

Abstract
Irradiation of vinyl and aryl azides with visible light in the presence of Ru photocatalysts results in the formation of reactive nitrenes, which can undergo a variety of C–N bond-forming reactions. The ability to use low-energy visible light instead of UV in the photochemical activation of azides avoids competitive photodecomposition processes that have long been a significant limitation on the synthetic utility of these reactions.
Keywords: azides, nitrenes, nitrogen heterocycles, photocatalysis, visible light
The use of visible light activated transition metal catalysts in synthesis is presently receiving increased attention.1 These methods are attractive both because visible light is easier to handle than high-energy UV light and because complex organic molecules are more stable towards photodecomposition under lower-energy visible light irradiation. Almost all of the reactions reported to date have involved activation by photoinduced electron transfer processes. Recently, we reported that the same class of transition metal photocatalysts can also promote cycloaddition reactions via a complementary energy transfer process.2,3 The ability to generate electronically excited organic molecules using visible light suggested to us that a much broader diversity of reactions traditionally conducted with high-energy UV light might be made more operationally accessible and more tolerant of complex functionality using transition metal photocatalysis. As a demonstration of this concept, we report herein the facile, room-temperature generation and rearrangement of vinyl nitrenes by visible light photocatalytic activation of azides.
Nitrenes are involved in a wide range of carbon–nitrogen bond-forming reactions, including several that produce heterocycles such as aziridines,4 indoles,5 and pyrroles.6 The photochemical generation of nitrenes from azides is an attractive strategy that liberates only dinitrogen as a stoichiometric byproduct; 7 however, the photolysis of azides with UV light generally results in poor functional group tolerance and competitive photodecomposition processes that can diminish the yield of these reactions.8 Liu has recently reported visible light induced photoreduction of aryl azides using Ru(bpy)32+ as a photocatalyst (Scheme 1, eq 1).9 The key intermediate in this reaction, however, was proposed to be a nitrene radical anion generated by one-electron reduction of the azide, and these intermediates do not exhibit the characteristic reactivity of neutral nitrenes.10 We wondered, therefore, if we might be able to access the powerful C–N bond-forming reactivity of nitrenes using visible light activation by employing transition metal photocatalysts as triplet sensitizers of azide decomposition.
Scheme 1.
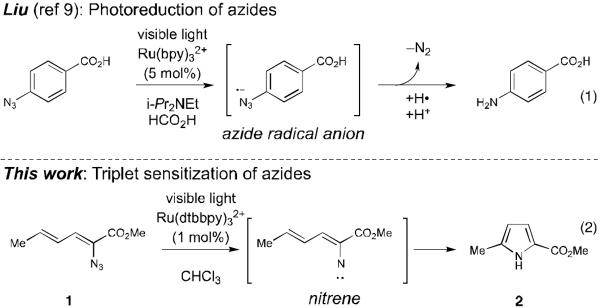
Photoredox vs energy transfer photocatalysis for activation of organic azides.
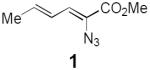
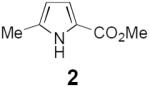
As a test of this hypothesis, we investigated the photocatalytic transformation of dienyl azides into pyrroles (Scheme 1, eq 2). Driver has reported that this overall transformation can be achieved by Lewis acid catalysis,11 although the method is limited to α-azidoesters and does not tolerate strongly Lewis basic substituents. Driver has proposed a mechanism involving chelate-controlled Schmidt-like cyclization rather than a discrete nitrene intermediate. This transformation thus offered an interesting opportunity to test our mechanistically distinct ideas about photocatalytic activation of azides. Dienyl azide 1 is insufficiently electron-deficient to be easily reduced by ruthenium photocatalysts; the reduction potentials of Ru*(bpy)32+ and 1, respectively, are −0.89 V12 and −1.81 V13 vs SCE. On the other hand, we calculated that the first electronically excited triplet state of 1 (ET = 45.4 kcal/mol)14 was energetically well poised for sensitization by the long-lived Ru*(bpy)32+ triplet state (ET = 46 kcal/mol).
Indeed, irradiation of 1 with blue light in the presence of several ruthenium polypyridyl photocatalysts (3a–e) resulted in formation of pyrrole 2 along with a non-isolable compound we assigned as azirine 4 (Table 1). Despite the fact that the excited state reduction potentials of the photocatalysts assayed span almost a volt (−0.05 V and −0.94 V vs SCE for 3b and 3c, respectively),15 similar levels of conversion were observed in each case (entries 1–5). This observation would be surprising for a mechanism involving photoinduced reduction of 1. On the other hand, it is consistent with a triplet energy transfer process, as the excited state triplet energies of these five photocatalysts are quite similar (45–47 kcal/mol).15 We also observed little dependence on solvent polarity; reactions in chloroform (ε = 4.81) and acetonitrile (ε = 36.6) gave similar results (entries 5–9). These data, too, support an energy transfer mechanism, as charged radical ion intermediates would be strongly destabilized in nonpolar media. We observed a modest increase in conversion upon decreasing the concentration of the reaction (entry 10), and a 5 h reaction time provided complete conversion to pyrrole 2 (entry 11), which confirmed that 4 is fully converted to 2 over the course of the reaction. Lowering the catalyst loading to 1 mol% had little effect on the yield of the reaction (entry 12), and under these optimized conditions, irradiation of dienyl azide 1 cleanly produced pyrrole 2 in 91% yield after 5 h of irradiation.
Table 1.
Initial investigations on triplet sensitization of dienyl azide 1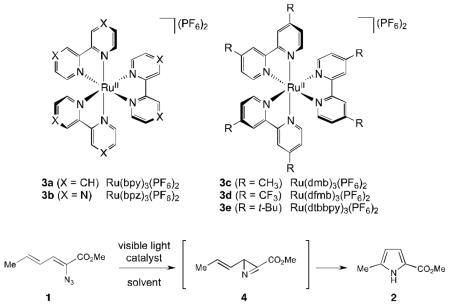
| entrya | catalyst (loading) | solvent (concentration) | time | yield 2 (yield 4)b |
|---|---|---|---|---|
| 1 | 3a (2.5 mol%) | MeCN (0.4 M) | 1.5 h | 12% (31%) |
| 2 | 3b (2.5 mol%) | MeCN (0.4 M) | 1.5 h | 9% (22%) |
| 3 | 3c (2.5 mol%) | MeCN (0.4 M) | 1.5 h | 21% (48%) |
| 4 | 3d (2.5 mol%) | MeCN (0.4 M) | 1.5 h | 17% (28%) |
| 5 | 3e (2.5 mol%) | MeCN (0.4 M) | 1.5 h | 25% (39%) |
| 6 | 3e (2.5 mol%) | DMSO (0.4 M) | 1.5 h | 22% (48%) |
| 7 | 3e (2.5 mol%) | acetone (0.4 M) | 1.5 h | 21% (41%) |
| 8 | 3e (2.5 mol%) | CH2CI2 (0.4M) | 1.5 h | 28% (32%) |
| 9 | 3e (2.5 mol%) | CHCI3 (0.4M) | 1.5 h | 30% (27%) |
| 10 | 3e (2.5 mol%) | CHCI3 (0.1 M) | 1.5 h | 34% (24%) |
| 11 | 3e (2.5 mol%) | CHCI3 (0.1 M) | 5 h | 94% (0%) |
| 12 | 3e (1 mol%) | CHCI3 (0.1 M) | 5 h | 91% (0%) |
| 13 | none | CHCI3 (0.1 M) | 5 h | 0% (0%) |
| 14c | 3e (1 mol%) | CHCI3 (0.1 M) | 5 h | 0% (0%) |
| 15d | 3e (1 mol%) | CHCI3 (0.1 M) | 5 h | 73% (12%) |
| 16e | none | MeCN (0.1 M) | 5 h | 49% (0%) |
Reactions conducted using 0.25 mmol of 1 under air-free conditions with irradiation from a blue LED strip unless otherwise noted.
Yields determined by 1H NMR analysis using an internal standard.
Reaction conducted in the dark.
Reaction irradiated with a 23 W compact fluorescent lightbulb.
Reaction irradiated at 254 nm in a Rayonet reactor.
Control experiments demonstrated that both the photocatalyst and light are required (entries 13 and 14). On the other hand, the reaction can be conveniently conducted using a variety of visible light sources; although blue LED strips provided somewhat faster rates, reactions irradiated with household light bulbs also provide good yields (entry 15). Direct photoreactions of 2 using 254 nm UV light suffered from significant photodecomposition, resulting in much lower yields and poor overall mass balance (entry 16), verifying our contention that the ability to use visible light in these reactions indeed provides a significant synthetic advantage.
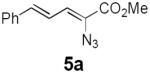
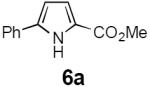
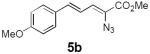
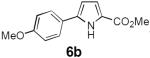
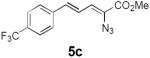
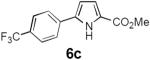
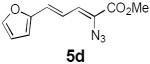
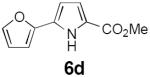
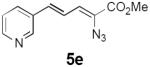
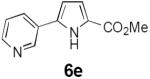
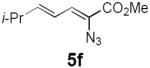
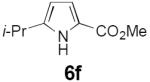
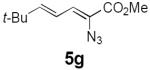
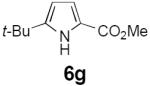
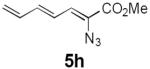
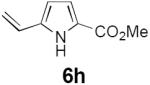
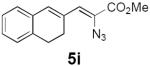
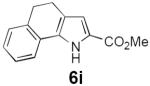
Table 2 summarizes our investigations into the scope of this transformation. Both electron-rich and electron-poor aryl substituents can be present on the dienyl unit, and the corresponding pyrroles can be isolated in nearly quantitative yields (entries 1–5). Although heterocyclic substituents are not well tolerated in Driver's Lewis acid-catalyzed process, these substrates were smoothly transformed to pyrroles 6d and 6e under photocatalytic conditions. As expected, aliphatic dienyl azides were compatible with the reaction conditions (entries 6–8). Vinyl substitution was also tolerated, affording 6h containing a versatile handle for further synthetic elaboration (entry 9). Tricyclic fused heterocycles were accessible from γ-substituted azidoacrylates (entry 10).
Table 2.
Pyrrole synthesis substrate scope study
| entry | substrate | product | time | yielda |
|---|---|---|---|---|
| 1 |
|
|
3 h | 99% |
| 2 |
|
|
4 h | 99% |
| 3 |
|
|
2.5 h | 95% |
| 4 |
|
|
4 h | 98% |
| 5 |
|
|
3 h | 86% |
| 6 |
|
|
8 h | 93% |
| 7 |
|
|
11 h | 89% |
| 8 |
|
|
14 h | 86% |
| 9 |
|
|
2 h | 89% |
| 10 |
|
|
4 h | 96% |
| 11 |
|
|
3 h | 92% |
| 12 |
|
|
36 h | 47% |
| 13b | 12 h | 70% | ||
| 14 |
|
|
20 h | 75% |
| 15 |
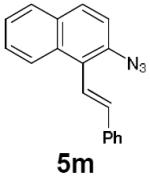
|
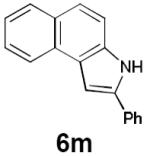
|
6 h | 93% |
Data represent the averaged isolated yields from two reproducible experiments
Conducted using [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) (1 mol%) as photocatalyst.
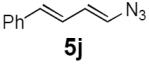
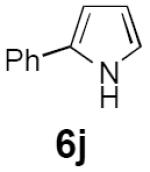
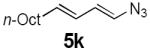
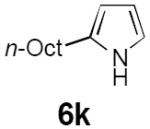
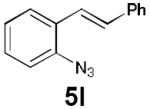
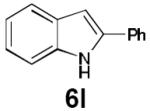
Dienyl azides lacking an α-ester substituent are also readily transformed into pyrroles (entries 11–13). We found that this class of substrates does not react under Lewis acid catalyzed conditions, even after prolonged reaction times, presumably due to the lack of the chelating moiety involved in the Schmidt-type activation of the azide. Conversely, 1-azido-4-phenylbutadiene (5j) undergoes cyclization to afford pyrrole 6j in excellent yield after 4 h of irradiation. Aliphatic dienyl azide 5k also underwent cyclization, although the conversion to pyrrole was modest after extended irradiation. We attribute the slow rate of reaction to the higher triplet energy of the less conjugated dienyl azide, which we computed to be 52 kcal/mol.14 This value is somewhat higher than the triplet excited state of Ru*(dtbbpy)32+ (ET = 47 kcal/mol), which would be consistent with inefficient triplet energy transfer. However, the [Ir(dF(CF3)ppy)2(dtbbpy)]+ complex we had found to be optimal for visible light triplet sensitization of styrenes has a significantly higher-energy triplet state (ET= 61 kcal/mol), 16 and using this photocatalyst, the cyclized pyrrole product is formed in 70% yield after 12 h. Furthermore, sensitization of aryl azides proceeded smoothly to afford indoles (entries 14–15). This is noteworthy because direct photolysis of aryl azides often produces complex mixtures of products from multiple reaction manifolds.17 Indeed, direct irradiation of 5m at 350 nm resulted in complete consumption of the starting azide within 1 h but produced only 51% yield of indole 6m as a part of a complex, inseparable mixture.
We were intrigued by the appearance of transitory intermediate 4 at partial conversions of dienyl azide 1. We assigned this intermediate as azirine 4 based upon analogy to the photochemical production of azirines by UV irradiation of β-styrenyl azides.18 The identity of azirine 4 was confirmed by adding 1 equiv of cyclopentadiene to a photosensitized reaction of 1, which produced the hetero-Diels–Alder cycloadduct expected from interception of the highly reactive C=N moiety in 71% isolated yield and as a single diastereomer. Notably, this trapping experiment, too, is markedly more efficient under photocatalytic conditions; the same experiment conducted by direct UV photolysis at 254 nm produced only 40% of the trapped hetero-Diels–Alder cycloadduct along with numerous inseparable decomposition products.
The azirine can be isolated in cases where subsequent ring expansion to the five-membered heterocycle is slow. For example, irradiation of the aryl-substituted vinyl azide 8 generates azirine 9 in 90% yield. Azirines are highly reactive intermediates that can be used in the synthesis of a variety of heterocyclic compounds.19 To highlight the utility of azirines generated under these conditions, we converted 9 to a variety of substituted aziridines by addition of nucleophiles and radicals to the C=N bond (10–12). Hetero-Diels–Alder cycloaddition with cyclopentadiene to afford cycloadduct 13 is also rapid and high-yielding. Finally, we found that 9 can be easily converted to highly substituted pyrrole 14 by reaction with a stabilized phosphonium ylide.
Scheme 3 outlines our working mechanism for the photocatalytic generation of pyrrole 2. Irradiation of Ru(dtbbpy)32+ with visible light produces its long-lived excited state; exergonic energy transfer to dienyl azide 1 produces triplet 1*, which expels dinitrogen to produce a nitrene intermediate. Ring-closure affords azirine 4, which undergoes slow but high-yielding rearrangement to pyrrole 2.
Scheme 3.

Energy transfer mechanism from [Ru] or [Ir] sensitizer to azidoacrylates
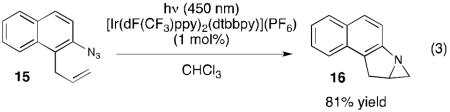
One of the most intriguing implications of these investigations is the possibility that visible light photocatalysis might provide convenient access to the diverse reactivity of free nitrenes under exceptionally mild conditions. As a preliminary demonstration of the generality of this approach, we examined the photocatalytic activation of azidonaphthalene 15 (eq 3). We calculated the triplet excited state energy of 15 to be 62.0 kcal/mol,14 which suggested that it could be efficiently sensitized by [Ir(dF(CF3)ppy)2(dtbbpy)]+. Indeed, visible light irradiation of 15 for 5 h in the presence of this Ir photocatalyst resulted in the clean production of aziridine 16, which presumably arises from intramolecular reaction of the photogenerated nitrene with the pendant allyl substituent.
In conclusion, we have demonstrated that photocatalytic activation of aryl and vinyl azides with visible light provides a convenient method to access the chemistry of nitrenes. The ability to use low-energy visible light in this application minimizes complications arising from photodecomposition and thus provides uniformly higher yields than direct photoexcitation with UV light. Given the importance of nitrene intermediates in a broad range of methods for the construction of heterocycles and C–N bonds, we anticipate that this approach could be useful in synthesis, drug discovery, and all of the various fields in which such structures are important.
Supplementary Material
Scheme 2.

Manipulation of the azirine intermediate. (a) [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) (1 mol %), CHCl3 (0.1 M), blue LEDs, rt, 8 h, 90% (b) Bu4NBH4, CH2Cl2, −78 °C, 15 min, 60% (c) NaOMe, MeOH, rt, 1 h, 93% (d) BEt3, EtI, CH2Cl2/hexanes, −40 °C, 30 min, 71% (e) Cp (5 equiv.), THF, rt, 1 h, 94% (f) 1-phenyl-2-(triphenylphosphanylidene)-ethanone (1 equiv.), CH2Cl2, rt, 24 h, 58%.
Acknowledgments
[**] This research was conducted using funds from the NIH (GM095666) and the Sloan Foundation. The NMR facilities at UW–Madison are funded by the NSF (CHE-1048642).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
REFERENCES
- 1.Narayanam JMR, Tucker JW, Stephenson CRJ. J. Am. Chem. Soc. 2009;131:8756–8757. doi: 10.1021/ja9033582. [DOI] [PubMed] [Google Scholar]; (b) Nagib DA, MacMillan DWC. Nature. 2011;480:224–228. doi: 10.1038/nature10647. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) DiRocco DA, Rovis T. J. Am. Chem. Soc. 2012;134:8094–8097. doi: 10.1021/ja3030164. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. J. Am. Chem. Soc. 2011;133:18566–18569. doi: 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Maity S, Zhu M, Shinabery RS, Zheng N. Angew. Chem. 2012;124:226–230. doi: 10.1002/anie.201106162. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012;51:222–226. [Google Scholar]
- 2.Lu Z, Yoon TP. Angew. Chem. Int. Ed. 2012;51:10329–10332. doi: 10.1002/anie.201204835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiao has also reported a photocatalytic [2+2] oxindole dimerization that they have suggested to occur via energy transfer: Zou Y-Q, Duan S-W, Meng X-G, Hu H-Q, Gao S, Chen JR, Xiao WJ. Tetrahedron. 2012;68:6914–6918.
- 4.(a) Nicholas PP. J. Org. Chem. 1975;40:3396–3398. [Google Scholar]; (b) Li Z, Quan RW, Jacobsen EN. J. Am. Chem. Soc. 1995;117:5889–5890. [Google Scholar]; (c) Bergmeier SC, Stanchina DM. J. Org. Chem. 1999;64:2852–2859. doi: 10.1021/jo9823893. [DOI] [PubMed] [Google Scholar]; (d) Muller P, Fruit C. Chem. Rev. 2003;103:2905–2919. doi: 10.1021/cr020043t. [DOI] [PubMed] [Google Scholar]
- 5.(a) Hemetsberger H, Knittel D, Weidmann H. Monatsh. Chem. 1970;101:161–165. [Google Scholar]; (b) Knittel D. Synthesis. 1985:186–188. [Google Scholar]; (c) Henn L, Hickey DMB, Moody CJ, Rees CW. J. Chem. Soc. Perkin Trans. 1984;1:2189–2196. [Google Scholar]; (d) Tichenor MS, Trzupek JD, Kastrinsky DB, Shiga F, Hwang I, Boger DL. J. Am. Chem. Soc. 2006;128:15683–15696. doi: 10.1021/ja064228j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Stokes BJ, Dong H, Leslie BE, Pumphrey AL, Driver TG. J. Am. Chem. Soc. 2007;129:7500–7501. doi: 10.1021/ja072219k. [DOI] [PubMed] [Google Scholar]
- 6.(a) Hemetsberger H, Spira I, Schoenfelder W. J. Chem. Res. Synop. 1977:247–249. [Google Scholar]; (b) Moody CJ, Warrellow GJ. J. Chem. Soc. Perkin Trans. 1990;1:2929–2936. [Google Scholar]; (c) O'Brien AG, Levesque F, Seeberger PH. Chem. Commun. 2011;47:2688–2690. doi: 10.1039/c0cc04481d. [DOI] [PubMed] [Google Scholar]
- 7.Scriven EFV, editor. Azides and Nitrenes, Reactivity and Utility. Academic; New York: 1984. pp. 205–246. [Google Scholar]
- 8.(a) Lwowski W, Mattingly TW. J. Am. Chem. Soc. 1964;87:1947–1958. [Google Scholar]; (b) Lewis FD, Saunders WH. J. Am. Chem. Soc. 1968;90:7033–7038. [Google Scholar]; (c) Marcinek A, Leyva E, Whitt D, Platz MS. J. Am. Chem. Soc. 1993;115:8609–8612. [Google Scholar]; (d) Borden WT, Gritsan NP, Hadad CM, Karney WL, Kemnitz CR, Platz MS. Acc. Chem. Res. 2000;33:765–771. doi: 10.1021/ar990030a. [DOI] [PubMed] [Google Scholar]; (e) Singh PND, Mandel SM, Sankaranarayanan J, Muthukrishnan S, Chang M, Robinson RM, Lahti PM, Ault BS, Gudmundsdόttir AD. J. Am. Chem. Soc. 2007;129:16263–16272. doi: 10.1021/ja077523s. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Kamlet AS, Steinman JB, Liu DR. Nature Chem. 2011;3:146–153. doi: 10.1038/nchem.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) McDonald RN, Chowdhury AK. J. Am. Chem. Soc. 1980;102:5119–5120. [Google Scholar]; (b) McDonald RN, Zhu Y, Schuster GB. J. Am. Chem. Soc. 1990;112:8583–8585. [Google Scholar]; (c) Wijeratne NR, Wenthold PG. J. Org. Chem. 2007;72:9518–9522. doi: 10.1021/jo701407n. [DOI] [PubMed] [Google Scholar]; (d) Adhikary A, Khanduri D, Pottiboyina V, Rice CT, Sevilla M. J. Phys. Chem. B. 2010;114:9289–9299. doi: 10.1021/jp103403p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong H, Shen M, Redford J, Stokes BJ, Pumphrey AL, Driver TG. Org. Lett. 2007;9:5191–5194. doi: 10.1021/ol702262f. [DOI] [PubMed] [Google Scholar]
- 12.Kalyanasundaram K. Coord. Chem. Rev. 1982;46:159–244. [Google Scholar]
- 13.Cyclic voltammetry was performed using a three-electrode setup: non-aqueous Ag/AgNO3 (n-Bu4NPF6 (0.1 M), Ag/AgNO3 (0.01 M)) reference electrode, Pt wire counter electrode, and glassy carbon working electrode. All experiments were carried out in deoxygenated CH3CN and referenced to SCE via Fc/Fc+ as the internal standard.
- 14.Theoretical calculations on dienyl azides 1 and 5k and azidonaphthalene 15 were performed using the Gaussian 09 software package. Geometrical optimization of the azide was carried out at the B3LYP/6-31G(d) level of theory prior to performing excited state calculations at the same level of theory.
- 15.(a) Bernhard S, Barron JA, Houston PL, Abruna HD, Ruglovsky JL, Gao X, Malliaras GG. J. Am. Chem. Soc. 2002;124:13624–13628. doi: 10.1021/ja0270524. [DOI] [PubMed] [Google Scholar]; (b) Rillema DP, Allen G, Meyer TJ, Conrad D. Inorg. Chem. 1983;22:1617–1622. [Google Scholar]
- 16.Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Jr., Malliaras GG, Bernhard S. Chem. Mater. 2005;17:5712–5719. [Google Scholar]
- 17.Burdzinski G, Hackett JC, Wang J, Gustafson TL, Hadad CM, Platz MC. J. Am. Chem. Soc. 2006;128:13402–13411. doi: 10.1021/ja061520i. [DOI] [PubMed] [Google Scholar]
- 18.(a) L'abbé G, Mathys G. J. Org. Chem. 1974;39:1778–1779. [Google Scholar]; (b) Isomura K, Ayabe G-I, Hatano S, Taniguchi H. J. Chem. Soc. Chem. Commun. 1980:1252–1253. [Google Scholar]
- 19.Palacios F, de Retana AMO, de Marigorta EM, de los Santos JM. Eur. J. Org. Chem. 2001:2401–2414. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.