a pilot grant from the Ohio Cancer Research Associates to MKS and by the US National Institutes of Health (R01CA138421 to Mark W Jackson).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26811
References
- 1.Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–3. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 2.Dobles M, Liberal V, Scott ML, Benezra R, Sorger PK. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 2000;101:635–45. doi: 10.1016/S0092-8674(00)80875-2. [DOI] [PubMed] [Google Scholar]
- 3.Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty VV, Benezra R. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 2001;409:355–9. doi: 10.1038/35053094. [DOI] [PubMed] [Google Scholar]
- 4.Babu JR, Jeganathan KB, Baker DJ, Wu X, Kang-Decker N, van Deursen JM. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J Cell Biol. 2003;160:341–53. doi: 10.1083/jcb.200211048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim HS, Park KH, Kim SA, Wen J, Park SW, Park B, Gham CW, Hyung WJ, Noh SH, Kim HK, et al. Frequent mutations of human Mad2, but not Bub1, in gastric cancers cause defective mitotic spindle checkpoint. Mutat Res. 2005;578:187–201. doi: 10.1016/j.mrfmmm.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 6.Percy MJ, Myrie KA, Neeley CK, Azim JN, Ethier SP, Petty EM. Expression and mutational analyses of the human MAD2L1 gene in breast cancer cells. Genes Chromosomes Cancer. 2000;29:356–62. doi: 10.1002/1098-2264(2000)9999:9999<::AID-GCC1044>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 7.Matsuura S, Matsumoto Y, Morishima K, Izumi H, Matsumoto H, Ito E, Tsutsui K, Kobayashi J, Tauchi H, Kajiwara Y, et al. Monoallelic BUB1B mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. Am J Med Genet A. 2006;140:358–67. doi: 10.1002/ajmg.a.31069. [DOI] [PubMed] [Google Scholar]
- 8.Hanks S, Coleman K, Reid S, Plaja A, Firth H, Fitzpatrick D, Kidd A, Méhes K, Nash R, Robin N, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet. 2004;36:1159–61. doi: 10.1038/ng1449. [DOI] [PubMed] [Google Scholar]
- 9.Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell. 2011;19:701–14. doi: 10.1016/j.ccr.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 11.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 14.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–8. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barber TD, McManus K, Yuen KW, Reis M, Parmigiani G, Shen D, Barrett I, Nouhi Y, Spencer F, Markowitz S, et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci U S A. 2008;105:3443–8. doi: 10.1073/pnas.0712384105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sotillo R, Schvartzman JM, Socci ND, Benezra R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 2010;464:436–40. doi: 10.1038/nature08803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hernando E, Nahlé Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald W, Benezra R, et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430:797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- 18.Sotillo R, Hernando E, Díaz-Rodríguez E, Teruya-Feldstein J, Cordón-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Habu T, Kim SH, Weinstein J, Matsumoto T. Identification of a MAD2-binding protein, CMT2, and its role in mitosis. EMBO J. 2002;21:6419–28. doi: 10.1093/emboj/cdf659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang M, Li B, Tomchick DR, Machius M, Rizo J, Yu H, Luo X. p31comet blocks Mad2 activation through structural mimicry. Cell. 2007;131:744–55. doi: 10.1016/j.cell.2007.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagan RS, Manak MS, Buch HK, Meier MG, Meraldi P, Shah JV, Sorger PK. p31(comet) acts to ensure timely spindle checkpoint silencing subsequent to kinetochore attachment. Mol Biol Cell. 2011;22:4236–46. doi: 10.1091/mbc.E11-03-0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jia L, Li B, Warrington RT, Hao X, Wang S, Yu H. Defining pathways of spindle checkpoint silencing: functional redundancy between Cdc20 ubiquitination and p31(comet) Mol Biol Cell. 2011;22:4227–35. doi: 10.1091/mbc.E11-05-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Westhorpe FG, Tighe A, Lara-Gonzalez P, Taylor SS. p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. J Cell Sci. 2011;124:3905–16. doi: 10.1242/jcs.093286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia G, Luo X, Habu T, Rizo J, Matsumoto T, Yu H. Conformation-specific binding of p31(comet) antagonizes the function of Mad2 in the spindle checkpoint. EMBO J. 2004;23:3133–43. doi: 10.1038/sj.emboj.7600322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varetti G, Guida C, Santaguida S, Chiroli E, Musacchio A. Homeostatic control of mitotic arrest. Mol Cell. 2011;44:710–20. doi: 10.1016/j.molcel.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 26.Fava LL, Kaulich M, Nigg EA, Santamaria A. Probing the in vivo function of Mad1:C-Mad2 in the spindle assembly checkpoint. EMBO J. 2011;30:3322–36. doi: 10.1038/emboj.2011.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teichner A, Eytan E, Sitry-Shevah D, Miniowitz-Shemtov S, Dumin E, Gromis J, Hershko A. p31comet Promotes disassembly of the mitotic checkpoint complex in an ATP-dependent process. Proc Natl Acad Sci U S A. 2011;108:3187–92. doi: 10.1073/pnas.1100023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miniowitz-Shemtov S, Eytan E, Ganoth D, Sitry-Shevah D, Dumin E, Hershko A. Role of phosphorylation of Cdc20 in p31(comet)-stimulated disassembly of the mitotic checkpoint complex. Proc Natl Acad Sci U S A. 2012;109:8056–60. doi: 10.1073/pnas.1204081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cipriano R, Kan CE, Graham J, Danielpour D, Stampfer M, Jackson MW. TGF-beta signaling engages an ATM-CHK2-p53-independent RAS-induced senescence and prevents malignant transformation in human mammary epithelial cells. Proc Natl Acad Sci U S A. 2011;108:8668–73. doi: 10.1073/pnas.1015022108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brenner AJ, Stampfer MR, Aldaz CM. Increased p16 expression with first senescence arrest in human mammary epithelial cells and extended growth capacity with p16 inactivation. Oncogene. 1998;17:199–205. doi: 10.1038/sj.onc.1201919. [DOI] [PubMed] [Google Scholar]
- 31.Garbe JC, Bhattacharya S, Merchant B, Bassett E, Swisshelm K, Feiler HS, Wyrobek AJ, Stampfer MR. Molecular distinctions between stasis and telomere attrition senescence barriers shown by long-term culture of normal human mammary epithelial cells. Cancer Res. 2009;69:7557–68. doi: 10.1158/0008-5472.CAN-09-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garbe JC, Holst CR, Bassett E, Tlsty T, Stampfer MR. Inactivation of p53 function in cultured human mammary epithelial cells turns the telomere-length dependent senescence barrier from agonescence into crisis. Cell Cycle. 2007;6:1927–36. doi: 10.4161/cc.6.15.4519. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Pan J, Li JL, Lee JH, Tunkey C, Saraf K, Garbe JC, Whitley MZ, Jelinsky SA, Stampfer MR, et al. Transcriptional changes associated with breast cancer occur as normal human mammary epithelial cells overcome senescence barriers and become immortalized. Mol Cancer. 2007;6:7. doi: 10.1186/1476-4598-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Novak P, Jensen TJ, Garbe JC, Stampfer MR, Futscher BW. Stepwise DNA methylation changes are linked to escape from defined proliferation barriers and mammary epithelial cell immortalization. Cancer Res. 2009;69:5251–8. doi: 10.1158/0008-5472.CAN-08-4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stampfer MR, Yaswen P. Culture systems for study of human mammary epithelial cell proliferation, differentiation and transformation. Cancer Surv. 1993;18:7–34. [PubMed] [Google Scholar]
- 36.Stampfer MR, Yaswen P. Culture models of human mammary epithelial cell transformation. J Mammary Gland Biol Neoplasia. 2000;5:365–78. doi: 10.1023/A:1009525827514. [DOI] [PubMed] [Google Scholar]
- 37.Yaswen P, Stampfer MR. Epigenetic changes accompanying human mammary epithelial cell immortalization. J Mammary Gland Biol Neoplasia. 2001;6:223–34. doi: 10.1023/A:1011364925259. [DOI] [PubMed] [Google Scholar]
- 38.Yaswen P, Stampfer MR. Molecular changes accompanying senescence and immortalization of cultured human mammary epithelial cells. Int J Biochem Cell Biol. 2002;34:1382–94. doi: 10.1016/S1357-2725(02)00047-X. [DOI] [PubMed] [Google Scholar]
- 39.Huschtscha LI, Noble JR, Neumann AA, Moy EL, Barry P, Melki JR, Clark SJ, Reddel RR. Loss of p16INK4 expression by methylation is associated with lifespan extension of human mammary epithelial cells. Cancer Res. 1998;58:3508–12. [PubMed] [Google Scholar]
- 40.Foster SA, Wong DJ, Barrett MT, Galloway DA. Inactivation of p16 in human mammary epithelial cells by CpG island methylation. Mol Cell Biol. 1998;18:1793–801. doi: 10.1128/mcb.18.4.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Binné UK, Classon MK, Dick FA, Wei W, Rape M, Kaelin WG, Jr., Näär AM, Dyson NJ. Retinoblastoma protein and anaphase-promoting complex physically interact and functionally cooperate during cell-cycle exit. Nat Cell Biol. 2007;9:225–32. doi: 10.1038/ncb1532. [DOI] [PubMed] [Google Scholar]
- 42.Luo X, Tang Z, Xia G, Wassmann K, Matsumoto T, Rizo J, Yu H. The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat Struct Mol Biol. 2004;11:338–45. doi: 10.1038/nsmb748. [DOI] [PubMed] [Google Scholar]
- 43.Ge S, Skaar JR, Pagano M. APC/C- and Mad2-mediated degradation of Cdc20 during spindle checkpoint activation. Cell Cycle. 2009;8:167–71. doi: 10.4161/cc.8.1.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nilsson J, Yekezare M, Minshull J, Pines J. The APC/C maintains the spindle assembly checkpoint by targeting Cdc20 for destruction. Nat Cell Biol. 2008;10:1411–20. doi: 10.1038/ncb1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan J, Chen RH. Spindle checkpoint regulates Cdc20p stability in Saccharomyces cerevisiae. Genes Dev. 2004;18:1439–51. doi: 10.1101/gad.1184204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma HT, Chan YY, Chen X, On KF, Poon RY. Depletion of p31comet protein promotes sensitivity to antimitotic drugs. J Biol Chem. 2012;287:21561–9. doi: 10.1074/jbc.M112.364356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chilà R, Celenza C, Lupi M, Damia G, Carrassa L. Chk1-Mad2 interaction: a crosslink between the DNA damage checkpoint and the mitotic spindle checkpoint. Cell Cycle. 2013;12:1083–90. doi: 10.4161/cc.24090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lohel M, Ibrahim B, Diekmann S, Dittrich P. The role of localization in the operation of the mitotic spindle assembly checkpoint. Cell Cycle. 2009;8:2650–60. doi: 10.4161/cc.8.16.9383. [DOI] [PubMed] [Google Scholar]
- 49.Diaz-Rodríguez E, Sotillo R, Schvartzman JM, Benezra R. Hec1 overexpression hyperactivates the mitotic checkpoint and induces tumor formation in vivo. Proc Natl Acad Sci U S A. 2008;105:16719–24. doi: 10.1073/pnas.0803504105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Varmark H, Kwak S, Theurkauf WE. A role for Chk2 in DNA damage induced mitotic delays in human colorectal cancer cells. Cell Cycle. 2010;9:312–20. doi: 10.4161/cc.9.2.10418. [DOI] [PubMed] [Google Scholar]
- 51.Stolz A, Ertych N, Kienitz A, Vogel C, Schneider V, Fritz B, Jacob R, Dittmar G, Weichert W, Petersen I, et al. The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nat Cell Biol. 2010;12:492–9. doi: 10.1038/ncb2051. [DOI] [PubMed] [Google Scholar]
- 52.Peart MJPM, Poyurovsky MV, Kass EM, Urist M, Verschuren EW, Summers MK, Jackson PK, Prives C. APC/C(Cdc20) targets E2F1 for degradation in prometaphase. Cell Cycle. 2010;9:3956–64. doi: 10.4161/cc.9.19.13162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Torres JZ, Miller JJ, Jackson PK. High-throughput generation of tagged stable cell lines for proteomic analysis. Proteomics. 2009;9:2888–91. doi: 10.1002/pmic.200800873. [DOI] [PMC free article] [PubMed] [Google Scholar]