

Abstract
Primary rodent cells undergo replicative senescence, independent from telomere shortening. We have recently shown that treatment with rapamycin during passages 3–7 suppressed replicative senescence in rat embryonic fibroblasts (REFs), which otherwise occurred by 10–14 passages. Here, we further investigated rapamycin-primed cells for an extended number of passages. Rapamycin-primed cells continued to proliferate without accumulation of senescent markers. Importantly, these cells retained the ability to undergo serum starvation- and etoposide-induced cell cycle arrest. The p53/p21 pathway was functional. This indicates that rapamycin did not cause either transformation or loss of cell cycle checkpoints. We found that rapamycin activated transcription of pluripotent genes, oct-4, sox-2, nanog, as well as further upregulated telomerase (tert) gene. The rapamycin-derived cells have mostly non-rearranged, near-normal karyotype. Still, when cultivated for a higher number of passages, these cells acquired a chromosomal marker within the chromosome 3. We conclude that suppression mTORC1 activity may prevent replicative senescence without transformation of rodent cells.
Keywords: aging, senescence, mTOR, gerosuppression, rapalogs
Introduction
Rat embryonic fibroblasts (REFs) have a limited lifespan in culture, which normally does not exceed 10–14 passages. Then, REFs undergo irreversible cell cycle arrest and expression of senescence-related markers. This phenomenon is known as replicative senescence.1-8 Senescent morphology is characterized by cell hypertrophy, cell flattening, hyper-secretory phenotype, and SA-βGal staining.1-3,5,9-19 This senescent morphology depends in part on the mTOR signaling pathway.20-25 Also, mTOR is involved in DDR, which could be activated by growth stimulation.26-28 It is important to emphasize that there are 2 processes involved in cellular senescence: cell cycle arrest and geroconversion.29,30 First, p53, p21, and p16 and cytostatic stresses induce cell cycle arrest.31-34 Second, cell cycle arrest is converted to senescence.29,35 Geroconversion is determined in part by the activity of the mTOR pathway. Agents and conditions that inhibit mTOR pathways all decelerate geroconversion.21,30,36-45 Cell cycle arrest and geroconversion can be dissociated.29 For example, p53 and other tumor suppressors are involved in senescence, because they cause cell cycle arrest.11,12,31,37,46-57 Yet, by inhibiting mTOR,58 they also can suppress geroconversion.59-69 Many oncogenes are aging promoters, whereas tumor suppressors are aging suppressors.70-72 Also, mTOR, p53 and other genes are involved in age-related diseases and aging.73-96 In replicative senescence of rodent cells the relationship between cell cycle arrest and geroconversion is more complex.1-5,97-99 Still, knockdown of mTOR gene by specific siRNAs and rapamycin treatment partially suppresses senescence and prevents expression of senescence-associated markers.100,101 In contrast to telomere-dependent replicative arrest in human cells, the length of telomeres is not limiting in dividing rodent cells, suggesting the mechanisms that limit their lifespan are telomere length-independent.5,26,102-104
We have recently shown that rapamycin, an inhibitor of both mTORC1 activity and geroconversion, prevented replicative senescence in rat embryonic fibroblasts (REFs).101 Here, we investigated the properties of these cell lines (named Rapa cells). Like normal cells, Rapa cell lines retain the checkpoints in response to serum withdrawal and etoposide treatment as well as the functional p53/p21 pathway. Noteworthy, expression of pluriponent genes oct-4, sox-2, and nanog as well as the telomerase gene was detected. The Rapa cells have mostly non-rearranged karyotype (modal value 42 chromosomes), albeit with a chromosomal marker within the chromosome 3. Thus, rapamycin-mediated suppression of mTOR, by abrogating geroconversion, ensures conditions to establish the cells with an increased lifespan that retain functional checkpoint control and near-normal karyotype.
Results
Rapamycin slows down cell senescence and restores proliferation of senescent REFs
As described previously, by the seventh passage, primary rat embryonic fibroblasts (REFs) slowed down proliferation rate and acquired a flat morphology (hypertrophy), SA-β-Gal staining, and accumulation of γH2AX foci.101 Rapamycin was added to REF cells at passage 7, and the cells were grown for 3 additional passages in rapamycin-containing media. Afterwards, rapamycin was removed, and during the subsequent passages the cells were becoming smaller in size, thereby reflecting a gradual restoration of the non-senescent phenotype as evidenced by Giemsa staining (Fig. 1). In parallel, the population lost such senescence markers as SA-β-Gal staining (Fig. S1) and restored the lost ability to proliferate as evidenced by an increase of S-phase cells (Fig. 2). We cultured the cells for a larger number of passages (25–30), analyzing the following parameters: growth of the cell population, the ability to grow in clonal density, and to undergo the cell cycle arrest in response to serum withdrawal and treatment with DNA-damaging agents (etoposide). According to analysis of cell growth data (Fig. 3A), rapamycin-derived cells demonstrate a higher proliferation rate as compared with REF cells. Thus, the doubling time decreases from 37.7 h in control REF cells to 26.8 h and 20.4 h in rapamycin-derived cells on passages 10 and 30, respectively (Fig. 3A) that correlate with the acquisition of the ability to grow in clonal density (Fig. 3B and table therein). In spite of high proliferation rate, the rapamycin-derived proliferating cells underwent cell cycle arrest after serum deprivation (LS) or etoposide (ETO) treatment accompanied by a decrease of S-phase cells (Fig. 4A). We have checked whether etoposide-induced cell cycle arrest was associated with activation of the p53/p21 pathway. In fact, etoposide increased p53 and p21 as well as p53 phosphorylation (Ser-15) (Fig. 4B).

Figure 1. Cell morphology. During senescence of REFs the cell size increases (hypertrophy), and the cells flatten over the substrate (the images are taken at the passages 3 and 5). Rapamycin treatment of senescent cells for 3 passages (from 7–10) decreases thereafter the size of cells cultured under rapamycin-free conditions (the images are taken from passages 18 and 30, Rapa); Rapamycin (200 nM) was present in cultured cells from passage 7 to passage 10. Then rapamycin was washed out, and the cells were cultured in rapamycin-free medium DMEM + 10% FCS. The rapamycin-treated cells gradually decrease in size and acquire a normal phenotype. Cells were stained by the Romanovsky–Giemsa to see change of cell morphology. Photomicrographs of cells were taken using a microscope Axioscope 40 × 10.

Figure 2. Flow cytometric analysis of cell cycle distribution of senescent REFs (passages 1 and 9 and REF cells treated by rapamycin (passage 19). Cells at different passages of culturing were prepared as described in the section “Flow cytometry” and analyzed for G1, S, and G2 phase ratio. (A) REF, passage 1 without rapamycin; (B) REF, passage 9 without rapamycin; (C) Rapamycin-derived cells, passage 19 (rapamycin treatment of REFs during passages from 7–10).

Figure 3. Growth curves of primary REF cells and rapamycin-derived cell lines. (A) Number of cells as a function of time (days). REF cells passage 3, rapamycin-derived cells (Rapa) passages 10 and 30 were seeded and counted every 24 h for 5 d. The population doubling time (PDT) Td was calculated by using formula Td = (log2)*t/[log2(Nt/N0)], where t is time in hours, Nt is the cell number at time t, and N0 is the cell number at the initial time. Each experiment was performed in triplicate. (B) Cloning efficiency of primary REF cells and rapamycin-derived cells. Ctrl, primary REF cells (passage 3); Rapa, rapamycin-derived cells (passages 10 and 30) were plated at clonal density (100 cells/60-mm plate) and left to grow for 14 d, when they formed eye-visible cell clones. Cells were stained by Giemsa dye to see and count visible clones in light microscope, magnification 10 × 40.

Figure 4. Rapamycin-derived cells demonstrate checkpoint control and functional p53Waf1 signaling pathway. (A) Flow cytometry. Rapa-cells were left untreated (left panel) or treated with etoposide (ETO, middle panel) for 1 h (12.5 M), the right panel shows data for the serum-starved cells (48 h). (B) Western blotting. Cells were left untreated, treated with etoposide (ETO) for 1 h (12.5 M) or serum-starved for 48 h. Cell extracts were prepared after 1 h-ETO treatment for p53 (Ser15) staining or 12 h post-ETO treatment for p21Waf1 staining. 1, REF (Ctrl); 2, Rapa, passage 8 (untreated); 3, Rapa, passage 21 (untreated); 4, REF (Etoposide); 5, Rapa, passage 8 (Etoposide); 6, Rapa, passage 21 (Etoposide).
Rapamycin activates autophagy in senescent REF cells
Senescent cells undergo cell cycle arrest and lose their ability to proliferate. Despite the cell cycle block, protein translation continues, cells accumulate various intracellular and secreted proteins and cytokines, creating a so-called “senescence-associated secretory phenotype”, or SASP.13,105,106 The abundant protein synthesis is ensured by the high activity of the mTOR signaling pathway (mTORC1 complexes), which negatively regulates autophagy.107-111 Recent data show that rapamycin can decelerate cellular senescence induced in tumor cells by overexpression of p21Waf1 and HDAC inhibitor treatment21-23 and delays manifestation of senescent phenotype of fibroblasts from patients with Hutchinson–Gilford progeria syndrome.108 Therefore, rapamycin-dependent inhibition of mTORC1 and subsequent resumption of proliferation requires the removal of excessive synthesized proteins that is likely provided by activation of autophagy. To assess autophagy, we used antibodies to the components of autophagolysosomes LC3 and LAMP1 proteins for immunofluorescent staining of senescent REFs and rapamycin-derived cells. Data presented in Figure 5 show that although LC3 gradually accumulates during senescence of rat fibroblasts, it does not form in the cytoplasm the well-defined punctate autophagic vesicles, being more or less evenly distributed throughout the cytoplasm. A key component of autophagolysosomes, LAMP1, neither accumulates nor co-localizes with LC3 in senescent cells (Fig. 5B). However, inhibition of mTORC1 caused colocalization of LC3/LAMP1 even in short-term rapamycin-treated senescent cells and resulted in a decrease in cell size (Fig. 5C and D). A marker of mTORC1 activity, target pS6 protein, accumulates in hypertrophic senescent cells, and then pS6 intensity decreases in rapamycin-derived cells (Fig. S2). Thus, rapamycin inhibits activity of mTORС1 that leads to a decrease of the cell size by means of autophagy, which likely helps to remove the excess of proteins.

Figure 5. Immunofluorescence of LC3/LAMP1 staining in senescent REFs and rapamycin-derived cells. Confocal microscopy images were taken from rapamycin-untreated senescent REF cells (A) passage 3, (B) passage7; (Cand D) REF cells of passage 7 after short-term rapamycin treatment for 5 h (C, magnification as in A and B) and a higher magnification (D). Coverslips were stained with LC3 rabbit polyclonal and anti-LAMP1 mouse monoclonal antibody (Cell Signaling). Nuclei were stained with DAPI.
Rapamycin-derived cells demonstrate transcription of pluripotent genes oct-4, sox-2, and nanog
The transition of senescent cells to a proliferation state may reflect a dramatic change in the transcription of genes involved in maintenance of self-renewal and pluripotency. Similar changes were shown to occur when somatic cells from the adult organism underwent the reprogramming induced by expression of pluripotent genes with subsequent formation of so-called induced pluripotent stem cells (iPSCs).112-119 Similarly, in the case of the rapamycin-induced trigger for senescent cells to proliferate, the driving force for proliferation may result from the pluripotent gene expression. We checked the expression of such pluripotent genes as oct-4, sox-2, and nanog in proliferating rapamycin-derived cells. Data presented in Figure 6 show that the analyzed genes in fact were upregulated in the cells, albeit at the lower levels as compared with mouse embryonic stem cells (positive control). Interestingly, transcription of telomerase gene (mtert) is additionally augmented in rapamycin-derived cells, implying that rapamycin treatment makes a positive impact on maintenance of telomeres. This is consistent with recent data showing that overexpression of telomerase gene (TERT) delays the physiological aging and extends longevity in normal mice already having the elevated levels of telomerase activity.117
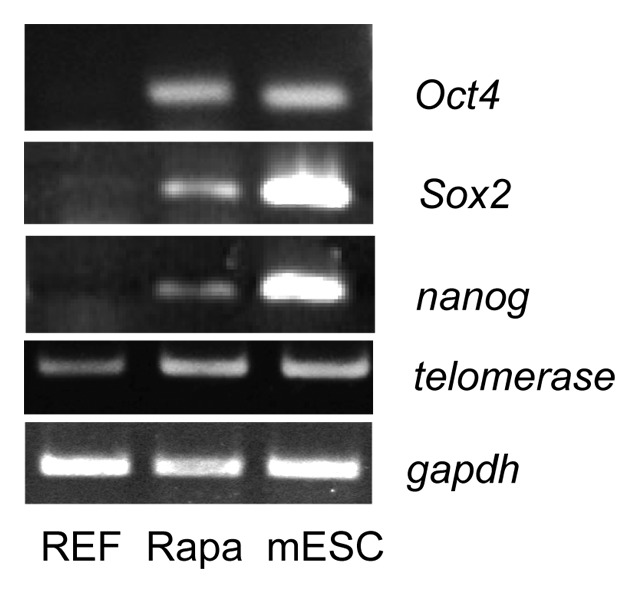
Figure 6. RT-PCR analysis of transcripts of pluripotent genes in rapamycin-derived cells.RNA was prepared from REFs (passage 3), rapamycin-derived cells (passage 21), and mESCs (positive control). RT-PCR step was performed by using primers to Oct4, Sox2, Nanog, and Tert genes. Gapdh mRNA level was taken as internal control.
Karyotype analysis
Cytogenetic analysis of rapamycin-derived cells as early as on passage 5 after rapamycin treatment showed that the cells have the diploid number of chromosomes, with the modal chromosome number 42 and a small proportion of polyploid cells (3%). By using a method of G-band staining, we found only one rearranged chromosome—an inversion of chromosome 3—inv (3) (p12q12 ~q21) (Fig. 7C). It was important to find out whether the chromosome rearrangements in Rapa cells are accompanied by a deletion of the nucleolar organizer(s), which are located on the short arm of the chromosome 3 and deletion of 3p12. We used silver staining of chromosomes to identify transcriptionally active nucleolar organizers. According to these data, the inversion in the chromosome 3 is pericentric, i.e., it includes the centromere and translocation of the short arm of the 3p12 locus, which includes a part of the nucleolar organizer, on the long arm of the chromosome. In addition, there is a transcriptionally active nucleolar organizer on both homologs of chromosome 11 and on one homolog of chromosome 12 (Fig. 7B). Karyotypic analysis of cells at the passage 17 showed an almost diploid number of chromosomes, but the modal value approached 43. The proportion of cells with 42 chromosomes is about 12% of population, while the number of polyploid cells is 4.7%. Among cells with 43 chromosomes there are cells with trisomy of chromosome 6 and the pericentric inversion of chromosome 3 (Fig. 7D). Cells with chromosome number 42 have a karyotype that is similar to those at passage 5 of Rapa cells. The obtained data evidences for preservation of normal karyotype at early passages followed by a gradual deviation of the chromosome number. It is currently unclear what events promote the chromosome rearrangements: either cultivation of cells in vitro for many passages or genomic instability caused by the rapamycin-induced conversion of senescent cells to immortal ones. The first outcome is often observed in cultured somatic or embryonic stem cells, and this phenomenon finds an explanation in accumulation of DNA damages induced by radical oxygen species (ROS) and other adverse factors.

Figure 7. Karyotype analysis of rapamycin-derived cells. (A) REF cells (passage 2) have normal karyotype with a modal chromosome number 42. (B) Chromosomes were stained with silver nitrate (AgNO3) to identify transcriptionally active nucleolar organizers (arrows) for confirmation of the pericentric inversion without loss of nuclear organizers. The insert below shows chromosome pairs with nucleolar organizers: the left chromosome in the pair: G-banding, the right chromosome in the pair: Ag-positive staining. The homolog of chromosome 12 is Ag-negative. (C) Rapamycin-derived cells (passage 5). Modal karyotype: 42 chromosomes, XX, inv (3) (pter → p12:: q21 → p12:: q21 → qter). The arrow shows the pericentric inversion in chromosome 3. (D) Rapamycin-derived cells (passage 17). Modal karyotype: 43XX, inv (3) (pter → p12:: q21 → p12:: q21 → qter). The arrow shows the pericentric inversion in chromosome 3.
Discussion
Both replicative senescence and Ras-induced senescence of primary human and mouse fibroblasts correlate with activation of DNA damage response signaling and mTORC1 activation.21,100,101 Attenuation of mTORC1 results in delay of cellular senescence.21,29 Depletion of TORC2 in human cells fails to affect the course of replicative or Ras-induce senescence implying a key role of mTORC1.100 Even pre-senescent fibroblasts can be returned to proliferation by introduction of pluripotent genes in the course of cell reprogramming, evidencing that cell senescence is not an irreversible barrier for proliferation.120The existence of tissue-specific cancer stem cells is an argument in favor of a transition of somatic cells to proliferating stem cells. Available data indicate that putative cancer stem cells do normally exist in a tissue, but by yet-unknown mechanisms may transform to real cancer stem cells, acquiring high proliferation potential.
Primary mouse and human fibroblasts after introduction of reprogramming factors (exemplified by Yamanaka cocktail Oct4, Sox2, Klf4, c-Myc) undergo a cell cycle arrest with multiple characteristics of senescence: SA-βGal activity, p16Ink4a accumulation, formation of senescence-associated heterochromatin foci (SAHFs).114,120-132 Then, by a yet-unknown stochastic process, the arrested cell, due to expression of the reprogramming factors and upregulation of their target genes, acquired an impulse to proliferate and become pluripotent.112,133,134 According to our data, rapamycin inhibits mTORC1 activity in cell cycle-arrested senescent REF cells, thereby preventing senescence and activating transcription of pluripotent genes, albeit at lower levels as compared with embryonic stem cells. Nevertheless, the elevated level of pluripotent genes expression is likely sufficient to slow down senescence and provide immortal characteristics of rapamycin-derived cells. The issues of how suppression of mTORC1 activity in senescent REF cells by rapamycin leads to activation of transcription of pluripotent genes, and whether modulation of mTORC1 activity takes place during reprogramming of somatic cells by pluripotent factors requires further investigation. Interestingly, in normal mice after 70% partial hepatectomy in vivo the reprogramming factors Oct4 and Nanog were found to be upregulated and therefore may play a role in a transition of quiescent hepatocytes to a proliferation state in the course of liver regeneration.135 This suggests that tissue regeneration in mice is mediated via formation of a stem cell-like phenotype.136 Two issues have to be further cleared: First, how rapamycin-mediated suppression of mTORC1, followed by a reversion of senescence program, promotes transcription of pluripotent genes, expression of which provides a stem-like cellular phenotype. To this end, it is still unclear what signaling pathways, in addition to mTORC1, are involved in rapamycin-induced transition of senescent cells to stem-like derivatives. Second, it would be important to find out whether a reversal of cellular senescence phenotype, which is initially induced by pluripotent factors during reprogramming of somatic cells (iPSC), obligates suppression of mTORC1 activity. It should be recalled that mTOR inhibitors rapamycin and pp242 enhance the efficiency of reprogramming to induced pluripotent stem cells.137 Apparently, these 2 processes are very functionally similar with respect to the order of events and the underlying mechanisms of the transition from the senescent to a stem-like state.
It is noteworthy that suppression of mTORC1 by rapamycin, in turn, leads to activation of autophagy, the process that seems to be protective in many types of normal and tumor cells and is a prerequisite for the establishment of stemness. Senescent cells do not proliferate. It has been recently shown that autophagy is highly activated during senescence.105,107,138 Autophagy usually makes cells smaller, but in Ras-induced senescent cells, both protein synthesis and autophagy are simultaneously activated. As follows from LC3/LAMP1 staining, replicative senescence is associated with augmented protein synthesis but decreased activity of autolysophagy (Fig. 5). Typically, when autophagy is activated, anabolic processes are suppressed, and thereafter the cell size becomes reduced. However, during Ras-induced senescence of human fibroblasts, the cells continue growing in size, even though they undergo proliferation arrest and eventually produce huge amounts of secretory proteins, termed as senescence-associated secretory phenotype (SASP). Recently, we have shown that mTORC1 suppression with rapamycin slows down senescence, thereby providing the conditions for continued proliferation of human and rodent tumor cells. The paradoxical activation of mTOR and autophagy in the process of cellular senescence needs further analysis.
Karyotype analysis of rapamycin-derived cells showed that cells taken from the early passages had normal karyotype with a modal chromosome number 42. During subsequent passages, the cells acquired a few chromosomal rearrangements, including an inversion of the chromosome 3, where a nucleolar organizer localizes. It is known that the majority of proliferating cells are unable to maintain the normal karyotype for a long time of culturing in vitro and acquire various chromosomal rearrangements. Moreover, even human and mouse embryonal stem cells demonstrate the same properties being cultured in vitro. Maintenance of the normal karyotype is a problem for using ESCs in cell therapy. In any case, resumption of proliferation in senescent cells induced by rapamycin-dependent mTORC1 suppression might be a promising approach for establishing cell lines from cells of adult patients.
Materials and Methods
Cell culture and reagents
Primary REF cells of the second passage were cultured in DMEM (Gibco) supplemented with 10% of FCS (HyClone) without antibiotics in a humidified incubator providing 5% CO2/95% air atmosphere. The early passage cells were split 1:3 twice a week by plating 5 × 105 cells per 60-mm dish. Mid-passage (7–10 passages) cells were split 1:2 weekly, and late-passage cultures were split 1:2 after confluence. In this way, several subsequent passages were performed, until the cells failed to undergo population doublings. Under the given conditions of cultivation, REF cells usually reached this state after 14–15 passages. Rapamycin (Calbiochem) was added to primary REFs at concentration 200 nM for cell passages 7–10. This concentration of rapamycin inhibited mTORC1 but did not completely inhibit proliferation of REF cells. After 3 passages in the presence of rapamycin, the inhibitor was removed, and cells were then cultured on fresh medium DMEM supplemented with 10% FCS.
Cell cycle analysis
Flow cytometry was peformed as described. Cells were trypsinized and washed with PBS and permeabilized for 30 min with 0.01% saponin. Then 105 cells were incubated with 40 μg/ml of propidium iodide (Sigma), 0.1 mg/ml RNase A (Sigma) for 15 min at 37 °C prior to analysis using an Odam cytofluorimeter (Brucker).
Growth curves
The growth rate of cells was determined by counting the number of cells as a function of time. The cells were seeded 3 × 104 per 30-mm dish and counted every 24 h for 5 d. The population doubling time (PDT) Td was calculated from the growth curve by using the formula Td = (log22)*t/[log2(Nt/N0)], where t is time in hours, Nt is the cell number at time t, and N0 is the cell number at the initial time. Each experiment was performed in triplicate.
Plating efficiency
Cells were plated at density 200 cells per 30-mm dish and incubated for 14 d to allow colony formation. Then cells were fixed and stained by crystal violet solution (10% acetic acid, 30% alcohol, and 0.4% crystal violet). The measurements were done in triplicate.
Cell size
The size of control and rapamycin-treated cells was compared by means of cytometric light scatering of propidium iodide-stained cells by using Win MDI program version 2.8.
Immunofluorescence microscopy
REF cells were seeded and grown on coverslips, washed in PBS, fixed in fresh 4% paraformaldehyde for 15 min at room temperature, and washed 3 times in PBS for 10 min each time. The cells were permeabilized with 0.2% Triton X-100 for 20 min, and blocked by incubation with 5% bovine serum albumin (BSA) (V fraction, Sigma) in PBS for 30 min. The coverslips were incubated with the following primary antibodies: anti-LAMP1 mouse monoclonal antibody (Cell Signaling), LC3 rabbit polyclonal (Cell Signaling), and anti-S6 mouse monoclonal or S6 polyclonal antibodies (Cell Signaling) for 1 h. The coverslips were washed 3 times in PBS-1% BSA for 10 min each time, and incubated with the secondary antibodies: rabbit anti-mouse fluorochrome-conjugated Alexa Fluor 488-conjugated or goat anti- rabbit Alexa Fluor 543-conjugated (Invitrogen) and DAPI or To-Pro III, (Molecular Probes) to stain nuclei for 1 h at room temperature. The images were obtained using Leica TCS SL confocal microscope (Leica Microsystems).
SA-β-Gal staining
Cells were fixed for 5 min in β-galactosidase fixative (2% formaldehyde; 0.2% glutaraldehyde in PBS), then washed in PBS and stained in β-galactosidase solution (1 mg/ml 5-bromo-4-chloro-3-indolyl-β-gal [X-gal] in 5 mM potassium ferricyamide, 5 mM potassium ferrocyamide, 2 mM MgCl2 in PBS) at 37 °C, until β-Gal staining become visible in either experiment or control plates. Thereafter, cells were washed in PBS, and the number of β-galactosidase-positive cells (blue staining) was counted under brightfield illumination.
RT-PCR
Total RNA from cells was isolated using TRIzol® (Invitrogen) according to the manufacturer’s instructions. Pellets were dissolved in DEPC-SQ. Reverse transcription was performed with 2 μg of RNA, using 1 g of random hexaprimers (Roche Applied Science) and MMLV (Moloney-murine leukemia virus) revertase (RevertAid; Fermentas). The amount of total RNA for RT-reactions was normalized by densitometry of 18 S rRNA band after electrophoresis in denaturing agarose gel. PCR step was made using DNA polymerase (Fermentas). The reaction parameters were according to the standard protocols (94 °С −40 s,Tm −40 s, 72 °С −40 s) with a hot start. The PCR step was performed in the presence of 100 ng of primers to the cDNA of rat genes: Oct3/4 (Oct3/4-F CAAGTTGGCGTGGAGACT, Oct3/4-R GCTCCTTCTG CAGGGCTTT, Tm = 57 °C, 35 cycles), Nanog (Nanog-F GAGACTGCCTCTCCTCCGCC TT, Nanog-R GTGCACACAACTGGGCCTGA, Tm = 59 °C, 35 cycles), Sox2 (Sox2-F ACATGAACGG CTGGAGCAAC G, Sox2-R CATGTAGGTC TGCGAGCTGG TC, Tm = 55 °C, 35 cycles), Tel (Tel-F TCTGAGTCTC ACCAGTACA, Tel-RTGGAGTAAA GGAAATGTCT G, Tm = 54 °C, 35 cycles). The data were normalized to gapdh mRNA levels.
Immunoblotting
For western blotting, cell lysates were obtained by incubating cells in RIPA buffer containing PBS solution, 1% Igepal, 0.5% sodium deoxycholate, 0.1% SDS (Sigma), protease and phosphatase inhibitors (CompleteTM protease inhibitor cocktail; Roche), 5 mM EGTA, 1 mM NaV, 10 mM NaF. Protein concentration was estimated by the Bradford method (Bradford, 1976). Fifty micrograms of each sample was loaded onto 12% or 15% polyacrylamide gels and transferred to PVDF membrane (Immobilon-P, Millipore). The membranes were blocked in a 3–5% of bovine serum albumin (BSA) or nonfat dry milk in PBS and 0.1% Tween 20 (PBS-T) for 1 h and then incubated overnight at 40 °C with primary antibodies diluted in the PBS-T buffer containing 1% BSA: p-p53 (#9284S, Cell Signaling), p53 (#2524S, Cell Signaling), p21Waf1 (Calbiochem OP079, Invitrogen AHZ0422) according to the manufacturer’s recommendations. The HRP (horseradish peroxidase)-conjugated goat anti-rabbit and rabbit anti-mouse antibodies (Sigma) diluted in PBS-T containing 5% BSA were used for 1 h at room temperature. Proteins on membranes were visualized by means of ECL (0.1 mM TRIS-HCl, 0.1 mM Luminol, 60 mM p-Coumaric acid, Sigma), and signal was fixed on X-ray Kodak film. The data were normalized to gapdh protein levels. For re-blotting, the membrane was washed with stripping buffer (62.5 mM Tris/HCl, pH 6.8, 2% SDS, and 100 mM 2-mercaptoethanol; Sigma) for 30 min at 56 °C.
Chromosome analysis
To obtain metaphase plates, cells were treated with colchicine (Sigma) at a final concentration of 0.06 g/ml for 1 h. Hypotonic treatment was performed by placing the cells into 0.55% KCl at 37 °C for 15 min, then cells were fixed with a mixture of methanol and glacial acetic acid (3:1) 3 times for 15 min. Suspension of fixed cells was applied to wet slides followed by drying in the air. To identify the individual chromosomes, we used a method of G-banding described in reference 139. Chromosome plates were treated with 0.02% trypsin (Difco) for 75−80 s at 22 °C, then chromosomes were stained with 2% Romanowsky-Giemsa dye in sodium–phosphate buffer (pH 6.8). To identify transcriptionally active nucleolar organizers, the modified method of selective staining with silver nitrate was used.140 Metaphase chromosomes were incubated in a solution containing 50% AgN03 and 2% gelatin for 8–10 min at 56 °C. After silver staining, the slides were washed thoroughly in tap water and dried in air. Chromosomes were successively dehydrated in 70% and 96% ethyl alcohol and then co-stained with Romanowsky–Giemsa stain.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was funded by Program of the Russian Academy of Sciences (MCB RAS) and grants from Russian Foundation for Basic Research (13-04-00552 and 12-04-01393), and supported by Program of St. Petersburg State University.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27396
References
- 1.Cristofalo VJ, Pignolo RJ. Molecular markers of senescence in fibroblast-like cultures. Exp Gerontol. 1996;31:111–23. doi: 10.1016/0531-5565(95)02018-7. [DOI] [PubMed] [Google Scholar]
- 2.Itahana K, Campisi J, Dimri GP. Mechanisms of cellular senescence in human and mouse cells. Biogerontology. 2004;5:1–10. doi: 10.1023/B:BGEN.0000017682.96395.10. [DOI] [PubMed] [Google Scholar]
- 3.Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113:3613–22. doi: 10.1242/jcs.113.20.3613. [DOI] [PubMed] [Google Scholar]
- 4.Bhaumik D, Scott GK, Schokrpur S, Patil CK, Orjalo AV, Rodier F, Lithgow GJ, Campisi J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging (Albany NY) 2009;1:402–11. doi: 10.18632/aging.100042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Russo I, Silver AR, Cuthbert AP, Griffin DK, Trott DA, Newbold RF. A telomere-independent senescence mechanism is the sole barrier to Syrian hamster cell immortalization. Oncogene. 1998;17:3417–26. doi: 10.1038/sj.onc.1202261. [DOI] [PubMed] [Google Scholar]
- 6.Di Micco R, Cicalese A, Fumagalli M, Dobreva M, Verrecchia A, Pelicci PG, di Fagagna Fd. DNA damage response activation in mouse embryonic fibroblasts undergoing replicative senescence and following spontaneous immortalization. Cell Cycle. 2008;7:3601–6. doi: 10.4161/cc.7.22.7152. [DOI] [PubMed] [Google Scholar]
- 7.Hubackova S, Krejcikova K, Bartek J, Hodny Z. IL1- and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘bystander senescence’. Aging (Albany NY) 2012;4:932–51. doi: 10.18632/aging.100520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harada H, Nakagawa H, Takaoka M, Lee J, Herlyn M, Diehl JA, Rustgi AK. Cleavage of MCM2 licensing protein fosters senescence in human keratinocytes. Cell Cycle. 2008;7:3534–8. doi: 10.4161/cc.7.22.7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hildebrand DG, Lehle S, Borst A, Haferkamp S, Essmann F, Schulze-Osthoff K. α-Fucosidase as a novel convenient biomarker for cellular senescence. Cell Cycle. 2013;12:1922–7. doi: 10.4161/cc.24944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Driscoll MK, Albanese JL, Xiong ZM, Mailman M, Losert W, Cao K. Automated image analysis of nuclear shape: what can we learn from a prematurely aged cell? Aging (Albany NY) 2012;4:119–32. doi: 10.18632/aging.100434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lahtela J, Corson LB, Hemmes A, Brauer MJ, Koopal S, Lee J, Hunsaker TL, Jackson PK, Verschuren EW. A high-content cellular senescence screen identifies candidate tumor suppressors, including EPHA3. Cell Cycle. 2013;12:625–34. doi: 10.4161/cc.23515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen YL, Chen YJ, Tsai WH, Ko YC, Chen JY, Lin SF. The Epstein-Barr virus replication and transcription activator, Rta/BRLF1, induces cellular senescence in epithelial cells. Cell Cycle. 2009;8:58–65. doi: 10.4161/cc.8.1.7411. [DOI] [PubMed] [Google Scholar]
- 13.Coppé JP, Kauser K, Campisi J, Beauséjour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. 2006;281:29568–74. doi: 10.1074/jbc.M603307200. [DOI] [PubMed] [Google Scholar]
- 14.Haferkamp S, Rizos H. Oncogene-induced senescence pathways in melanocytes. Cell Cycle. 2010;9:4778–9. doi: 10.4161/cc.9.24.14248. [DOI] [PubMed] [Google Scholar]
- 15.Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123:966–72. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Acosta JC, O’Loghlen A, Banito A, Raguz S, Gil J. Control of senescence by CXCR2 and its ligands. Cell Cycle. 2008;7:2956–9. doi: 10.4161/cc.7.19.6780. [DOI] [PubMed] [Google Scholar]
- 17.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 18.Serrano M, Blasco MA. Putting the stress on senescence. Curr Opin Cell Biol. 2001;13:748–53. doi: 10.1016/S0955-0674(00)00278-7. [DOI] [PubMed] [Google Scholar]
- 19.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008;7:3355–61. doi: 10.4161/cc.7.21.6919. [DOI] [PubMed] [Google Scholar]
- 21.Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009;8:4112–8. doi: 10.4161/cc.8.24.10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romanov VS, Abramova MV, Svetlikova SB, Bykova TV, Zubova SG, Aksenov ND, Fornace AJ, Jr., Pospelova TV, Pospelov VA. p21(Waf1) is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle. 2010;9:3945–55. doi: 10.4161/cc.9.19.13160. [DOI] [PubMed] [Google Scholar]
- 23.Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–95. doi: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- 24.Darzynkiewicz Z. Forever young, slim and fit: rapamycin to the rescue. Cell Cycle. 2009;8:1818–22. doi: 10.4161/cc.8.12.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garbers C, Kuck F, Aparicio-Siegmund S, Konzak K, Kessenbrock M, Sommerfeld A, Häussinger D, Lang PA, Brenner D, Mak TW, et al. Cellular senescence or EGFR signaling induces Interleukin 6 (IL-6) receptor expression controlled by mammalian target of rapamycin (mTOR) Cell Cycle. 2013;12:3421–32. doi: 10.4161/cc.26431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodríguez-Jiménez FJ, Moreno-Manzano V, Mateos-Gregorio P, Royo I, Erceg S, Murguia JR, Sánchez-Puelles JM. FM19G11: A new modulator of HIF that links mTOR activation with the DNA damage checkpoint pathways. Cell Cycle. 2010;9:2875–85. doi: 10.4161/cc.9.14.12250. [DOI] [PubMed] [Google Scholar]
- 27.Perucca P, Cazzalini O, Madine M, Savio M, Laskey RA, Vannini V, Prosperi E, Stivala LA. Loss of p21 CDKN1A impairs entry to quiescence and activates a DNA damage response in normal fibroblasts induced to quiescence. Cell Cycle. 2009;8:105–14. doi: 10.4161/cc.8.1.7507. [DOI] [PubMed] [Google Scholar]
- 28.Monasor A, Murga M, Lopez-Contreras AJ, Navas C, Gomez G, Pisano DG, Fernandez-Capetillo O. INK4a/ARF limits the expansion of cells suffering from replication stress. Cell Cycle. 2013;12:1948–54. doi: 10.4161/cc.25017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY) 2012;4:159–65. doi: 10.18632/aging.100443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leontieva OV, Blagosklonny MV. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle. 2013;12:3063–9. doi: 10.4161/cc.26130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jee HJ, Kim AJ, Song N, Kim HJ, Kim M, Koh H, Yun J. Nek6 overexpression antagonizes p53-induced senescence in human cancer cells. Cell Cycle. 2010;9:4703–10. doi: 10.4161/cc.9.23.14059. [DOI] [PubMed] [Google Scholar]
- 32.Campaner S, Doni M, Verrecchia A, Fagà G, Bianchi L, Amati B. Myc, Cdk2 and cellular senescence: Old players, new game. Cell Cycle. 2010;9:3655–61. doi: 10.4161/cc.9.18.13049. [DOI] [PubMed] [Google Scholar]
- 33.Yamakuchi M, Lowenstein CJ. MiR-34, SIRT1 and p53: the feedback loop. Cell Cycle. 2009;8:712–5. doi: 10.4161/cc.8.5.7753. [DOI] [PubMed] [Google Scholar]
- 34.Itahana K, Dimri G, Campisi J. Regulation of cellular senescence by p53. Eur J Biochem. 2001;268:2784–91. doi: 10.1046/j.1432-1327.2001.02228.x. [DOI] [PubMed] [Google Scholar]
- 35.Blagosklonny MV. Cell cycle arrest is not senescence. Aging (Albany NY) 2011;3:94–101. doi: 10.18632/aging.100281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Demidenko ZN, Blagosklonny MV. At concentrations that inhibit mTOR, resveratrol suppresses cellular senescence. Cell Cycle. 2009;8:1901–4. doi: 10.4161/cc.8.12.8810. [DOI] [PubMed] [Google Scholar]
- 37.Leontieva OV, Gudkov AV, Blagosklonny MV. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010;9:4323–7. doi: 10.4161/cc.9.21.13584. [DOI] [PubMed] [Google Scholar]
- 38.Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle. 2009;8:1896–900. doi: 10.4161/cc.8.12.8809. [DOI] [PubMed] [Google Scholar]
- 39.Blagosklonny MV. Aging-suppressants: cellular senescence (hyperactivation) and its pharmacologic deceleration. Cell Cycle. 2009;8:1883–7. doi: 10.4161/cc.8.12.8815. [DOI] [PubMed] [Google Scholar]
- 40.Lee JH, Bodmer R, Bier E, Karin M. Sestrins at the crossroad between stress and aging. Aging (Albany NY) 2010;2:369–74. doi: 10.18632/aging.100157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Menendez JA, Joven J, Aragonès G, Barrajón-Catalán E, Beltrán-Debón R, Borrás-Linares I, Camps J, Corominas-Faja B, Cufí S, Fernández-Arroyo S, et al. Xenohormetic and anti-aging activity of secoiridoid polyphenols present in extra virgin olive oil: a new family of gerosuppressant agents. Cell Cycle. 2013;12:555–78. doi: 10.4161/cc.23756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle. 2009;8:88–96. doi: 10.4161/cc.8.1.7499. [DOI] [PubMed] [Google Scholar]
- 43.Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010;107:9660–4. doi: 10.1073/pnas.1002298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci U S A. 2012;109:13314–8. doi: 10.1073/pnas.1205690109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leontieva OV, Demidenko ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ. 2013;20:1241–9. doi: 10.1038/cdd.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baker DJ, Jin F, van Deursen JM. The yin and yang of the Cdkn2a locus in senescence and aging. Cell Cycle. 2008;7:2795–802. doi: 10.4161/cc.7.18.6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pfeuty B, David-Pfeuty T, Kaneko K. Underlying principles of cell fate determination during G1 phase of the mammalian cell cycle. Cell Cycle. 2008;7:3246–57. doi: 10.4161/cc.7.20.6853. [DOI] [PubMed] [Google Scholar]
- 48.Choong ML, Yang H, Lee MA, Lane DP. Specific activation of the p53 pathway by low dose actinomycin D: a new route to p53 based cyclotherapy. Cell Cycle. 2009;8:2810–8. doi: 10.4161/cc.8.17.9503. [DOI] [PubMed] [Google Scholar]
- 49.Jung-Hynes B, Ahmad N. Role of p53 in the anti-proliferative effects of Sirt1 inhibition in prostate cancer cells. Cell Cycle. 2009;8:1478–83. doi: 10.4161/cc.8.10.8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hubackova S, Novakova Z, Krejcikova K, Kosar M, Dobrovolna J, Duskova P, Hanzlikova H, Vancurova M, Barath P, Bartek J, et al. Regulation of the PML tumor suppressor in drug-induced senescence of human normal and cancer cells by JAK/STAT-mediated signaling. Cell Cycle. 2010;9:3085–99. doi: 10.4161/cc.9.15.12521. [DOI] [PubMed] [Google Scholar]
- 51.Akakura S, Nochajski P, Gao L, Sotomayor P, Matsui S, Gelman IH. Rb-dependent cellular senescence, multinucleation and susceptibility to oncogenic transformation through PKC scaffolding by SSeCKS/AKAP12. Cell Cycle. 2010;9:4656–65. doi: 10.4161/cc.9.23.13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang B, Li D, Kovalchuk O. p53 Ser15 phosphorylation and histone modifications contribute to IR-induced miR-34a transcription in mammary epithelial cells. Cell Cycle. 2013;12:2073–83. doi: 10.4161/cc.25135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang B, Xiao Z, Ko HL, Ren EC. The p53 response element and transcriptional repression. Cell Cycle. 2010;9:870–9. doi: 10.4161/cc.9.5.10825. [DOI] [PubMed] [Google Scholar]
- 54.Ha L, Merlino G, Sviderskaya EV. Melanomagenesis: overcoming the barrier of melanocyte senescence. Cell Cycle. 2008;7:1944–8. doi: 10.4161/cc.7.13.6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prencipe M, McGoldrick A, Perry AS, O’Grady A, Phelan S, McGrogan B, Fitzpatrick P, Watson JA, Furlong F, Brennan DJ, et al. MAD2 downregulation in hypoxia is independent of promoter hypermethylation. Cell Cycle. 2010;9:2828–37. doi: 10.4161/cc.9.14.12362. [DOI] [PubMed] [Google Scholar]
- 56.Miller KR, Kelley K, Tuttle R, Berberich SJ. HdmX overexpression inhibits oncogene induced cellular senescence. Cell Cycle. 2010;9:3396–3402. doi: 10.4161/cc.9.16.12779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lehmann BD, Brooks AM, Paine MS, Chappell WH, McCubrey JA, Terrian DM. Distinct roles for p107 and p130 in Rb-independent cellular senescence. Cell Cycle. 2008;7:1262–8. doi: 10.4161/cc.7.9.5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feng Z, Hu W, Rajagopal G, Levine AJ. The tumor suppressor p53: cancer and aging. Cell Cycle. 2008;7:842–7. doi: 10.4161/cc.7.7.5657. [DOI] [PubMed] [Google Scholar]
- 59.Blagosklonny MV. Hypoxia, MTOR and autophagy: converging on senescence or quiescence. Autophagy. 2013;9:260–2. doi: 10.4161/auto.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Serrano M. Shifting senescence into quiescence by turning up p53. Cell Cycle. 2010;9:4256–7. doi: 10.4161/cc.9.21.13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Santoro R, Blandino G. p53: The pivot between cell cycle arrest and senescence. Cell Cycle. 2010;9:4262–3. doi: 10.4161/cc.9.21.13853. [DOI] [PubMed] [Google Scholar]
- 62.Demaria M, Campisi J. Matters of life and breath: A role for hypoxia in determining cell state. Aging (Albany NY) 2012;4:523–4. doi: 10.18632/aging.100480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Long JS, Ryan KM. p53 and senescence: A little goes a long way. Cell Cycle. 2010;9:4050–1. doi: 10.4161/cc.9.20.13747. [DOI] [PubMed] [Google Scholar]
- 64.Galluzzi L, Kepp O, Kroemer G. TP53 and MTOR crosstalk to regulate cellular senescence. Aging (Albany NY) 2010;2:535–7. doi: 10.18632/aging.100202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Halicka HD, Zhao H, Li J, Lee YS, Hsieh TC, Wu JM, Darzynkiewicz Z. Potential anti-aging agents suppress the level of constitutive mTOR- and DNA damage- signaling. Aging (Albany NY) 2012;4:952–65. doi: 10.18632/aging.100521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steelman LS, McCubrey JA. Intriguing novel abilities of Nutlin-3A: induction of cellular quiescence as opposed to cellular senescence--implications for chemotherapy. Cell Cycle. 2009;8:3634–5. doi: 10.4161/cc.8.22.10165. [DOI] [PubMed] [Google Scholar]
- 67.Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010;2:344–52. doi: 10.18632/aging.100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jackson TR, Salmina K, Huna A, Inashkina I, Jankevics E, Riekstina U, Kalnina Z, Ivanov A, Townsend PA, Cragg MS, et al. DNA damage causes TP53-dependent coupling of self-renewal and senescence pathways in embryonal carcinoma cells. Cell Cycle. 2013;12:430–41. doi: 10.4161/cc.23285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tavana O, Benjamin CL, Puebla-Osorio N, Sang M, Ullrich SE, Ananthaswamy HN, Zhu C. Absence of p53-dependent apoptosis leads to UV radiation hypersensitivity, enhanced immunosuppression and cellular senescence. Cell Cycle. 2010;9:3348–56. doi: 10.4161/cc.9.16.12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Longo VD. Ras: the other pro-aging pathway. Sci Aging Knowledge Environ. 2004;2004:pe36. doi: 10.1126/sageke.2004.39.pe36. [DOI] [PubMed] [Google Scholar]
- 71.Hasty P. Is NHEJ a tumor suppressor or an aging suppressor? Cell Cycle. 2008;7:1139–45. doi: 10.4161/cc.7.9.5807. [DOI] [PubMed] [Google Scholar]
- 72.Blagosklonny MV. Tumor suppression by p53 without apoptosis and senescence: conundrum or rapalog-like gerosuppression? Aging (Albany NY) 2012;4:450–5. doi: 10.18632/aging.100475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Blagosklonny MV. TOR-driven aging: speeding car without brakes. Cell Cycle. 2009;8:4055–9. doi: 10.4161/cc.8.24.10310. [DOI] [PubMed] [Google Scholar]
- 74.Blagosklonny MV. Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program. Cell Cycle. 2010;9:3171–6. doi: 10.4161/cc.9.16.13120. [DOI] [PubMed] [Google Scholar]
- 75.Blagosklonny MV. Rapamycin and quasi-programmed aging: four years later. Cell Cycle. 2010;9:1859–62. doi: 10.4161/cc.9.10.11872. [DOI] [PubMed] [Google Scholar]
- 76.Blagosklonny MV. Increasing healthy lifespan by suppressing aging in our lifetime: preliminary proposal. Cell Cycle. 2010;9:4788–94. doi: 10.4161/cc.9.24.14360. [DOI] [PubMed] [Google Scholar]
- 77.Blagosklonny MV. Calorie restriction: decelerating mTOR-driven aging from cells to organisms (including humans) Cell Cycle. 2010;9:683–8. doi: 10.4161/cc.9.4.10766. [DOI] [PubMed] [Google Scholar]
- 78.Blagosklonny MV. Hormesis does not make sense except in the light of TOR-driven aging. Aging (Albany NY) 2011;3:1051–62. doi: 10.18632/aging.100411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blagosklonny MV. Prospective treatment of age-related diseases by slowing down aging. Am J Pathol. 2012;181:1142–6. doi: 10.1016/j.ajpath.2012.06.024. [DOI] [PubMed] [Google Scholar]
- 80.Blagosklonny MV. Once again on rapamycin-induced insulin resistance and longevity: despite of or owing to. Aging (Albany NY) 2012;4:350–8. doi: 10.18632/aging.100461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Blagosklonny MV. Answering the ultimate question “what is the proximal cause of aging?”. Aging (Albany NY) 2012;4:861–77. doi: 10.18632/aging.100525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bauer JH, Helfand SL. Sir2 and longevity: the p53 connection. Cell Cycle. 2009;8:1818–22. doi: 10.4161/cc.8.12.9010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Harikumar KB, Aggarwal BB. Resveratrol: a multitargeted agent for age-associated chronic diseases. Cell Cycle. 2008;7:1020–35. doi: 10.4161/cc.7.8.5740. [DOI] [PubMed] [Google Scholar]
- 84.Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature. 2007;448:767–74. doi: 10.1038/nature05985. [DOI] [PubMed] [Google Scholar]
- 85.Schoppy DW, Ruzankina Y, Brown EJ. Removing all obstacles: a critical role for p53 in promoting tissue renewal. Cell Cycle. 2010;9:1313–9. doi: 10.4161/cc.9.7.11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu Y, Elf SE, Asai T, Miyata Y, Liu Y, Sashida G, Huang G, Di Giandomenico S, Koff A, Nimer SD. The p53 tumor suppressor protein is a critical regulator of hematopoietic stem cell behavior. Cell Cycle. 2009;8:3120–4. doi: 10.4161/cc.8.19.9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Richardson RB. p53 mutations associated with aging-related rise in cancer incidence rates. Cell Cycle. 2013;12:2468–78. doi: 10.4161/cc.25494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Herrera-Merchan A, Cerrato C, Luengo G, Dominguez O, Piris MA, Serrano M, Gonzalez S. miR-33-mediated downregulation of p53 controls hematopoietic stem cell self-renewal. Cell Cycle. 2010;9:3297–3305. doi: 10.4161/cc.9.16.12598. [DOI] [PubMed] [Google Scholar]
- 89.Gems D, de la Guardia Y. Alternative Perspectives on Aging in Caenorhabditis elegans: Reactive Oxygen Species or Hyperfunction? Antioxid Redox Signal. 2013;19:321–9. doi: 10.1089/ars.2012.4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gems D, Partridge L. Genetics of longevity in model organisms: debates and paradigm shifts. Annu Rev Physiol. 2013;75:621–44. doi: 10.1146/annurev-physiol-030212-183712. [DOI] [PubMed] [Google Scholar]
- 91.Kaeberlein M, Kapahi P. The hypoxic response and aging. Cell Cycle. 2009;8:2324. doi: 10.4161/cc.8.15.9126. [DOI] [PubMed] [Google Scholar]
- 92.Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–45. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tower J. The genetic architecture of aging: sexual antagonistic pleiotropy of p53 and foxo. Cell Cycle. 2010;9:3840–1. doi: 10.4161/cc.9.19.13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Benson EK, Zhao B, Sassoon DA, Lee SW, Aaronson SA. Effects of p21 deletion in mouse models of premature aging. Cell Cycle. 2009;8:2002–4. doi: 10.4161/cc.8.13.8997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bojesen SE, Nordestgaard BG. The common germline Arg72Pro polymorphism of p53 and increased longevity in humans. Cell Cycle. 2008;7:158–63. doi: 10.4161/cc.7.2.5249. [DOI] [PubMed] [Google Scholar]
- 96.Roemer K. Are the conspicuous interdependences of fecundity, longevity and cognitive abilities in humans caused in part by p53? Cell Cycle. 2010;9:3438–41. doi: 10.4161/cc.9.17.13001. [DOI] [PubMed] [Google Scholar]
- 97.Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–7. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tang DG, Tokumoto YM, Apperly JA, Lloyd AC, Raff MC. Lack of replicative senescence in cultured rat oligodendrocyte precursor cells. Science. 2001;291:868–71. doi: 10.1126/science.1056780. [DOI] [PubMed] [Google Scholar]
- 99.Zhang H, Hoff H, Marinucci T, Cristofalo VJ, Sell C. Mitogen-independent phosphorylation of S6K1 and decreased ribosomal S6 phosphorylation in senescent human fibroblasts. Exp Cell Res. 2000;259:284–92. doi: 10.1006/excr.2000.4965. [DOI] [PubMed] [Google Scholar]
- 100.Kolesnichenko M, Hong L, Liao R, Vogt PK, Sun P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle. 2012;11:2391–401. doi: 10.4161/cc.20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pospelova TV, Leontieva OV, Bykova TV, Zubova SG, Pospelov VA, Blagosklonny MV. Suppression of replicative senescence by rapamycin in rodent embryonic cells. Cell Cycle. 2012;11:2402–7. doi: 10.4161/cc.20882. [DOI] [PubMed] [Google Scholar]
- 102.Bazarov AV, Hines WC, Mukhopadhyay R, Beliveau A, Melodyev S, Zaslavsky Y, Yaswen P. Telomerase activation by c-Myc in human mammary epithelial cells requires additional genomic changes. Cell Cycle. 2009;8:3373–8. doi: 10.4161/cc.8.20.9856. [DOI] [PubMed] [Google Scholar]
- 103.Harari Y, Romano GH, Ungar L, Kupiec M. Nature vs nurture: Interplay between the genetic control of telomere length and environmental factors. Cell Cycle. 2013;12:3465–70. doi: 10.4161/cc.26625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kaminker PG, Kim SH, Desprez PY, Campisi J. A novel form of the telomere-associated protein TIN2 localizes to the nuclear matrix. Cell Cycle. 2009;8:931–9. doi: 10.4161/cc.8.6.7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Narita M, Young AR, Narita M. Autophagy facilitates oncogene-induced senescence. Autophagy. 2009;5:1046–7. doi: 10.4161/auto.5.7.9444. [DOI] [PubMed] [Google Scholar]
- 106.Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira M, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332:966–70. doi: 10.1126/science.1205407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med. 2011;3:89ra58. doi: 10.1126/scitranslmed.3002346. [DOI] [PubMed] [Google Scholar]
- 109.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 110.Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, Rubinsztein DC. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol. 2006;76:89–101. doi: 10.1016/S0070-2153(06)76003-3. [DOI] [PubMed] [Google Scholar]
- 111.Chang YY, Juhász G, Goraksha-Hicks P, Arsham AM, Mallin DR, Muller LK, Neufeld TP. Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochem Soc Trans. 2009;37:232–6. doi: 10.1042/BST0370232. [DOI] [PubMed] [Google Scholar]
- 112.Blum B, Benvenisty N. The tumorigenicity of diploid and aneuploid human pluripotent stem cells. Cell Cycle. 2009;8:3822–30. doi: 10.4161/cc.8.23.10067. [DOI] [PubMed] [Google Scholar]
- 113.Li Y, Shen Z, Shelat H, Geng YJ. Reprogramming somatic cells to pluripotency: A fresh look at Yamanaka’s model. Cell Cycle. 2013;12 doi: 10.4161/cc.26952. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Corominas-Faja B, Cufí S, Oliveras-Ferraros C, Cuyàs E, López-Bonet E, Lupu R, Alarcón T, Vellon L, Iglesias JM, Leis O, et al. Nuclear reprogramming of luminal-like breast cancer cells generates Sox2-overexpressing cancer stem-like cellular states harboring transcriptional activation of the mTOR pathway. Cell Cycle. 2013;12:3109–24. doi: 10.4161/cc.26173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Thompson LH, Whiston RA, Rakhimov Y, Taccioli C, Liu CG, Croce C, Metcalfe SM. A LIF/Nanog axis is revealed in T lymphocytes that lack MARCH-7, a RINGv E3 ligase that regulates the LIF-receptor. Cell Cycle. 2010;9:4213–21. doi: 10.4161/cc.9.20.13543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–7. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 117.Bernardes de Jesus B, Vera E, Schneeberger K, Tejera AM, Ayuso E, Bosch F, Blasco MA. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med. 2012;4:691–704. doi: 10.1002/emmm.201200245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–6. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 119.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 120.Lluis F, Cosma MP. Somatic cell reprogramming control: signaling pathway modulation versus transcription factor activities. Cell Cycle. 2009;8:1138–44. doi: 10.4161/cc.8.8.8206. [DOI] [PubMed] [Google Scholar]
- 121.Menendez S, Camus S, Izpisua Belmonte JC. p53: guardian of reprogramming. Cell Cycle. 2010;9:3887–91. doi: 10.4161/cc.9.19.13301. [DOI] [PubMed] [Google Scholar]
- 122.Marzi I, Cipolleschi MG, D’Amico M, Stivarou T, Rovida E, Vinci MC, Pandolfi S, Dello Sbarba P, Stecca B, Olivotto M. The involvement of a Nanog, Klf4 and c-Myc transcriptional circuitry in the intertwining between neoplastic progression and reprogramming. Cell Cycle. 2013;12:353–64. doi: 10.4161/cc.23200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, Khalil A, Rheinwald JG, Hochedlinger K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009;460:1145–8. doi: 10.1038/nature08285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Leon J, Ferrandiz N, Acosta JC, Delgado MD. Inhibition of cell differentiation: a critical mechanism for MYC-mediated carcinogenesis? Cell Cycle. 2009;8:1148–57. doi: 10.4161/cc.8.8.8126. [DOI] [PubMed] [Google Scholar]
- 125.Lagarkova MA, Shutova MV, Bogomazova AN, Vassina EM, Glazov EA, Zhang P, Rizvanov AA, Chestkov IV, Kiselev SL. Induction of pluripotency in human endothelial cells resets epigenetic profile on genome scale. Cell Cycle. 2010;9:937–46. doi: 10.4161/cc.9.5.10869. [DOI] [PubMed] [Google Scholar]
- 126.Roberts RM, Telugu BP, Ezashi T. Induced pluripotent stem cells from swine (Sus scrofa): why they may prove to be important. Cell Cycle. 2009;8:3078–81. doi: 10.4161/cc.8.19.9589. [DOI] [PubMed] [Google Scholar]
- 127.Marión RM, Strati K, Li H, Murga M, Blanco R, Ortega S, Fernandez-Capetillo O, Serrano M, Blasco MA. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149–53. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huang D, Wei Z, Lu W. Genome organization by Klf4 regulates transcription in pluripotent stem cells. Cell Cycle. 2013;12:3351–2. doi: 10.4161/cc.26577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ben-David U, Benvenisty N, Mayshar Y. Genetic instability in human induced pluripotent stem cells: classification of causes and possible safeguards. Cell Cycle. 2010;9:4603–4. doi: 10.4161/cc.9.23.14094. [DOI] [PubMed] [Google Scholar]
- 130.Lako M, Neganova I, Armstrong L. G1 to S transition and pluripotency: two sides of the same coin? Cell Cycle. 2009;8:1105–11. doi: 10.4161/cc.8.8.8237. [DOI] [PubMed] [Google Scholar]
- 131.Edel MJ, Izpisua Belmonte JC. The cell cycle and pluripotency: Is there a direct link? Cell Cycle. 2010;9:2694–5. doi: 10.4161/cc.9.14.12456. [DOI] [PubMed] [Google Scholar]
- 132.Brown KE, Bagci H, Soza-Ried J, Fisher AG. Atypical heterochromatin organization and replication are rapidly acquired by somatic cells following fusion-mediated reprogramming by mouse ESCs. Cell Cycle. 2013;12:3253–61. doi: 10.4161/cc.26223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Soufi A, Zaret KS. Understanding impediments to cellular conversion to pluripotency by assessing the earliest events in ectopic transcription factor binding to the genome. Cell Cycle. 2013;12:1487–91. doi: 10.4161/cc.24663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Salomonis N, Conklin BR. Stem cell pluripotency: alternative modes of transcription regulation. Cell Cycle. 2010;9:3133–4. doi: 10.4161/cc.9.16.12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bhave VS, Paranjpe S, Bowen WC, Donthamsetty S, Bell AW, Khillan JS, Michalopoulos GK. Genes inducing iPS phenotype play a role in hepatocyte survival and proliferation in vitro and liver regeneration in vivo. Hepatology. 2011;54:1360–70. doi: 10.1002/hep.24507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bedelbaeva K, Snyder A, Gourevitch D, Clark L, Zhang XM, Leferovich J, Cheverud JM, Lieberman P, Heber-Katz E. Lack of p21 expression links cell cycle control and appendage regeneration in mice. Proc Natl Acad Sci U S A. 2010;107:5845–50. doi: 10.1073/pnas.1000830107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chen T, Shen L, Yu J, Wan H, Guo A, Chen J, Long Y, Zhao J, Pei G. Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell. 2011;10:908–11. doi: 10.1111/j.1474-9726.2011.00722.x. [DOI] [PubMed] [Google Scholar]
- 138.Wu JJ, Quijano C, Wang J, Finkel T. Metabolism meets autophagy. Cell Cycle. 2010;9:4780–1. doi: 10.4161/cc.9.24.14273. [DOI] [PubMed] [Google Scholar]
- 139.Ozkinay C, Mitelman F. A simple trypsin-Giemsa technique producing simultaneous G- and C-banding in human chromosomes. Hereditas. 1979;90:1–4. doi: 10.1111/j.1601-5223.1979.tb01287.x. [DOI] [PubMed] [Google Scholar]
- 140.Howell WM, Black DA. Controlled silver-staining of nucleolus organizer regions with a protective colloidal developer: a 1-step method. Experientia. 1980;36:1014–5. doi: 10.1007/BF01953855. [DOI] [PubMed] [Google Scholar]
- 141.Lapasset L, Milhavet O, Prieur A, Besnard E, Babled A, Aït-Hamou N, Leschik J, Pellestor F, Ramirez JM, De Vos J, et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011;25:2248–53. doi: 10.1101/gad.173922.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.