

Abstract
Glioblastoma is a highly aggressive malignant disease with notable resistance to chemotherapy. In this study, we found that leptin receptor (ObR)-positive glioblastoma cells were resistant to temozolomide (TMZ), and TMZ-resistant cells exhibited high expression of ObR. ObR can serve as a marker to enrich glioblastoma cells with some stem/progenitor cell traits, which explained the reason for TMZ resistance of ObR+ cells. STAT3-mediated SOX2/OCT4 signaling axis maintained the stem/progenitor cell properties of ObR+ cells, which indirectly regulated glioblastoma TMZ resistance. These findings gain insight into the molecular link between obesity and glioblastoma, and better understanding of this drug-resistant population may lead to the development of more effective therapeutic interventions for glioblastoma.
Keywords: glioblastoma stem cells, leptin receptor, chemoresistance, STAT3
Introduction
Glioblastoma multiforme (GBM) is the most malignant primary tumors of the central nervous system.1,2 The prognosis of patients with GBM is very poor, mainly because it remains therapeutic challenge, and the conventional postsurgical chemotherapeutic agents exhibit limited effects.3,4 Temozolomide (TMZ), as currently the most promising chemotherapeutic drug, is applied to treat malignant glioma, including GBM.5,6 However, TMZ is not always effective in patients with GBM, and the intrinsic or acquired chemoresistance of GBM cells always results in this unsatisfactory outcome.7
A large number of epidemiological studies on cancer incidence link the propensity of cancers with the individual’s obesity.8,9 Obesity may also be associated with adverse treatment outcome of cancer, including resistance to chemotherapies.10 Leptin, encoded by the obesity gene,11 directly contributes to leptin receptor (ObR)-positive cancer cell proliferation, migration, and invasion.12 Zheng et al. concluded that leptin was a mammary tumor-initiating factor on the basis of its ability to stimulate cancer stem cells (CSCs) survival, and ObR was defined as breast CSCs surface marker.13 Recently, CSCs were reported to be enriched within chemotherapy- resistant subpopulations.14 However, whether ObR-positive GBM cells are responsible for cancer chemoresistance needs further validation.
In the present study, we report that ObR-positive glioblastoma cells are resistant to TMZ, as well as U87R cells, an established TMZ-resistant cell, expressing high levels of ObR. These findings are highly provocative but will require a more in-depth evaluation of the reason. Here we demonstrate that ObR+ cells possess stem/progenitor cell properties and selective inhibition of STAT3 can abrogate self-renewal, which suggests that this selective targeting would be valuable to inhibit TMZ resistance at the level of primitive cells. The purpose of this study focuses on the CSCs-like properties of TMZ-resistant ObR+ cells. A better understanding of this drug-resistant population may lead to the development of more effective therapeutic interventions for glioblastoma.
Results
Gliobalastoma cells represent TMZ-resistant characteristic with high expression of ObR
To identify the effect of ObR in glioma cells’ chemoresistance, we first used flow cytometry to sort ObR+ cells; the flow cytometry analysis showed that approximately 16.7 ± 0.2% of U87 gliobalastoma cells expressed ObR (Fig. 1A). The ObR+ and ObR− cells were exposed to the indicated concentration of TMZ for 72 h; chemosensitivity was determined by MTT assay. Compared with ObR− U87 cells, ObR+ gliobalastoma cells exhibited a growth advantage in the presence of TMZ, especially with the low dose (Fig. 1B). To further validate the chemoresistance of ObR+ U87 cells, TMZ-induced apoptosis was assessed. Annexin V and PI staining demonstrated that when treated with 50 or 100 μM TMZ, the apoptosis rate of ObR− U87 cells was more than 30%, while ObR+ cells had no significant change (Fig. 1C). Subsequently, we treated U87 cells with a low dose of TMZ in culture media for 4 weeks to establish TMZ-resistant cells designated as U87R. To ensure the TMZ resistance of U87R cells, nude mice bearing U87 and U87R cells xenografts were treated with 100 μM TMZ, as shown in Figure 1D, the weight of U87R cells xenografts was larger than that of the U87 cells xenografts (P < 0.05). According to the increased resistance, the mRNA and protein expression levels of ObR were elevated in U87R cells (Fig. 1E and F). These results suggested that the cells with high expression of ObR represented TMZ-resistant characteristic.

Figure 1. Gliobalastoma cells with high expression of ObR represent TMZ-resistant characteristic. (A) Flow cytometry to sort ObR+ cells in U87 gliobalastoma cells. ObR+ and ObR− cells were cultured for 72 h in the presence of indicated concentration of TMZ. MTT assay was performed to assess the anti-proliferative effects of TMZ (B), Annexin V and PI staining FACS was used to assess apoptosis of cells (C). (D) U87 cells with a low dose of TMZ in culture media for 4 wk to establish TMZ-resistant cells, and the image showed that nude mice bearing U87 and U87R cells xenografts were treated with 100 μM TMZ; 4 wk later, the xenografts were weighted. (E) The mRNA and (F) protein expression levels of ObR were elevated in U87R cells.
ObR+ gliobalastoma cells possess stem/progenitor cell properties
Preclinical studies suggested that glioblastoma stem/progenitor cells were highly resistant to conventional chemotherapeutic drugs, including TMZ.15,16 We therefore hypothesized that ObR+ cells might exhibit intrinsic properties of stem/progenitor cells. For this purpose, we first evaluated the expression of CD133, Nestin, and GFAP in ObR+ and U87R cells by using immunofluorescence. Interestingly, both ObR+ cells and U87R cells coexpressed ObR, CD133, and Nestin, whereas negative for GFAP, a biomaker for mature astrocyte. Consistent with the immunofluorescence data, western blot analysis showed that CD133 and Nestin were high expression significantly in ObR+ cells and U87R cells, but GFAP displayed almost no expression in both cells (Fig. 2A and B). The isolated ObR+ U87 cells formed clones efficiently, whereas ObR− cells failed to do so (Fig. 2C). In addition, ObR+ cells were much more invasive than ObR− cells (P < 0.05, Fig. 2D). We next inoculated nude mice subcutaneously with 104 ObR+ cells and ObR− cells to investigate their tumorigenicity in vivo. A significant difference in tumor incidence was observed between the mice inoculated with the 2 cells. The ObR+ cells produced tumors in 100% mice, whereas ObR− cells produced tumors in only 25% mice 5 wk after injection (Fig. 2E). Collectively, these results suggested that ObR+ cells displayed gliobalastoma stem/progenitor cells properties.

Figure 2. Stem/progenitor cell properties of ObR+ gliobalastoma cells. (A) Immunofluorescence analysis of ObR+ and U87R cells stained with ObR, CD133, Nestin, and GFAP. (B) Western blot analysis of CD133, Nestin, and GFAP in ObR+ and U87R cells. (C) Representative image of the plates containing colonies derived from 1000 ObR− and ObR+ cells. (D) Cells invasiveness of ObR+ and ObR− cells using the Matrigel invasion assay. (E) Representative mice with subcutaneous tumors and tumorigenicity of 104 sorted cells derived from ObR+ and ObR− cells.
It has been shown previously that gliobalastoma stem/progenitor cells have self-renewal properties. When the ObR+ cells and ObR− cells were cultured in DMEM supplemented with 10% FBS, 7 d later, the percentage of ObR+ cells remained more than 80%, and after 2 wk, ObR+ fraction remained almost 60% by FACS analysis. In contrast, ObR− cells maintained their ObR status (Fig. 3A). To evaluate the effect of leptin on U87R cells oncogenesis, nude mice implanted with U87R cells (4 × 104) were randomly assigned and received i.p. injections of either PBS or leptin (2 μg/g body weight) every other day for a total 5 times. Two weeks later, the tumor volume of leptin-treated mice grew more rapidly compared with the PBS group (Fig. 3B), suggesting that leptin could aggravate the cells with high expression of ObR-mediated malignancy.
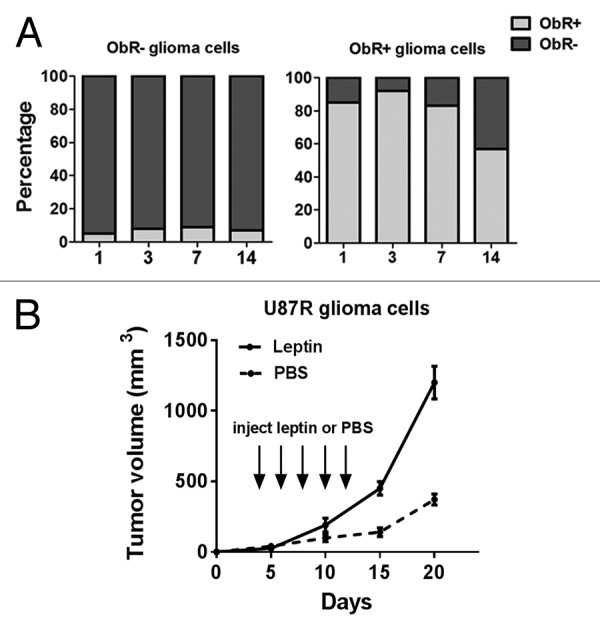
Figure 3. The maintenance of self-renewal in ObR+ cells. (A) Percentage of sorted ObR+ and ObR− cells after culturing for various times as analyzed by flow cytometry. (B) Leptin stimulated tumor development by implanted U87R cells. nude mice implanted with U87R (4 × 104) cells were randomly assigned and received i.p. injections of either PBS or leptin (2 μg/g body weight) every other day for 2 wk.
Correlation of ObR and CD133 expression in glioma tissues
To investigate the clinical significance, immunohistochemical analysis was conducted by using 15 sets of malignant glioma as primary and recurrence tumors. These primary tumors were surgically resected, TMZ treated, and then the recurrent tumors were surgically resected again. The scores of ObR were evaluated as 0 to 3 and increased significantly in the recurrence tumors compared with the primary tumors (Fig. 4A and B). Enrichment of ObR+ cells in the recurrence tumors suggested their involvement in the tumor regeneration. Furthermore, coimmunofluorescence staining of ObR and CD133 highlighted the colocalization of CD133+ and ObR+ cells in glioma tissues (Fig. 4C). Noticeably, as shown in Figure 4D, ObR expression was significantly associated with CD133 in the primary as well as recurrence tumors (P < 0.001). Thus, ObR+ cells may serve as a target to eliminate glioblastoma cells with stem/progenitor features.
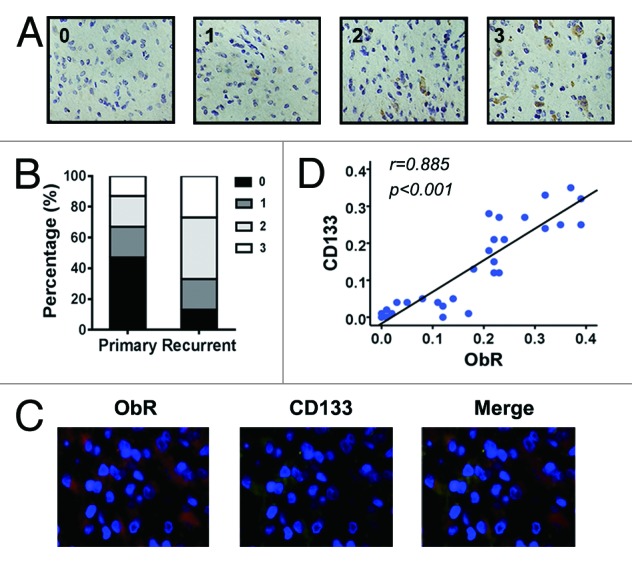
Figure 4. Correlation of ObR and CD133 expression in glioma tissues. (A) Representative photographs of immunohistochemical analysis for ObR in surgical specimens of malignant glioma as score 0 to 3 (×200). (B) Immunohistochemical analysis of 15 cases of recurrent malignant glioma. The score of ObR in primary or recurrent tumor was shown as graph. (C) Immunofluorescence staining of glioma tissues stained with anti-CD133, anti-ObR, and DAPI (×400). (D) Correlation between ObR and CD133 in 30 cases of primary or recurrent glioma specimens.
ObR+ cells maintains self-renewal and chemoresistance dependent on STAT3 signaling pathway
Having documented that ObR+ cells possessed stem/progenitor cell properties, we sought to elucidate the mechanism regulating self-renewal. Our previous study has shown that leptin can act as a mitogen, and STAT3 is rapidly activated in gliobalastoma cells proliferation.17 In this study, the expression of STAT3 and p-STAT3 in ObR+ cells and U87R cells was kept a higher level. SOX2 and OCT4, the pluripotency-associated transcription factors, were also upregualted in both cells. We used NSC74859, the STAT3 inhibitor, to reduce phosphorylated levels of STAT3. The efficacy of STAT3 inhibitor was validated by the downregulation of p-STAT3 in a dose-dependent manner, and the protein expression of SOX2 and OCT4 was also diminished as p-STAT3 depletion in ObR+ cells and U87R cells (Fig. 5A), similar to the previous study that had explained as STAT3 binding directly to the proximal promoter regions of OCT4 and SOX2. Therefore, ObR+ cells may have retained their stem/progenitor cell properties through p-STAT3-SOX2/OCT4 signaling axis. We next examined whether SOX2 and OCT4 were required for expression of ObR; siRNA-mediated knockdown of SOX2 or OCT4 was confirmed by western blot and resulted in an apparent reduction of ObR protein expression in ObR+ cells and U87R cells (Fig. 5B). The involvement of SOX2 and OCT4 in expression of ObR indicated the correspondence between elevated ObR expression and the pluripotent state, which raised the possibility that leptin maintained self-renewal and mediated malignancy of ObR+ cells.
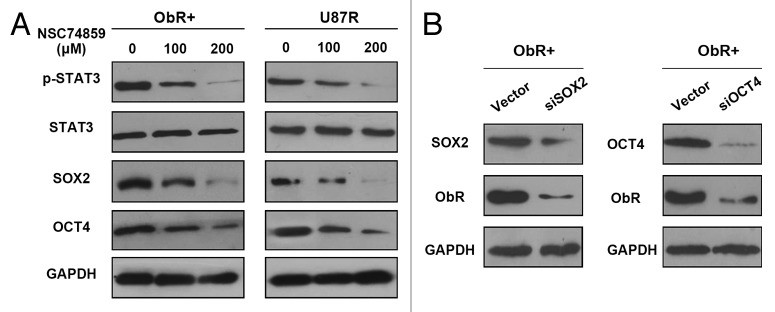
Figure 5. ObR+ cells maintains self-renewal dependent on STAT3 signaling pathway. (A) Western blot analysis of p-STAT3, SOX2, and OCT4 in ObR+ cells and U87R cells treated with NSC74859-STAT3 inhibitor. Does of STAT3 inhibitor was indicated on the top. (B) Western blot analysis of SOX2, OCT4, and ObR in ObR+ cells expressing SOX2 or OCT4 siRNA.
Glioblastoma stem/progenitor cells were closely related to chemoresistance. We treated the ObR+ cells and U87R cells with TMZ alone or with TMZ and STAT3 inhibitor. Both cells, after 72 h of treatment with TMZ, had no significant growth inhibition, whereas combined treatment of TMZ with STAT3 inhibitor was effective compared with TMZ or NSC74859 alone (Fig. 6A and B). Further to confirm these results, we used FACS to analyze apoptosis of ObR+ U87 cells, and found that combined treatment of TMZ (100 μM) and NSC74859 (200 μM) exhibited a strong apoptotic effect (Fig. 6C), which implied that STAT3 inhibition could attenuate TMZ resistance of ObR+ glioblastoma cells.

Figure 6. STAT3 inhibitor potentiated TMZ efficacy in glioma cells with high expression of ObR. (A) MTT assay of ObR+ cells treated by STAT3 inhibitor and TMZ. Error bars represent SD of 3 independent experiments.*P < 0.05 vs. TMZ (0). (B) MTT assay of U87R cells treated by STAT3 inhibitor and TMZ. Error bars represent SD of 3 independent experiments.*P < 0.05 vs. TMZ (0). (C) FACS was used to analyze the apoptosis of ObR+ cells treated by STAT3 inhibitor (200 μM) and TMZ (100 μM). *P < 0.05 iii vs. i, # P < 0.05 iv vs. iii.
Discussion
Glioblastoma is a highly aggressive malignant disease with notable resistance to chemotherapy. In this study, we found that ObR+ glioblastoma cells were resistant to TMZ, and TMZ-resistant cells exhibited high expression of ObR. ObR can serve as a marker to enrich glioblastoma cells with some stem/progenitor cell traits, which explained the reason for TMZ resistance of ObR+ cells. STAT3-mediated SOX2/OCT4 signaling axis maintained the stem/progenitor cell properties of ObR+ cells, which indirectly regulated glioblastoma TMZ resistance.
It has become increasingly recognized that obesity is a critical public health problem, as it is a risk factor for many human diseases, including diabetes, atherosclerosis, and cancer.18 Adipokines represent likely candidates to mediate, at least in part, the increased cancer risk and enhanced progression associated with adipocyte-derived secretory proteins.19 Among the adipokines, leptin is the most intensively studied factor due to its involvement in tumor initiation, primary tumor growth, invasion, and metastasis.20 However leptin contributes to cancer initiation or whether its role is restricted to promoting the growth of existing tumor cells may primarily depend on receptor levels of ObR among those cells.13 Diverse cancer cells derived from different tissues, such as brain, breast, colon or prostate, were reported to have ObR expression.21-25 Furthermore, Ob-R overexpression not only correlated with the degree of malignancy, but also was an independent prognostic variable to predict poor progression-free survival in brain tumor, ovarian cancer, and breast cancer,24,26,27 which suggested that ObR+ cells might be closely related with tumor initiation and development. In this study, we claimed that ObR+ glioblastoma cells presented chemoresistance, which showed that TMZ could not inhibit cell proliferation and could not induce apoptosis effectively. To identify the responsible of ObR+ cells for TMZ resistance, U87 cells was treated with a low dose of TMZ in culture media to establish TMZ-resistant cells. With overexpression of ObR on U87R cells, ObR was confirmed to be responsible for chemoresistance of glioblastoma again.
Many studies demonstrated that selection of chemotherapy-resistant cells by using anticancer drugs would be an effective means to enrich the CSC-like subpopulation,28,29 and CSCs indeed possessed intrinsic properties that mediated their resistance to chemotherapy.30,31 Lzumiya et al. found that 5-FU-pretreated pancreatic cancer cells expressed enhanced levels of the stemness genes OCT4 and NANOG,32 and exhibited a so-called CSC-like phenotype, including a sphere-forming capability.14 Interestingly, we found that the ObR+ glioblastoma cells, as well as TMZ-resistant U87R cells, also exhibited stem/progenitor cell properties. ObR expression in glioblastoma cells overlapped with the expression of reported glioblastoma CSCs markers, such as CD133 and Nestin; however, very few ObR+ cells expressed GFAP, a biomarker for mature astrocyte. As shown in Figure 4, ObR expression was significantly associated with CD133 in the primary as well as recurrence tumors (P < 0.001). Additionally, ObR+ cells possessed a better clones-forming capability in vitro and tumorigenicity in vivo. In line with our data, Feldman et al. indicated that ObR was strongly coexpressed in a majority of CD133+ liver tumor cells; selective expression of ObR was a characteristic feature of tumor-initiating stem cells.33 Therefore, with the stem/progenitor cell properties, ObR+ cells played a critical role in resistance of chemotherapeutic agent inevitability.
Sherry et al. reported that STAT3 is required for proliferation and maintenance of multipotency in GBM stem cells.34 Villalva et al. further indicated that stem cells derived from GBM expressed a constitutively phosphorylated STAT3, which was essential for the proliferative and the self-renewal capacity.35 Thus, STAT3 activation appears as a common feature of CSC-GBM. Furthermore, ObR had full signaling capabilities and was able to activate the JAK/STAT3 pathway, the major pathway used by leptin to exert its effects.20 In our study, both ObR+ cells and U87R cells exhibited active STAT3 and increased levels of SOX2 and OCT4, when STAT3 activity was inhibited, the pluripotency-associated transcription factors were downregulated. Additionally, the self-reinforcing leptin-signaling module active within ObR+ cells resulted in maintaining cells self-renewal and malignancy. All of the data suggested that STAT3 might be a therapeutic target for TMZ resistance in glioblastoma. The combination therapy for ObR+ cells and U87R cells with TMZ and STAT3 inhibitor presented significant growth inhibition. Chemoresistance of glioblastoma cancer stem cells exhibited much more complex than expected.36 MGMT (O6-methylguanine-methyltransferase) methylation is considered as one of the principal mechanisms contributing to TMZ sensitivity of glioblastoma stem-like cells,37 which may be also the mechanism for TMZ resistance of ObR+ cells.
In conclusion, we identify overexpression of ObR cells are resistant to TMZ due to the stem/progenitor cell properties, and STAT3 signaling as a novel pathway that keeps self-renewal and mediates resistance of TMZ in ObR+ glioblastoma cells. These findings gain insight into the molecular link between obesity and glioblastoma.
Materials and Methods
Glioblastoma cells and samples
U87 human glioma cell line was purchased from the Chinese Academy of Sciences Cell Bank and cultured in DMEM supplemented with 10% fetal bovine serum (FBS). The human glioblastoma samples were obtained from Changhai Hospital of Second Military Medical University after obtaining informed consent from a patient diagnosed with glioma.
Sorting and analysis by Fluorescence Activated Cell Sorting (FACS)
For cell sorting, 107 U87 glioblastoma cells were labeled with 10 μl anti-human ObR-allophycocyanin (APC) (Miltenyi Biotec) for 10 min in the dark at 4 °C. Mouse IgG monoclonal antibody was used as a control. After ObR staining, cells were sorted using a BD FACS Aria II with Cell Quest Pro software (BD Bioscience). The purity of the ObR cells was analyzed using a similar protocol, and the cells were maintained in stem cell medium as previously described.38 The cells’ apoptosis was detected by using Annexin V-FITC Apoptosis Detection Kit (BD Biosciences) according to the manufacturer’s instructions. Briefly speaking, the pretreated cells were harvested and stained with PI and Annexin V-FITC, the apoptosis rate was assayed using FACS at 488 nm.
MTT Assay
MTT assay was used to determine cell viability following the manufacturer’s protocols (Cayman Chemical Company). 104 cells were plated in 96-well plates for 72 h, MTT solution was incubated at 37 °C for 4 h, and then the crystal dissolving solution was added to solubilize the formazan crystals. The absorbance of the plates was measured on a microplates reader at a wavelength of 570 nm. Each sample was performed in triplicate, and the experiment was repeated 3 times.
Colony formation assay
For the colony formation assays, 103 ObR+ cells and ObR− cells were seeded in 6-well plates after FACS sorting, respectively. After 14 d of culture, the cells were fixed by 100% methanol and stained with methylene blue for 20 min, and the colonies of more than 50 cells were counted.
Invasion assays
Invasion assays were performed using BD Bio Coat Matrigel Matrix Cell Culture Inserts and Control Inserts (BD Biosciences) essentially as previously described.39 Briefly, upper inserts were coated with Matrigel (1:1 dilution; BD Biosciences) and then incubated at 37 °C for 30 min. 104 cells were resuspended in 200 μl of serum-free DMEM and added to the corresponding upper inserts, respectively. DMEM with 10% FBS was added to the lower chamber. After 24 h, invaded cells were fixed and stained with crystal violets for 20 min, and then counted in 5 different fields for each inserts. The experiment was repeated at least 3 times.
Real-time polymerase chain reaction and western blot
Total RNA extraction, cDNA (cDNA) synthesis and qPCR were performed as described.40 The primer sequences used for Ob-R were sense: 5′-CTTTCCACTG TTGCTTTCGG-3′; antisense: 5′-TCTGTGATTT CCATATGCAA ACC-3′. For western blot analysis, cell lysates were boiled and reduced in sodium dodecyl sulfate PAGE (SDS-PAGE) sample buffer, and proteins were separated on 10% standard SDS-PAGE and transferred onto nitrocellulose membrane that were blocked and incubated with rabbit antibodies of ObR (1:1000; Abcam), CD133(1:100; Cell Signaling Technology), Nestin(1:200; Abcam), GFAP9 (1:1000, Dako), p-STAT3(1:500, Cell Signaling Technology), STAT3(1:500, Cell Signaling Technology), SOX2(1:500; Abcam), OCT4(1:200; Abcam), GAPDH(1:500, Cell Signaling Technology), and followed by horseradish peroxidase-conjugated anti-rabbit IgG (1:2,000; Invitrogen). Enhanced chemiluminescence (ECL; Amersham/GE Healthcare) was used for the detection of bound antibody.
Immunohistochemistry and immunofluorescence staining
Immunohistochemistry (IHC) and immunofluorescence staining were performed on paraffin sections of glioma specimens to investigate ObR and CD133 expression. The protocol was performed as previously described.41 Sections deparaffinized and rehydrated in graded alcohols. After antigen retrieval, sections were blocked with 2% goat serum in PBS for 1 h and then incubated overnight with antibodies to ObR (1:1000; Abcam) and CD133 (1:100; Cell Signaling Technology). Twenty-four h later, sections were washed and incubated for 2 h with secondary antibodies, and the fluorescence-conjugated antibodies were used for immunofluorescence.
Tumorigenicity in nude mice
All procedures involving animals were performed in accordance with the institutional animal welfare guidelines of Second Military Medical University. Cells suspension was prepared as single-cell type suspension, and 8-wk-old nude mice (BALB/c strain) were injected subcutaneously (s.c.) with 200 μL suspension. The size and incidence of subcutaneous tumors were recorded. Tumor size was measured by use of a caliper and volume was calculated as length × height × width × 0.5236 with reference to a previous report.42
Statistical analysis
All data, expressed as mean ± SEM, were from at least three separate experiments. Groups were compared by analysis of variance (ANOVA) with a posteriori contrast by least significant difference; or by Student t test using the Microsoft Excel Analysis Tool Pak (Microsoft). P < 0.05 was considered significant.
Acknowledgments
This study was supported by National Natural Science Fund (Grants 81101906, 81271271).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26809
References
- 1.Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005;109:93–108. doi: 10.1007/s00401-005-0991-y. [DOI] [PubMed] [Google Scholar]
- 2.Guillard S, Clarke PA, Te Poele R, Mohri Z, Bjerke L, Valenti M, Raynaud F, Eccles SA, Workman P. Molecular pharmacology of phosphatidylinositol 3-kinase inhibition in human glioma. Cell Cycle. 2009;8:443–53. doi: 10.4161/cc.8.3.7643. [DOI] [PubMed] [Google Scholar]
- 3.Palanichamy K, Erkkinen M, Chakravarti A. Predictive and prognostic markers in human glioblastomas. Curr Treat Options Oncol. 2006;7:490–504. doi: 10.1007/s11864-006-0024-7. [DOI] [PubMed] [Google Scholar]
- 4.Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60:166–93. doi: 10.3322/caac.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mrugala MM, Chamberlain MC. Mechanisms of disease: temozolomide and glioblastoma--look to the future. Nat Clin Pract Oncol. 2008;5:476–86. doi: 10.1038/ncponc1155. [DOI] [PubMed] [Google Scholar]
- 6.Quann K, Gonzales DM, Mercier I, Wang C, Sotgia F, Pestell RG, Lisanti MP, Jasmin JF. Caveolin-1 is a negative regulator of tumor growth in glioblastoma and modulates chemosensitivity to temozolomide. Cell Cycle. 2013;12:1510–20. doi: 10.4161/cc.24497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan Q, Yang XJ, Wang HM, Dong XT, Wang W, Li Y, Li JM. Chemoresistance to temozolomide in human glioma cell line U251 is associated with increased activity of O6-methylguanine-DNA methyltransferase and can be overcome by metronomic temozolomide regimen. Cell Biochem Biophys. 2012;62:185–91. doi: 10.1007/s12013-011-9280-7. [DOI] [PubMed] [Google Scholar]
- 8.Bianchini F, Kaaks R, Vainio H. Overweight, obesity, and cancer risk. Lancet Oncol. 2002;3:565–74. doi: 10.1016/S1470-2045(02)00849-5. [DOI] [PubMed] [Google Scholar]
- 9.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–38. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 10.Renehan AG, Dive C. Obesity, insulin and chemoresistance in colon cancer. J Gastrointest Oncol. 2011;2:8–10. doi: 10.3978/j.issn.2078-6891.2011.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frederich RC, Löllmann B, Hamann A, Napolitano-Rosen A, Kahn BB, Lowell BB, Flier JS. Expression of ob mRNA and its encoded protein in rodents. Impact of nutrition and obesity. J Clin Invest. 1995;96:1658–63. doi: 10.1172/JCI118206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sierra-Honigmann MR, Nath AK, Murakami C, García-Cardeña G, Papapetropoulos A, Sessa WC, Madge LA, Schechner JS, Schwabb MB, Polverini PJ, et al. Biological action of leptin as an angiogenic factor. Science. 1998;281:1683–6. doi: 10.1126/science.281.5383.1683. [DOI] [PubMed] [Google Scholar]
- 13.Zheng Q, Dunlap SM, Zhu J, Downs-Kelly E, Rich J, Hursting SD, Berger NA, Reizes O. Leptin deficiency suppresses MMTV-Wnt-1 mammary tumor growth in obese mice and abrogates tumor initiating cell survival. Endocr Relat Cancer. 2011;18:491–503. doi: 10.1530/ERC-11-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Izumiya M, Kabashima A, Higuchi H, Igarashi T, Sakai G, Iizuka H, Nakamura S, Adachi M, Hamamoto Y, Funakoshi S, et al. Chemoresistance is associated with cancer stem cell-like properties and epithelial-to-mesenchymal transition in pancreatic cancer cells. Anticancer Res. 2012;32:3847–53. [PubMed] [Google Scholar]
- 15.Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, Pilozzi E, Larocca LM, Peschle C, De Maria R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006;13:1238–41. doi: 10.1038/sj.cdd.4401872. [DOI] [PubMed] [Google Scholar]
- 17.Han G, Wang L, Zhao R, Yue Z, Zhou X, Hu X, Cao Y, Dai D, Liu J. Leptin promotes human glioblastoma growth through activating Signal Transducers and Activators of Transcription 3 signaling. Brain Res Bull. 2012;87:274–9. doi: 10.1016/j.brainresbull.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Maya-Monteiro CM, Bozza PT. Leptin and mTOR: partners in metabolism and inflammation. Cell Cycle. 2008;7:1713–7. doi: 10.4161/cc.7.12.6157. [DOI] [PubMed] [Google Scholar]
- 19.Park J, Scherer PE. Leptin and cancer: from cancer stem cells to metastasis. Endocr Relat Cancer. 2011;18:C25–9. doi: 10.1530/ERC-11-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cirillo D, Rachiglio AM, la Montagna R, Giordano A, Normanno N. Leptin signaling in breast cancer: an overview. J Cell Biochem. 2008;105:956–64. doi: 10.1002/jcb.21911. [DOI] [PubMed] [Google Scholar]
- 21.Ratke J, Entschladen F, Niggemann B, Zänker KS, Lang K. Leptin stimulates the migration of colon carcinoma cells by multiple signaling pathways. Endocr Relat Cancer. 2010;17:179–89. doi: 10.1677/ERC-09-0225. [DOI] [PubMed] [Google Scholar]
- 22.Frankenberry KA, Skinner H, Somasundar P, McFadden DW, Vona-Davis LC. Leptin receptor expression and cell signaling in breast cancer. Int J Oncol. 2006;28:985–93. [PubMed] [Google Scholar]
- 23.Frankenberry KA, Somasundar P, McFadden DW, Vona-Davis LC. Leptin induces cell migration and the expression of growth factors in human prostate cancer cells. Am J Surg. 2004;188:560–5. doi: 10.1016/j.amjsurg.2004.07.031. [DOI] [PubMed] [Google Scholar]
- 24.Riolfi M, Ferla R, Del Valle L, Piña-Oviedo S, Scolaro L, Micciolo R, Guidi M, Terrasi M, Cetto GL, Surmacz E. Leptin and its receptor are overexpressed in brain tumors and correlate with the degree of malignancy. Brain Pathol. 2010;20:481–9. doi: 10.1111/j.1750-3639.2009.00323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lang K, Ratke J. Leptin and Adiponectin: new players in the field of tumor cell and leukocyte migration. Cell Commun Signal. 2009;7:27. doi: 10.1186/1478-811X-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia XH, Gu JC, Bai QY, Yu W. Overexpression of leptin and leptin receptors in breast cancer positively correlates with clinicopathological features. Chin Med J (Engl) 2009;122:3078–81. [PubMed] [Google Scholar]
- 27.Uddin S, Bu R, Ahmed M, Abubaker J, Al-Dayel F, Bavi P, Al-Kuraya KS. Overexpression of leptin receptor predicts an unfavorable outcome in Middle Eastern ovarian cancer. Mol Cancer. 2009;8:74. doi: 10.1186/1476-4598-8-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steiniger SC, Coppinger JA, Krüger JA, Yates J, 3rd, Janda KD. Quantitative mass spectrometry identifies drug targets in cancer stem cell-containing side population. Stem Cells. 2008;26:3037–46. doi: 10.1634/stemcells.2008-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–68. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 30.Carra E, Barbieri F, Marubbi D, Pattarozzi A, Favoni RE, Florio T, Daga A. Sorafenib selectively depletes human glioblastoma tumor-initiating cells from primary cultures. Cell Cycle. 2013;12:491–500. doi: 10.4161/cc.23372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle. 2009;8:3274–84. doi: 10.4161/cc.8.20.9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiou SH, Wang ML, Chou YT, Chen CJ, Hong CF, Hsieh WJ, Chang HT, Chen YS, Lin TW, Hsu HS, et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010;70:10433–44. doi: 10.1158/0008-5472.CAN-10-2638. [DOI] [PubMed] [Google Scholar]
- 33.Feldman DE, Chen C, Punj V, Tsukamoto H, Machida K. Pluripotency factor-mediated expression of the leptin receptor (OB-R) links obesity to oncogenesis through tumor-initiating stem cells. Proc Natl Acad Sci U S A. 2012;109:829–34. doi: 10.1073/pnas.1114438109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells. 2009;27:2383–92. doi: 10.1002/stem.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Villalva C, Martin-Lannerée S, Cortes U, Dkhissi F, Wager M, Le Corf A, Tourani JM, Dusanter-Fourt I, Turhan AG, Karayan-Tapon L. STAT3 is essential for the maintenance of neurosphere-initiating tumor cells in patients with glioblastomas: a potential for targeted therapy? Int J Cancer. 2011;128:826–38. doi: 10.1002/ijc.25416. [DOI] [PubMed] [Google Scholar]
- 36.Beier D, Schulz JB, Beier CP. Chemoresistance of glioblastoma cancer stem cells--much more complex than expected. Mol Cancer. 2011;10:128. doi: 10.1186/1476-4598-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Villalva C, Cortes U, Wager M, Tourani JM, Rivet P, Marquant C, Martin S, Turhan AG, Karayan-Tapon L. O6-Methylguanine-Methyltransferase (MGMT) Promoter Methylation Status in Glioma Stem-Like Cells is Correlated to Temozolomide Sensitivity Under Differentiation-Promoting Conditions. Int J Mol Sci. 2012;13:6983–94. doi: 10.3390/ijms13066983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yi L, Zhou ZH, Ping YF, Chen JH, Yao XH, Feng H, Lu JY, Wang JM, Bian XW. Isolation and characterization of stem cell-like precursor cells from primary human anaplastic oligoastrocytoma. Mod Pathol. 2007;20:1061–8. doi: 10.1038/modpathol.3800942. [DOI] [PubMed] [Google Scholar]
- 39.Kohsaka S, Wang L, Yachi K, Mahabir R, Narita T, Itoh T, Tanino M, Kimura T, Nishihara H, Tanaka S. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol Cancer Ther. 2012;11:1289–99. doi: 10.1158/1535-7163.MCT-11-0801. [DOI] [PubMed] [Google Scholar]
- 40.Fava G, Alpini G, Rychlicki C, Saccomanno S, DeMorrow S, Trozzi L, Candelaresi C, Venter J, Di Sario A, Marzioni M, et al. Leptin enhances cholangiocarcinoma cell growth. Cancer Res. 2008;68:6752–61. doi: 10.1158/0008-5472.CAN-07-6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, Xiao HL, Wang B, Yi L, Wang QL, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J Immunol. 2012;189:444–53. doi: 10.4049/jimmunol.1103248. [DOI] [PubMed] [Google Scholar]
- 42.Jing Y, Han Z, Liu Y, Sun K, Zhang S, Jiang G, Li R, Gao L, Zhao X, Wu D, et al. Mesenchymal stem cells in inflammation microenvironment accelerates hepatocellular carcinoma metastasis by inducing epithelial-mesenchymal transition. PLoS One. 2012;7:e43272. doi: 10.1371/journal.pone.0043272. [DOI] [PMC free article] [PubMed] [Google Scholar]