

Abstract
Heat shock factor 1 (HSF1) protects neurons from death caused by the accumulation of misfolded proteins. It is believed that this protective effect is mediated by the transcriptional stimulation of genes encoding heat shock proteins (HSPs), a family of chaperones that refold or degrade misfolded proteins. Whether HSF1 is protective when neuronal death is not caused by protein misfolding has not been studied. Here, we report that HSF1 expression is necessary for the survival of rat neurons and that HSF1 mRNA and protein expression is reduced in neurons primed to die. Knock-down of HSF1 induces death of otherwise healthy neurons, whereas reestablishment of elevated levels of HSF1 protects neurons even when death is not due to accumulation of misfolded proteins. Neuroprotection by HSF1 does not require its trimerization, an event obligatory for the binding of HSF1 to heat shock elements within HSP gene promoters. Moreover, knock-down of HSP70 or blockade of HSP90 signaling does not reduce neuroprotection by HSF1. Although several neuroprotective molecules and signaling pathways, including CaMK, PKA, Casein kinase-II, and the Raf-MEK-ERK and PI-3K-Akt pathways, are not required for HSF1-mediated neuroprotection, protection is abrogated by inhibition of classical histone deacetylases (HDACs). We report that the novel mechanism of neuroprotection by HSF1 involves cooperation with SIRT1, an HDAC with well documented neuroprotective effects. Using a cell culture model of Huntington's disease, we show that HSF1 trimerization is not required for protection against mutant huntingtin-induced neurotoxicity, suggesting that HSF1 can protect neurons against both proteinopathic and nonproteinopathic death through a noncanonical pathway.
Introduction
Eukaryotic cells respond to heat shock by activating the production of chaperones called heat shock proteins (HSPs). This conserved protective response, referred to as the heat-shock response, facilitates the refolding of denatured proteins and the degradation of severely damaged proteins (Lindquist, 1986; Morimoto, 1998; Björk and Sistonen, 2010; Fujimoto and Nakai, 2010). Members of the HSP family include the HSP10, HSP27, HSP40, HSP70, HSP90, and HSP110 proteins. The increased production of these HSPs is mediated at the transcriptional level primarily by heat shock factor 1 (HSF1; Lindquist, 1986; Morimoto, 1998; Björk and Sistonen, 2010; Fujimoto and Nakai, 2010).
In most cell types, HSF1 is cytoplasmic in a monomeric form kept inactive in a protein complex containing HSP90 and various other HSPs (Lindquist, 1986; Morimoto, 1998; Björk and Sistonen, 2010; Fujimoto and Nakai, 2010). Upon exposure to heat or protein-damaging stress, the HSPs are diverted to the newly misfolded proteins, allowing HSF1 to translocate to the nucleus, where it trimerizes. Trimeric HSF1 binds to a sequence called the heat shock element (HSE) in the promoters of genes encoding HSPs to turn on transcription (Lindquist, 1986; Morimoto, 1998; Björk and Sistonen, 2010; Fujimoto and Nakai, 2010).
Aggregation of misfolded proteins is a pathological hallmark of many neurodegenerative diseases. As in heat shock-induced protein damage, HSF1 protects against neuronal death in diverse models of proteinopathic neurodegenerative disease. Indeed, knock-down of HSF1 expression enhances the neuropathological effects of toxic misfolded proteins (Nollen et al., 2004; Kraemer et al., 2006; Wang et al., 2009), whereas overexpression protects in models of diverse proteinopathic disorders (Fujimoto et al., 2005; Hayashida et al., 2010; Liangliang et al., 2010; Zhang et al., 2011). Several compounds have been identified that activate HSF1 by promoting the disassociation of the inhibitory HSP-containing complex normally sequestering HSF1. Such pharmacological activators of HSF1, which also induce HSP synthesis, suppress degeneration in invertebrate and mouse models of proteinopathic neurodegenerative diseases (Auluck and Bonini, 2002; Kieran et al., 2004; Waza et al., 2005; Fujikake et al., 2008). These results, along with the observations that direct overexpression of HSPs by themselves suppress neurodegeneration (for review, see Bonini, 2002), has led to the conclusion that the protective effect of neurons by HSF1 is mediated through HSP stimulation.
Although protection of neurons by HSF1 against misfolded protein accumulation is amply documented, it is not clear whether HSF1 can also protect neurons when death is not caused by protein misfolding or aggregation. In this study, we report that HSF1 can protect neurons even under situations when degeneration is the result of nonproteotoxic insults. Indeed, we show that HSF1 expression is necessary for the survival of neurons normally and that suppression of HSF1 expression induces death of otherwise healthy neurons. Interestingly, this neuroprotective effect of HSF1 is not mediated by the canonical HSP-dependent pathway. Our results suggest the existence of a novel mechanism by which HSF1 protects neurons. We suggest that this mechanism involves cooperation with the class III histone deacetylase (HDAC) SIRT1, which is known to have strong neuroprotective effects.
Materials and Methods
Materials.
All reagents and cell culture media were obtained from Invitrogen. All chemicals were purchased from Sigma-Aldrich. Poly-l-Lysine for tissue culture was obtained from Trevigen. The antibodies used in this paper were as follows: Flag (catalog #F1804; Sigma-Aldrich), HA (Y-11 catalog #sc-805, F-7 catalog #sc-7392; Santa Cruz Biotechnology), α-Tubulin (TU-02, catalog #sc-8035; Santa Cruz Biotechnology), GFP (B-2, catalog #sc-9996, FL catalog #sc-8334; Santa Cruz Biotechnology), Myc (9B11, catalog #2276; Cell Signaling Technology), HSF1 (E-4, catalog #sc-17757; Santa Cruz Biotechnology), HSP70 (W27, catalog #sc-24; Santa Cruz Biotechnology), HSP90α/β (F-8, catalog #sc-13119; Santa Cruz Biotechnology), HSP40 (catalog #H4038; Sigma-Aldrich), HSP27/25 (catalog #H2289; Sigma-Aldrich), and SIRT1 (D1D7, catalog #9745; Cell Signaling Technology). Primary antibodies were used at concentrations ranging from 1:250 to 1:1000 in 5% bovine serum albumin. Secondary antibodies (from Pierce) were used at concentrations of 1:10,000. Enhanced PVDF membrane was from Bio-Rad.
Plasmids.
Flag-tagged HSF1 (HSF1-Flag) plasmid donated by Carl Wu (National Institutes of Health, Bethesda, MD) was purchased from Addgene. Dominant-negative HSP90 (DnHSP90) plasmid donated by William Sessa at Yale University was purchased from Addgene. Myc-tagged HSF1 (HSF1-Myc) and HSF1S230A plasmids were a kind gift from Dr. Lea Sistonen at the Centre for Biotechnology Tykistökatu (Turku, Finland). Flag-tagged SIRT1 (SIRT1-Flag) and SIRT1 deletions constructs (SIRT1-Δ1–10) were a kind gift from Dr. Zhenkun Lou at the Mayo Clinic. GFP-tagged mutant HTT plasmids (HTT-Q138) were a kind gift from Dr. Troy Littleton (Massachusetts Institute of Technology, Cambridge, MA). The HSF1 deletion constructs were generated from the HSF1-Flag plasmid by site-directed mutagenesis using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene) following the manufacturer's instructions. The following primers were used to create site-directed mutants:
The trimerization mutant, HSF1Δ156–226: HSF1AB-4: 5′-CAGGGAGAACTGCCGGTCCATGCACTCCTG-3′; HSF1AB-3: 5′-CAGGAGTGCATGGACCGGCAGTTCTCCCTG-3′. The DNA-binding mutants, HSF1Δ15–56, Δ15–90: HSF1 DBD 15–56 F 5′-GCGGGGCCCAGCAACCTGCCCAAGTACTTC-3′; HSF1 DBD 15–56 R 5′-GAAGTACTTGGGCAGGTTGCTGGGCCCCGC-3′; HSF1 DBD 15–90 F 5′-GCGGGGCCCAGCAACAAGCCAGAGAGAGAC-3′; HSF1 DBD 15–90 R 5′-GTCTCTCTCTGGCTTGTTGCTGGGCCCCGC-3′.
The transcription activation domain mutants HSF1Δ505–530 and HSF1Δ370–430: HSF1 TA 505–530 F 5′-TCCGAAGGGGACGGCGACTACAAGGACGAC-3′; HSF1 TA 505–530 R 5′-GTCGTCCTTGTAGTCGCCGTCCCCTTCGGA-3′; HSF1 TA 370–430 F 5′-CCGCCCACCTCCACCGACCTTGACAGCAGC-3′; HSF1 TA 370–430 R 5′-GCTGCTGTCAAGGTCGGTGGAGGTGGGCGG-3′.
All constructs were sequenced to confirm deletion, checked against mutations, and expressed in HEK293T cells to check for protein expression and to verify proper size. Efficiency of expression for all the mutants was similar to wild-type HSF1-Flag.
Neuronal culture, treatments, and viability assay.
Cerebellar granule neuronal (CGN) cultures obtained from the cerebellum of 7- to 8-d-old rat or 5- to 6-d-old mice of both sexes were plated in Basal Minimal Eagle's medium supplemented with 10% fetal bovine serum (FBS), 25 mm KCl, 2 mm glutamine, and 0.2% gentamycin, as described previously (D'Mello et al., 1993), in 24-well dishes (1 ×106 cells/well for viability assay) or 60 mm dishes (12 × 106 cells/dish for immunoprecipitation and Western blotting; D'Mello et al., 1993). Cytosine arabinoforanoside (10 μm) was added to the culture medium 18–22 h after plating to prevent replication of non-neuronal cells. Cultures were maintained for 4–5 d before transient transfection was performed using the calcium phosphate method, as described previously (Koulich et al., 2001; Yalcin et al., 2003). In vitro, cultures were switched to serum-free Basal Minimal Eagle's medium supplemented with 25 mm KCl (high potassium, HK) or without KCl (low potassium, LK) 8 h after transfection. For pharmacological inhibitor studies, at the time of media switch, cells were treated with LK medium or LK medium supplemented with inhibitors of the following concentrations: PD98059 at 50 μm, U0126 at 20 μm, Wortmannin at 200 nm, LY294002 at 10 μm, Akt inhibitor X at 5 μm, KN62 at 50 μm, IC261 at 10 μm, H89 at 2 μm, Trichostatin A (TSA) at 1 μm, TBB at 10 μm, D4476 at 10 μm, Nicotinamide at 5 μm, and Sirtinol at 100 μm. All pharmacological inhibitors were purchased from Calbiochem. The ability of these inhibitors to inhibit their targets at the doses listed above was confirmed in control experiments. After 24 h of treatment, cells were fixed, immunocytochemistry was performed, and cell viability was quantified by cell morphology using 4′6-diamidino-2-phenylindole hydrochloride (DAPI) staining. Cells with condensed or fragmented nuclei were scored as dead. Unless mentioned otherwise, all viability experiments were performed in duplicate and each experiment repeated three times. For each experiment, ≥200 transfected cells were counted.
Rat cortical cultures were obtained from the cerebral cortex of Wistar rats (day 17 of gestation) and treated with homocysteic acid (HCA) to induce oxidative stress, as described previously (Chen et al., 2004; Ratan et al., 1994; Langley et al., 2008; Haskew-Layton et al., 2010). On day 2 in vitro, transient transfections were performed using the calcium phosphate method and, 8 h later, were treated for 15–18 h with 1 mm HCA, as described previously (Bardai et al., 2012; Dastidar et al., 2012). HCA was made as a 100 mm stock solution that was adjusted to pH 7.5. Viability was quantified as described for CGNs.
Heat shock experiments.
For CGNs, cells were subjected to heat shock on day 8 in vitro by placing them in an incubator at 44°C for 30, 60, and 90 min and then recovered at 37°C for 60 min, as described previously (Chen et al., 2004). For cortical neurons, heat shock was performed 1 d after plating by subjecting cells to 44°C for 30, 60, and 90 min and then recovered at 37°C for 6–8 h.
RNA preparation and RT-PCR.
RNA was extracted from cultured neurons or the HT22 cell line by using TRIzol (Invitrogen) according to the manufacturer's instructions. cDNA was prepared from 3 μg of RNA using the Superscript First Strand Synthesis System for RT-PCR kit (Invitrogen) according to the manufacturer's instructions. PCR was performed with GoTaq Green Master Mix (Promega). The primers used for PCR amplification were as follows: HSF1 Forward: 5′-CACCCTGAAGAGTGAGGACATAA-3′; HSF1 Reverse: 5′-GGCTGGAGATGGAGCTGAGTA-3′; Actin Forward: 5′-AGGACTCCTATGTGGGTGACGA-3′; Actin Reverse: 5′-CGTTGCCAATAGTGATGACCTG-3′; c-Jun Forward: 5′-GATGGAAACGACCTTCTACG-3′; c-Jun Reverse: 5′-GTTGAAGTTGCTGAGGTTGG-3′; 18s Forward: 5′-GCTACCACATCCAAGGAAGG-3′; 18s Reverse: 5′-GGCCTCGAAAGAGTCCTGTA-3′; HSP27 Forward: 5′-CGTGCCCTTCTCGCTACT-3′; HSP27 Reverse: 5′-ATCCTGCCTTTCTTCGTG-3′; HSP60 Forward: 5′-CACTGGCTCCTCATCTCA-3′; HSP60 Reverse: 5′-GTCTTTGTCTCCGTTTGC-3′; HSP10 Forward: 5′-AGTTTCTTCCGCTATTTG-3′; HSP10 Reverse: 5′-ACTTTGGTGCCTCCGTGT-3′; HSP110 Forward:5′-AGCAGGACCTCCCGAATG-3′; HSP110 Reverse: 5′-CCCAAAGAATCTGGCAAT-3′; HSP40 Forward: 5′-CAGTGGTGGTGCTAATGG-3′; HSP40 Reverse: 5′-CTTGAGTAGGGCGGGAAC-3′; HSP70 Forward: 5′-CGCCACTTCGTAGCAT-3′; HSP70 Reverse: 5′-TAGCAGGATCTGGATAGGG-3′; HSP90 Forward: 5′-ACATTCTCAGTTTATTGGCTAC-3′; HSP90 Reverse: 5′-TGTTCTTCCCAGTCGTTG-3′; CRYAB Forward: 5′-CGTCCCTTCTTTCCTTTC-3′; CRYAB Reverse: 5′-GGGTGATGGGAATGGTGC-3′; PDZK3 Forward: 5′-GAGCCACTTTGGACGAGA-3′; PDZK3 Reverse: 5′-CCTCACTCTGGCTTTCACT-3′; NFAT3 Forward: 5′-TATGATAACCTCCACCCATTTGC-3′; NFAT3 Reverse: 5′-GACCTGTGACCTGGGCTGCTACT-3′.
Immunoblot analysis.
The cells were lysed in 1× cell lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 μg/ml leupeptin, and protease inhibitor mixture tablets), incubated at −80°C for 1 h, thawed, and centrifuged at 14,000 rpm for 10 min at 4°C. The supernatant was collected as the whole-cell soluble lysate and protein concentration was determined by the Bradford assay. Equal concentrations of protein were mixed with 6× SDS sample buffer (375 mm Tris-HCl, pH 6.8, 12% SDS, 60% glycerol, 300 mm dithiothreitol, and 0.012% bromophenol blue). The samples were then boiled at 95°C for 5 min and subjected to SDS-PAGE. The proteins were electrophoretically transferred from the gel to a PVDF membrane and then incubated in blocking buffer (1× TBS, 0.5% Tween 20 with 5% w/v nonfat dry milk) at room temperature for 1 h. The membrane was subsequently incubated at 4°C overnight with primary antibodies, followed by secondary antibody for 1 h at 25°C. Immunoreactivity was developed by enhanced chemiluminescence and visualized by autoradiography using ECL reagent (GE Healthcare).
shRNA knock-down experiments.
For the knock-down of HSF1, 5 shRNAs were obtained from Sigma-Aldrich: TRCN0000008502, TRCN0000008503, TRCN0000008504, TRCN0000008506, and TRCN0000280517 denoted as shRNA1, shRNA2, shRNA3, shRNA4, and shRNA5, respectively. The pLKO.1-TRC control vector, which contains a non-hairpin, 18 bp insert, was used as a transfection control. Suppression experiments were performed in HT22 cells using shRNA1 and shRNA2. HT22 cells were transfected with pLKO.1 or with HSF1 shRNAs plasmids for 72 h, after which protein lysates were made and subjected to Western blotting. Knock-down experiments in neuronal cultures were performed by cotransfecting in vitro day 4 rat or day 3 mice CGN cultures with the shRNA-expressing plasmids or the control vector with CMV-GFP in a ratio of 6.5: 1. Two days later, the neurons were treated with HK/LK media followed by immunocytochemistry after 24 h. Viability of the GFP-expressing cells were quantified by DAPI staining. For HSP70 shRNA experiments, five separate shRNAs were purchased from Signa-Aldrich: TRCN0000008512, TRCN0000008515, TRCN0000008513, TRCN0000008514, and TRCN0000008516, which for the purposes of this report are designated as shRNA-A, shRNA-B, shRNA-C, shRNA-D, and shRNA-E, respectively. Two of these shRNAs, shRNA-A and shRNA-B, were chosen to cotransfect with HSF1 based on efficacy.
Cell lines and transfection.
Mouse neuroblastoma HT22 cells were a kind gift from Dr. Rajiv Ratan (Burke Medical Research Institute, White Plains, NY). Human embryonic kidney cells (HEK293T) were purchased from ATCC. Both cell lines were grown in DMEM supplemented with 10% FBS and transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Immunoprecipitation.
Cell lysates were prepared by using 300 μl of 1× cell lysis buffer and centrifuged at 14,000 rpm for 10 min at 4°C. An aliquot of the cell lysate (25 μl) was mixed with 6× SDS, boiled at 95°C for 5 min, and subjected to Western blot analysis to check for protein overexpression (in case of transfected cell lines) and for equal protein usage for immunoprecipitation. Then, 1 μg of pull-down antibody was added to 30 μl of Protein A/G Plus-Agarose beads (Santa Cruz Biotechnology) in 300 μl of 1× cell lysis buffer for 1 h at 4°C with constant rocking. Beads bound with pull-down antibody were precipitated by centrifugation at 4000 rpm for 2 min and the supernatant was discarded. Then, 250 μl of the precleared protein lysate was added to the beads and incubated overnight at 4°C with constant rocking. Immunoprecipitate was collected by centrifugation at 4000 rpm for 2 min and washed three times with 500 μl of 1× cell lysis buffer. Then, 30 μl of 3× SDS was added to the immunoprecipitate, boiled for 5 min at 95°C, and then subjected to Western blot analysis.
Analysis of HSF1 trimerization by crosslinking.
Evaluation of HSF1 trimerization by crosslinking was performed as described previously (Neef et al., 2010). Cell lysates from HEK293T cells transfected with HSF1-Flag were harvested using 150 μl of 1× cell lysis buffer. Lysates were centrifuged at 14,000 rpm for 10 min at 4°C. To 50 μg of lysates, 0.5 mm ethylene glycol bis-succinimidylsuccinate was added for 30 min at 24°C. The crosslinking reaction was quenched by addition of 0.025 mm Tris, pH 7.5, for 15 min at 24°C. The crosslinking reactions were then electrophoresed on a 7.5% SDS PAGE gel and subjected to Western blot analysis to detect HSF1-Flag.
Chromatin immunoprecipitation.
For the chromatin immunoprecipitation (ChIP) assay, HEK293T cells were grown to 80–90% confluency and then transfected with pLKO.1, HSF1-FLAG, and HSF1Δ156–226 for 24 h. The cultures were then fixed with formaldehyde and incubated for 10 min on a shaker at room temperature. The medium was aspirated and cells were washed with ice-cold PBS. The cells were then harvested with ice-cold PBS and the lysates were sonicated to yield fragments 200–1000 bp in size. The samples were then diluted in L3 buffer (1 mm EDTA, pH 8, 0.5 mm EGTA, pH 8, 10 mm Tris HCl, pH 8.1, 0.3 m NaCl, 1% Triton X-100, and 0.1% deoxycholate), followed by immunoprecipitation with IgG or Flag antibody. Immunocomplex was then washed, eluted using elution buffer (10 mm Tris HCl, pH 8, 1 mm EDTA, pH 8, 1% SDS), and then reverse crosslinking of the eluates was done at 65°C overnight. Protein was then degraded by addition of 33 μl of 10× proteinase K buffer (0.1M Tris, pH 8, 0.05 EDTA, pH 8) and 2 μl of proteinase K solution at 55°C for 2 h. DNA was then extracted by phenol/chloroform/isoamyl extraction, followed by precipitation with ethanol, and then subjected to PCR. Binding of HSF1-Flag and HSF1Δ156–226 was evaluated using primers spanning the HSE of the HSP70 gene promoter (Rossi et al., 2012; Kim et al., 2013). The primers used were as follows: Forward 5′-CACTCCCCCTTCCTCTCAG-3′ and Reverse 5′-TTCCCTTCTGAGCCAATCAC-3′.
Biotin-streptavidin pull-down assay.
This assay was performed as described previously (Deng et al., 2003; Kodeboyina et al., 2010). The probe targeting the HSE was prepared by annealing 5′-Biotin-AACGAGAATCTTCGAGAATGGCT-3′ and 5′-Biotin AGCCATTCTCGAAGATTCTCGTT-3′ oligonucleotides at 95°C for 5 min, followed by cooling at room temperature. As a negative control, a probe lacking the HSE sequence but with the same flanking regions was prepared by annealing 5′-Biotin AACGACGGTCGCTCCGCCTGGCT-3′ and 5′-Biotin AGCCAGGCGGAGCGACCGTCGTT-3′ oligonucleotides. HEK293T cells were transfected with pLKO.1, HSF1-Flag, and HSF1Δ156–226 for 24 h, and lysates were then collected. Next, 600 μg of lysates were incubated with 2 μg of the probe and 10 μg of salmon sperm DNA in pull-down buffer (10 mm HEPES, pH7.9, 25 mm KCl, 500 μm EDTA, 500 μm DTT, 2.5% glycerol, and protease inhibitors) on ice for 90 min. Streptavidin agarose beads were then added to the mixture and incubated overnight at 4°C. The pelleted beads were then washed three times with pull-down buffer. Next, 30 μl of 3×SDS was added to the washed beads and then boiled at 95°C for 5 min. The proteins were analyzed by immunoblotting by probing the membranes with Flag antibody; 30 μl of whole-cell lysates were also run to check for protein expression.
Statistical analysis.
All the graphs in this study were generated using GraphPad Prism 5 software. Unless otherwise mentioned in the figure legends, statistical analysis was done using unpaired two-tailed t test (Student's t test) and the results are shown as mean ± SD. p < 0.05 was deemed statistically significant. Asterisks in the figure legends indicate the following: *p < 0.05; **p < 0.01; ***p < 0.001.
Results
HSF1 is necessary for neuronal survival and protects against protein-aggregation-independent neuronal death
When CGNs are switched from medium containing depolarizing levels of potassium (HK) to nondepolarizing levels of potassium (LK), they undergo apoptosis (D'Mello et al., 1993). Survival in HK mimics the well documented survival-promoting effect of neuronal activity in vivo. We found that the expression of HSF1 mRNA and protein in CGNs was reduced within 6 h of LK treatment (Fig. 1A,B). Previous work from us and others has found that irreversible commitment to death occurs between 4 and 6 h after LK treatment, although cell death itself begins after 12 h (Galli et al., 1995; Borodezt and D'Mello, 1998). The downregulation of HSF1 coincident to the commitment of the neurons to death raises the possibility that HSF1 is necessary for the normal survival of neurons and that reduced HSF1 expression triggers cell death. To investigate this possibility, we knocked down HSF1 expression using shRNAs. Two shRNAs, shRNA1 and shRNA2, were identified that substantially knocked down HSF1 protein expression when tested in HT22 mouse neuroblastoma cells (Fig. 2A,B). When used in rat CGNs, expression of shRNA2 reduced the viability of otherwise healthy neurons (maintained in HK) and enhanced the extent of cell death in LK (Fig. 2C,E). The inability of shRNA1 to produce the same effect can be explained by its inability to suppress HSF1 expression because of a mismatch in the sequence between mouse and rat HSF1 mRNAs in the region targeted by shRNA1 (Fig. 2A). Indeed, no knock-down was observed with shRNA1 in rat CGNs. However, when expressed in CGNs cultured from mice, shRNA1 reduced neuronal viability to a similar extent as shRNA2 (Fig. 2D).
Figure 1.
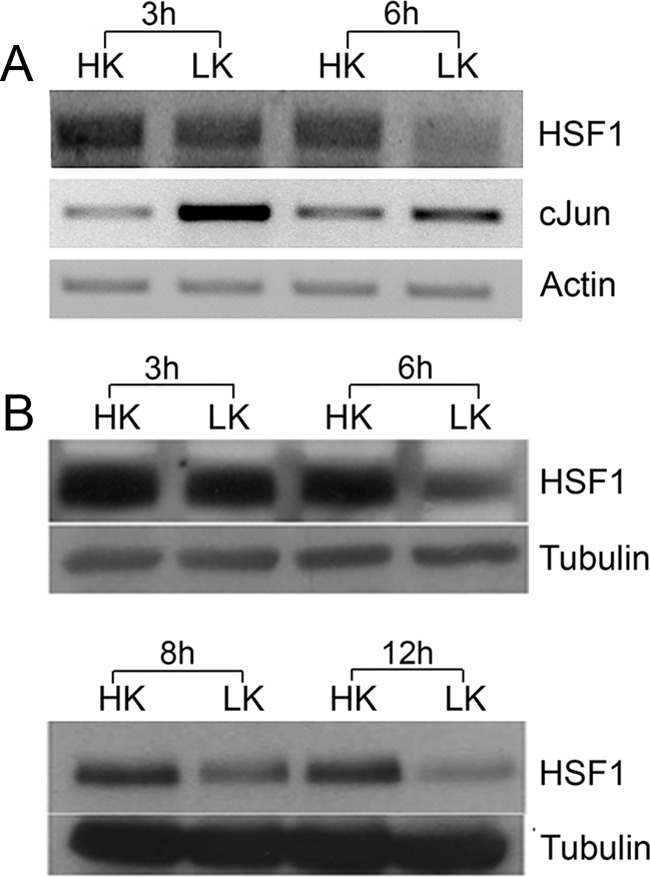
HSF1 RNA and protein expression is reduced in CGNs primed to undergo apoptosis. A, RNA was obtained from CGNs treated with HK or LK for 3 and 6 h and RT-PCR analysis was performed using HSF1 primers. β-Actin served as a loading control. c-Jun was used as a positive control to ensure that cells were primed to die by LK treatment. B, Protein lysates were prepared from CGNs treated with HK or LK for 3, 6, 8, or 12 h and subjected to Western blotting using HSF1 antibody. Tubulin served as a loading control.
Figure 2.

Elevated HSF1 expression is necessary for neuronal survival. A, Schematic showing the mismatch between mouse and rat HSF1 mRNA sequences in the region targeted by shRNA1. B, Whole-cell lysates from HT22 cells transfected with shRNA1 and shRNA2, two shRNAs constructs against HSF1 mRNA, and a control plasmid pLKO.1 (NC) and then subjected to Western blot analysis with a HSF1 antibody. The membrane was also probed with tubulin, which served as a loading control. C, Rat CGNs cotransfected with plasmids expressing pLKO.1, shRNA1, or shRNA2 with GFP in a 6.5:1 ratio were treated with HK or LK medium. Viability of the transfected neurons was quantified 24 h later by DAPI staining and normalized to pLKO.1- and GFP-cotransfected cultures treated with HK. Data represents the mean ± SD from three independent experiments. ***p ≤ 0.001 compared with neurons cotransfected with pLKO.1 and GFP under HK conditions; **p ≤ 0.01 compared with neurons cotransfected with pLKO.1 and GFP under LK conditions. D, Mouse CGNs were cotransfected with plasmids expressing pLKO.1, shRNA1, or shRNA2 and GFP in a 6.5:1 ratio were treated with HK or LK medium. Viability of the transfected neurons was quantified 24 h later by DAPI staining and was normalized to pLKO.1- and GFP-cotransfected cultures treated with HK. Data represent the mean ± SD from three independent experiments. **p ≤ 0.01 compared with neurons cotransfected with pLKO.1 and GFP under HK conditions. E, Appearance of neurons transfected with HSF1 shRNA2. Cells transfected with the shRNA underwent apoptosis and were visualized by immunocytochemistry. Viability of neurons was quantified by DAPI staining.
If reduction in HSF1 expression plays a causal role in neuronal death, then elevated HSF1 levels could be expected to prevent death. As shown in Figure 3A, ectopic expression of HSF1 protected CGNs against LK-induced death. Although HSF1 has been described to reside in the cytoplasm of many cell types under unstressed conditions, in CGNs, ectopically expressed HSF1 localizes exclusively to the nucleus in both HK and LK conditions, suggesting that nuclear-cytoplasmic shuttling is not relevant to the regulation of neuronal survival by HSF1 (Fig. 3B). As observed with ectopically expressed HSF1, endogenous HSF1 is also localized in the nucleus of primary neurons (Fig. 3C) and the two cell lines used in this study, HEK293T and HT22 cells (data not shown).
Figure 3.

Overexpression of HSF1 in CGNs. A, CGNs transfected with GFP or HSF1-Flag were switched to HK or LK medium for 24 h. Transfected cells were visualized by immunocytochemistry with GFP or Flag antibody and their viability was quantified by DAPI staining. The survival rates were normalized to GFP-transfected cultures treated with HK. Data represent the mean ± SD from three independent experiments. ***p ≤ 0.001 compared with neurons transfected with GFP under LK conditions. B, HSF1 localization was analyzed in HK and LK CGNs using Flag antibody and immunocytochemistry. Quantification revealed that 99.7% of the cells displayed nuclear localization in HK. C, Endogenous localization of HSF1 was analyzed in CGNs using the HSF1 antibody. HSF1 showed nuclear localization in primary neurons.
As observed in CGNs, the knock-down of HSF1 expression also induces cell death of primary embryonic rat cortical neurons (Fig. 4A). Treatment of cortical neurons with HCA results in death by oxidative stress (Ratan et al., 1994; Langley et al., 2008; Haskew-Layton et al., 2010). As observed in LK-treated CGNs, HSF1 levels are reduced by HCA treatment (Fig. 4B). Forced expression of HSF1 protects cortical neurons against HCA-induced death (Fig. 4C). A similar protection against HCA-induced death is seen in HT22 neuroblastoma cells (Fig. 4D). Together, these results show that HSF1 is necessary for the survival of healthy neurons and that elevated expression of HSF1 is protective even when neuronal death is not the result of protein aggregation.
Figure 4.
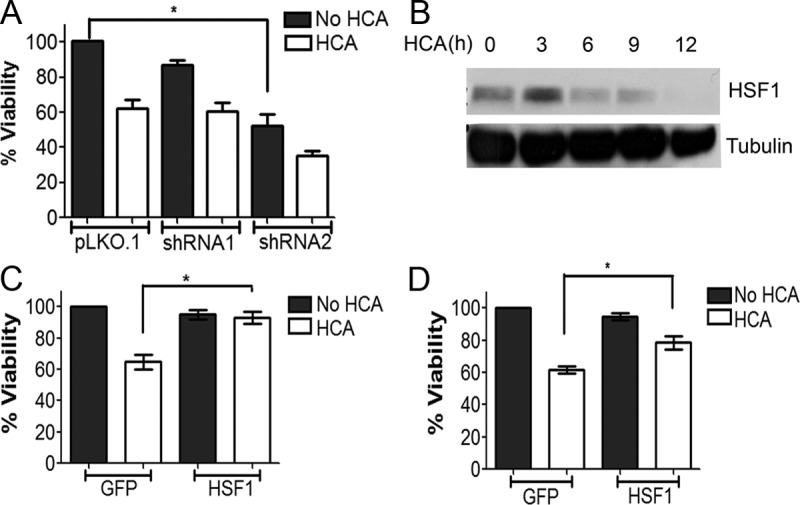
Knock-down and overexpression of HSF1 in cortical neurons. A, Rat cortical neuronal cultures cotransfected with pLKO.1, shRNA1, or shRNA2 and GFP in a 6.5:1 ratio were treated with medium with or without HCA. Viability of transfected neurons was quantified 24 h later by DAPI staining. The survival rates were normalized to pLKO.1- and GFP-cotransfected cultures treated without HCA. Data represent the mean ± SD from three independent experiments. *p ≤ 0.05 compared with neurons transfected with GFP and treated with HCA. B, Lysates from rat cortical neuron cultures treated with 1 mm HCA for 0, 1, 3, 6, and 9 h were analyzed by Western blotting using an HSF1 antibody. Tubulin served as a loading control. HSF1 levels decreased with time from 3 to 9 h. C, Rat cortical cultures transfected with GFP or HSF1-Flag were treated with 1 mm HCA for 18 h. Transfected cells were visualized by immunocytochemistry with a GFP or Flag antibody and their viability quantified by DAPI staining. Results represent mean ± SD from three independent experiments. The survival rates were normalized to GFP-transfected cultures treated without HCA. *p ≤ 0.05 compared with cortical neurons transfected with GFP and treated with HCA. D, HT22 cells transfected with GFP or HSF1-Flag were treated with 1 mm HCA for 18 h. Transfected cells were visualized by immunocytochemistry with a GFP or Flag antibody and their viability was quantified by DAPI staining, as described in Materials and Methods. Result represents mean ± SD from three independent experiments. *p ≤ 0.05 compared with HT22 cells transfected with GFP and treated with HCA.
Promotion of neuronal survival by HSF1 does not involve the canonical HSP-dependent pathway
Although genes encoding HSPs are the primary targets of HSF1, LK-induced downregulation of HSF1 does not result in a general reduction in the expression of major HSPs (Fig. 5A). For example, expression of HSP90 mRNA is unchanged, whereas expression of HSP70 mRNA is increased in LK. Results for HSP70 and HSP90 were also analyzed at the protein level, which showed a small increase in HSP90 levels at 12 h after LK treatment (Fig. 5B). The lack of correlation between HSF1 expression and the expression of its canonical targets suggests that the survival-promoting effect of HSF1 in neurons that are not under proteotoxic stress is mediated through a mechanism unrelated to HSP activation. A lack of correlation is also observed in cortical neurons induced to die by HCA treatment. As shown in Figure 5C, expression of HSP27, HSP40, and HSP70 is increased at 3 and 6 h after HCA treatment.
Figure 5.

Changes in levels of HSPs in CGNs and cortical neurons primed to die. A, RNA from rat CGNs treated with HK or LK for 6 h was subjected to RT-PCR analysis using primers against HSF1 and several major HSPs. c-Jun expression was evaluated to confirm that the cultures were primed to die. Actin served as a loading control. B, Whole-cell lysates from CGNs treated with HK or LK for 3, 6, 8, and 12 h were analyzed by Western blotting using antibodies specific to Hsp70 and Hsp90. Tubulin served as a loading control. C, Rat cortical neurons were treated with HCA for 0, 3, 6, and 9 h and then protein lysates were collected. Immunoblotting experiments were performed using antibodies against different HSPs. Tubulin served as a loading control and c-Jun was used to ensure cells were induced to die by HCA. HSP70 was slightly induced at 3 and 6 h, but then reduced at 9 h. HSP40 showed an increase in 3, 6, and 9 h. HSP27 increased at 3 and 6 h and remained elevated at 9 h.
To determine whether stimulation of HSPs could protect neurons under nonproteinopathic conditions, we used 17-AAG, a well established and widely used chemical activator of HSF1. 17-AAG binds to HSP90, disrupting the repressive association between HSP90 and HSF1 and thus freeing HSF1 to activate expression of HSPs. Treatment with 17-AAG and other related HSP90 inhibitors have impressive protective effects in cell culture and in in vivo models of protein aggregation-associated neurodegenerative disease (Auluck and Bonini, 2002; Katsuno et al., 2005; Waza et al., 2005; Batulan et al., 2006; Fujikake et al., 2008) [for review, see (Nagai et al., 2010)]. Unexpectedly, in view of its impressive protective effects against proteinopathic neuronal death, 17-AAG is incapable of preventing LK-induced death of CGNs (Fig. 6A); on the contrary, 17-AAG itself displays some neurotoxicity. As reported previously, 17-AAG does induce HSP production in CGNs, as evidenced by increased expression of HSP70 in 17-AAG-treated neurons (Fig. 6B).
Figure 6.
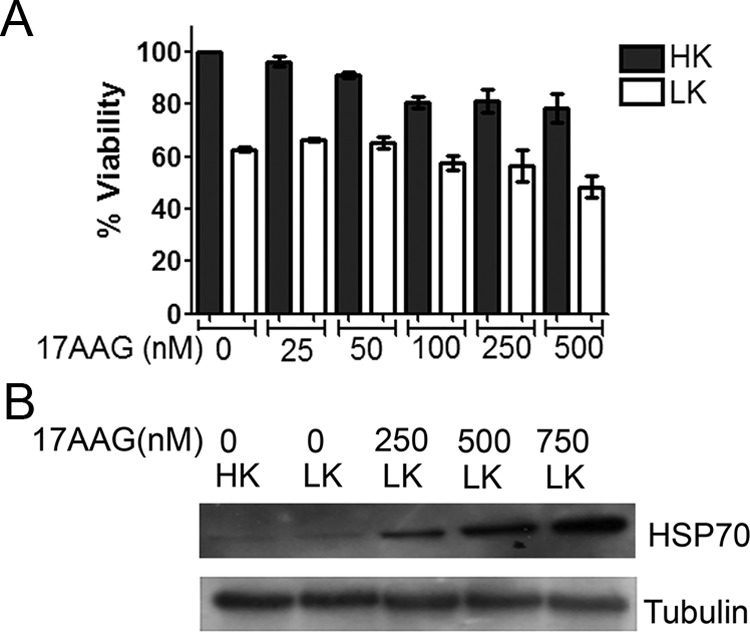
17-AAG induces HSP70, but does not protect CGNs undergoing apoptosis. A, CGNs were treated with no additives or 17-AAG at increasing concentrations from 25 to 500 nm for 16 h and then switched to HK or LK medium for 24 h. Cell viability was quantified by DAPI staining. The survival rates were normalized to cultures treated with HK. Results represent mean ± SD from three independent experiments. No significant protection was found with treatment of 17-AAG. B, CGNs were treated with no additives or 250, 500, or 750 mm 17-AAG for 16 h and then treated with HK or LK medium for 8 h. Whole-cell lysates were collected and proteins were analyzed by Western blotting using an antibody specific to HSP70. Tubulin served as a loading control.
It was possible that the neurotoxicity observed with 17-AAG was due to off-target effects. To stimulate HSP production more physiologically, we induced heat shock. Others have found that heat shock protects CGN and cortical neurons against glutamate toxicity (Lowenstein et al., 1991; Rordorf et al., 1991). In addition, heat shock has been shown to protect CGNs against LK-induced death (Chen et al., 2004). As described by these other investigators, the exposure of CGNs to heat shock stimulates expression of HSP70, a key player in the heat shock response and protection against proteotoxic neurodegeneration (Warrick et al., 1999; Aridon et al., 2011; Turturici et al., 2011; Fig. 7A). Although a robust increase in HSP70 production is seen within 30 min of heat shock, statistically significant protection against LK-induced neuronal death requires 60 min of heat shock (Fig. 7B). In addition, HSP70 induction is highest at 60 min of heat shock, whereas the extent of protection against LK-induced death is higher at 90 min compared with 60 min of heat shock (Fig. 7B). Increases, albeit to a smaller extent, are also seen in the expression of HSP27 and HSP40 by 30 min of heat shock (Fig. 7A). In contrast, the expression of HSP90, another important HSP in the context of proteotoxic neurodegeneration (Luo et al., 2010; Aridon et al., 2011), was not increased (Fig. 7A). The discordance in the pattern of HSP stimulation versus neuroprotection suggests that the protective effect of heat shock is not mediated by the canonical HSP-dependent mechanism. Given that heat shock stimulates the expression of non-HSP proteins, it is likely that neuroprotection is mediated by one or more of such non-HSP proteins that are induced relatively late after heat shock. A similar conclusion was reached by Cao et al. (2007), who reported that heat shock activates Akt and inhibits GSK3β in LK-treated CGNs. Pharmacological inhibitors of PI-3 kinase-Akt signaling reduced the protective effect of heat shock, but had no effect on the stimulation of HSP expression (Cao et al., 2007). As observed in CGNs, heat shock of cortical neurons using conditions known to stimulate HSPs does not protect the neurons from HCA-induced toxicity (Fig. 7C).
Figure 7.

Heat shock of CGNs induces HSP70 expression but not HSP90. A, CGNs were heat shocked at 44°C for 0, 15, 30, 60, and 90 min and then recovered at 37°C for 60 min. The cultured CGNs were then treated with HK or LK for 6 h and cell lysates were analyzed by Western blotting using antibodies against HSP70, HSP90, HSP27, and HSP40. Tubulin served as a loading control. B, CGNs were heat shocked as described above and then treated with HK or LK for 24 h before cell viability was quantified by DAPI staining. The survival rates were normalized to HK cultures that were not heat shocked. Result represents mean ± SD from three independent experiments. C, Cortical cultures were heat shocked at 44°C for 0, 15, 30, 60, and 90 min and then recovered at 37°C for 6–8 h. These cultures were then treated with HCA or left untreated for 15–18 h before cell viability was quantified by DAPI staining. The survival rates were normalized to untreated cultures that were not heat shocked. Result represents mean ± SD from three independent experiments. *p < 0.05 compared with neurons treated with LK that were not heat shocked. D, Rat CGNs were transfected with pLKO.1 or HSP70 shRNAs, shRNA-A, shRNA-B, shRNA-C, shRNA-D, and shRNA-E in a 6.5:1 ratio with GFP and their ability to reduce the protection by HSP70 was evaluated. shRNA-A and shRNA-B showed robust effects in knocking down HSP70. Viability was quantified by DAPI staining. The survival rates were normalized to pLKO.1- and GFP-cotransfected cultures treated with HK. Data represent the mean ± SD from three independent experiments. ***p ≤ 0.001 compared with neurons cotransfected with pLKO.1 and GFP under HK conditions. E, Rat CGNs were cotransfected with GFP and pLKO.1 or HSF1 and pLKO.1, shRNA-A, or shRNA-B in a 1:2 ratio. Cells were treated 48 h later with HK or LK for 24 h followed by immunocytochemistry. Cell viability was quantified by DAPI staining. The survival rates were normalized to GFP- and pLKO.1-cotransfected cells treated with HK. Result represents mean ± SD from three independent experiments. *p ≤ 0.05 and **p ≤ 0.01 compared with neurons cotransfected with GFP and pLKO.1 and treated with LK. HSP70 shRNAs were unable to bring down the neuroprotective effect of HSF1 in both HK and LK conditions. F, Rat CGNs were transfected with GFP, HSF1-Flag, or DnHSP90 or cotransfected with HSF1-Flag and DnHSP90. Cells were treated 8 h later with HK or LK medium. After a 24 h treatment, cells were fixed and immunocytochemistry was performed. Viability was quantified by DAPI staining. Data represent the mean ± SD from three independent experiments. DnHSP90 was unable to reduce the protective effect of HSF1 in both HK and LK conditions. ***p ≤ 0.001 compared with neurons transfected with GFP and treated with HK; ***p ≤ 0.001 compared with neurons transfected with GFP and treated with LK.
To confirm that HSPs were not involved in HSF1-mediated neuroprotection, we knocked down HSP70 expression using two separate shRNAs. Although reducing the normal viability of neurons in HK medium, knocking down of HSP70 had no effect on neuroprotection by HSF1 (Fig. 7D,E). To examine this issue further, we used a dominant-negative form of HSP90 that was shown previously to interfere with HSP90 signaling (Miao et al., 2008). Coexpression of DnHSP90 reduced survival of normal neurons, but not the ability of HSF1 to protect (Fig. 7F).
Trimerization of HSF1 and domains necessary for transcriptional stimulation of HSP expression are not required for neuroprotection
It has been amply demonstrated that HSF1 binds to HSE sequences within promoters of its target genes as a homotrimer (Lindquist, 1986; Morimoto, 1998; Björk and Sistonen, 2010; Fujimoto and Nakai, 2010). Trimerization is believed to be necessary for the transcriptional activity of HSF1 because deletion and point-mutant forms of HSF1 that are unable to oligomerize are transcriptionally inactive when analyzed using HSE-containing promoters (Orosz et al., 1996; Farkas et al., 1998; Lu et al., 2008; Lu et al., 2009). Indeed, monomeric HSF1 has generally been considered to be transcriptionally and functionally inactive. HSF1 trimerization requires an oligomerization domain comprised of hydrophobic heptad repeats, often referred to as the trimerization domain (Fig. 8A). Deletion of this domain renders HSF1 incapable of assembling into a trimer (Inouye et al., 2007). To determine whether trimerization was necessary for the protective effect of HSF1 in LK-treated CGNs, we generated a deletion mutant lacking the entire trimerization domain, HSF1Δ156–226. We confirmed that HSF1Δ156–226 is unable to self-associate using coimmunoprecipitation analysis (Fig. 8B). Similarly, results from crosslinking analysis showed that HSF1Δ156–226 was unable to oligomerize (Fig. 8C). Despite being unable to oligomerize, HSF1Δ156–226 is fully capable of protecting neurons (Fig. 8D).
Figure 8.

Evaluation of the neuroprotective effects of various mutant HSF1 constructs. A, Schematic diagram of the full-length HSF1 protein and the different deletion constructs of HSF1 used during this study. The different domains of HSF1 are DNA binding domain (DBD), Trimerization domain (TD), regulatory domain (RD), and transcription activation domain (TAD). B, HEK293T cells were transfected with HSF1-Myc and either HSF1-Flag or HSF1-FlagΔ156–226 for 48 h. GFP transfection was used as a negative control. The HSF1-Flag and HSF1-FlagΔ156–226 were immunoprecipitated using Flag antibody. The immunoprecipitate and aliquots of preimmunoprecipitation lysate (WCL) were analyzed by Western blotting using Myc antibody. HSF1-Flag interacted with HSF1-Myc, but no interaction was found between HSF1-FlagΔ156–226 and HSF1-Myc. C, HEK293T cells were transfected with wild-type HSF1-Flag and HSF1-FlagΔ156–226. HSF1 multimerization was evaluated after ethylene glycol bis-succinimidylsuccinate crosslinking and Western blotting using a Flag antibody. Trimers were formed by HSF1, but not by HSF1Δ156–226. D, CGNs transfected with GFP or different mutant HSF1 constructs were treated with HK or LK and viability of transfected cells was quantified by DAPI staining. The survival rates were normalized to GFP-transfected cultures treated with HK. Data represent the mean ± SD from three independent experiments. ***p ≤ 0.001 compared with neurons transfected with GFP and treated with LK. E, HEK293T cells were transfected with GFP, HSF1-Flag, HSF1Δ15–90, HSF1Δ15–56, HSF1Δ156–226, HSF1Δ370–430, HSF1Δ505–530, HSF1 221–530, and HSF1 371–530 for 24 h. Cell lysates were collected and immunoblotting was performed to compare the expression levels of full-length HSF1-Flag and all deletion constructs. All HSF1 deletion constructs expressed. GFP was used as a positive control for transfection. Tubulin served as a loading control.
We also generated and tested other HSF1 mutants that have deletions in other functional domains (Fig. 8A). Although partial deletion of the DNA-binding domain (HSF1Δ15–56) has no effect on neuroprotection, deletion of a larger region of the DNA-binding domain (HSF1 Δ15–90) abrogates the neuroprotective activity of HSF1 (Fig. 8D). Control experiments confirmed that the DNA-binding mutants are expressed at levels that are equal to or higher than wild-type HSF1 (Fig. 8E). This suggests that DNA-binding is required for neuroprotection by HSF1. Because DNA binding is necessary but trimerization is not, it is possible that the neuroprotective activity of HSF1 involves binding to DNA in a monomeric form at a sequence different from the HSE.
To garner evidence that neuroprotection by HSF1 does not involve binding to an HSE, we compared the binding of wild-type HSF1 with the trimerization-deficient mutant HSF1Δ156–226 in a ChIP assay using the promoter region of the HSP70 gene as a template. As described previously, the HSP70 promoter contains an HSE that is bound by HSF1 in ChIP assays (Rossi et al., 2012; Kim et al., 2013). Whereas wild-type HSF1 associates with the HSE element, HSF1Δ156–226 does not, which is consistent with the need for trimerization for HSE binding (Fig. 9A). We confirmed this finding using a separate biotin-streptavidin pull-down assay in which streptavidin was used to pull down biotinylated double-stranded oligonucleotides containing an HSE sequence that had been incubated with lysates containing wild-type HSF1 or HSF1Δ156–226. Although the HSE probe pulls down wild-type HSF1 efficiently, it does not pull-down HSF1Δ156–226 (Fig. 9B). Because stimulation of HSP gene transcription requires the binding of trimeric HSF1 to HSEs, the ability of HSF1Δ156–226 to protect neurons suggests an HSP-independent mechanism.
Figure 9.
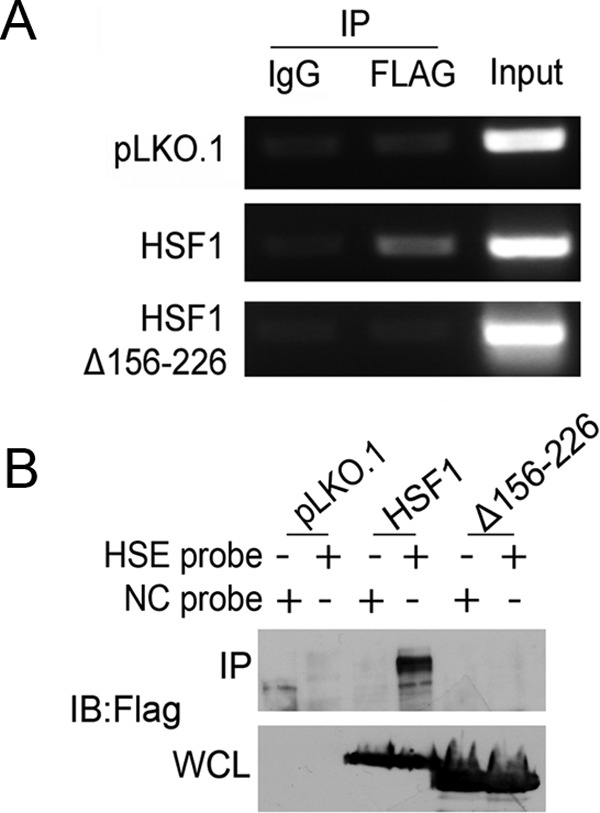
Evaluation of binding of trimerization-deficient HSF1 to HSE sequences. A, HEK293T cells were transfected with pLKO.1, HSF1-Flag, or HSF1Δ156–226 for 24 h. ChIP-enriched DNA was obtained by using either mouse IgG or Flag antibody. The quantification of binding of each of the constructs was determined by PCR using HSE primers targeting the HSP70 promoter, as described in the Materials and Methods. HSF1 showed binding to the HSE region, but not HSF1Δ156–226. pLKO.1 was used as a negative control. B, Biotin-streptavidin pull-down was performed using HEK293T cells transfected with pLKO.1, HSF1-Flag, or HSF1Δ156–226 for 24 h. Lysates were incubated with either the biotin-labeled, HSE or negative control (NC) probes, followed by pull-down with streptavidin beads, as described in the Material and Methods. The pre-pull-down whole-cell lysates and the pull-down products were analyzed by immunoblotting using a Flag antibody.
Two activation domains (ADs, labeled 1 and 2) have been mapped within the transactivation domain (TAD) of HSF1 that are essential for transcription of HSP genes. Surprisingly, deletion of AD1 (HSF1Δ370–430) has no effect on the ability of HSF1 to protect neurons (Fig. 8D). Partial deletion of AD2 (HSF1Δ505–530) also has no detrimental effect (Fig. 8D), indicating that domains of HSF1 necessary for HSP stimulation are dispensable for protecting neurons. A residue that is important for the stimulation of HSP expression by HSF1 is Ser230. Mutation of Ser230 to a nonphosphorylatable alanine severely diminishes the ability of HSF1 to induce expression of HSP70 (Holmberg et al., 2001). However, HSF1S230A is as effective as wild-type HSF1 in protecting CGNs against LK-induced death (Fig. 8D). These results strengthen the conclusion that neuroprotection by HSF1 is independent of HSP stimulation. In contrast to the above-described HSF1 mutants, which are protective, a mutant containing only the TAD domain, HSF1 371–529, or another containing the regulatory domain and transcription activation domain but nothing else, HSF1 221–529, fail to protect neurons.
Several molecules and signaling pathways that protect neurons are not involved in HSF1-mediated neuroprotection
To investigate the novel mechanism by which HSF1 protects neurons, we treated HSF1-transfected neuronal cultures with pharmacological inhibitors against well established neuroprotective signaling pathways and molecules. Pharmacological inhibition of the PI3 kinase-Akt pathway blocks the protective effect of heat shock on LK-induced death of CGNs (Cao et al., 2007). However, these inhibitors did not reduce the protective effect of HSF1. Similarly, inhibition of other signaling pathways and molecules involved in neuroprotection in other paradigms, including the MEK-ERK pathway (with U0126 and PD98059), CaMKII (with KN62), Casein kinase-I (with IC261 and D4476), Casein kinase-II (with TBB), and PKA (with H89), all failed to inhibit the ability of HSF1 to protect against neuronal death (Fig. 9). Control experiments confirmed that these inhibitors inhibited their targets at the concentrations used (data not shown). Interestingly HSF1-mediated neuroprotection was completely abolished by TSA, an inhibitor of classical histone deacetylases (Fig. 10). In contrast, two separate sirtuin inhibitors, nicotinamide and sirtinol, did not reduce neuroprotection (Fig. 10).
Figure 10.

Pharmacological inhibitors do not affect HSF1 mediated neuroprotection in CGNs. CGNs transfected with HSF1-Flag were treated with LK medium or LK medium supplemented with the following inhibitors: 200 mm Wortmannin (Wort), 5 μm AKT inhibitor X (AktT-X), 10 μm LY294002 (LY), 20 μm U0126, 50 μm PD98059, 10 μm IC261, 10 μm D4476, 2 μm H89, 50 μm KN62 (KN), 1 μm TSA, 10 μm TBB, 5 μm Nicotinamide, or 100 μm Sirtinol for 24 h. Cells treated with only LK medium served as a control. Viability was determined by immunocytochemistry and DAPI staining and compared with GFP-transfected cultures treated with HK. Data represent the mean ± SD from three independent experiments. ***p ≤ 0.001 compared with neurons transfected with HSF1 and treated under LK conditions.
Expression of non-HSP transcriptional targets of HSF1 identified in other systems does not correlate with HSF1 expression in neurons
Although HSPs are the best-characterized targets of HSF1, particularly in the context of heat or proteotoxic stress, a large number of genes unconnected to HSPs or chaperone function have been identified as potential transcriptional targets of HSF1. One of these is PDZK3/Papin (Fujimoto et al., 2010). PDZK3 expression is induced by heat shock in mouse embryonic fibroblasts (MEFs) overexpressing HSF1, but not in HSF1−/− MEFs (Fujimoto et al., 2010). Furthermore, HSF1 binds to the PDZK3 promoter in MEFs (Fujimoto et al., 2010; Hayashida et al., 2010). It was possible, therefore, that the survival-promoting activity of HSF1 is mediated by maintaining elevated PDZK3 expression and that neuronal death is caused by reduced PDZK3 expression as a consequence of HSF1 downregulation. However, although HSF1 expression is reduced by LK treatment, the expression of PDZK3 is robustly increased in CGNs induced to die by LK treatment (Fig. 11). Another gene shown to be upregulated in HSF1-overexpressing HeLa cells is α-crystallin, B (CRYAB; Hayashida et al., 2006). As shown in Figure 11, the expression of CRYAB is not reduced in CGNs after LK treatment. Two other genes identified as HSF1 targets in the same microarray screen were PROM2, a cholesterol-binding membrane glycoprotein, and CSRP2 (Hayashida et al., 2010), a transcriptional coactivator that plays an important role in the cardiovascular system (Jain et al., 1996; Jain et al., 1998). However, expression of PROM2 and CSRP2 is undetectable in the brain (Jain et al., 1996; Jain et al., 1998). Interesting results were also obtained by these scientists with another putative target, NFATc2 /NFAT1, a member of the NFAT family of transcription factors. NFATc2 overexpression strongly suppresses polyglutamine (polyQ) aggregation in MEFs (Hayashida et al., 2010). Despite much effort, however, we have failed to detect NFATc2 expression in CGNs (data not shown), arguing against its being an essential mediator of the survival-promoting effect of HSF1 in these neurons. Examination of NFAT3, a related member of the NFAT family that is expressed in CGNs and that has been implicated in promoting neuronal survival (Benedito et al., 2005), showed no change in expression after LK treatment. Together, these results indicate that the non-HSP targets of HSF1 in neurons, particularly with regard to the maintenance of neuronal survival, are likely to be distinct from those in other cell types such as HeLa cells and MEFS.
Figure 11.

Expression of previously identified non-HSP transcriptional targets of HSF1 do not correlate with HSF1 expression in neurons. RNA was obtained from CGNs treated with HK or LK for 6 h and RT-PCR analysis was performed using primers for PDZK3, CRYAB and NFAT3. These genes were previously suggested to be targets of HSF1 in non-neuronal cells and systems (Benedito et al., 2005; Hayashida et al., 2006; Fujimoto et al., 2010; Hayashida et al., 2010). β-Actin served as a loading control.
SIRT1 may be involved in HSF1 neuroprotection
Several groups, including our own, have reported that the class III HDAC SIRT1 has strong neuroprotective effects (Qin et al., 2006; Kim et al., 2007; Pfister et al., 2008; Tang and Chua, 2008; Jeong et al., 2012; Jiang et al., 2011). We previously reported HSF1 and SIRT1 are both nuclear proteins in neurons. As was also observed by others, we found that the two proteins interact both in neurons and when coexpressed in HEK293T cells (Westerheide et al., 2009) and at endogenous levels in CGNs (Fig. 12A,B). Moreover, the two proteins colocalize in the nuclei of neurons and HEK293T cells (Fig. 12C,D).
Figure 12.

HSF1 might interact with SIRT1 to bring about neuroprotection. A, HEK293T cells were transfected with GFP, SIRT1-Flag, or cotransfected with SIRT1-Flag and HSF1-Myc for 48 h. GFP transfection was used as a negative control. SIRT1-Flag was immunoprecipitated using the Flag antibody. The immunoprecipitate as well as the whole-cell lysate (WCL) were analyzed by Western blotting using Myc and Flag antibodies. SIRT1-Flag interacted with HSF1-Myc. B, Rat CGNs lysates were collected after treatment with HK or LK medium for 8 h. HSF1 antibody was used to pull down endogenous HSF1 and IgG was used as a control. The immunoprecipitates and WCL were analyzed by immunoblotting using a SIRT1 antibody. HSF1 was found to interact with SIRT1 at the endogenous level in CGNs. Unt, Rat CGNs untreated with HK or LK. C, CGNs were transfected with SIRT1-Flag and HSF1-Myc for 8 h and then treated with HK or LK medium for 24 h. Transfected cells were visualized by immunocytochemistry with Myc and Flag antibodies. SIRT1 and HSF1 were found to colocalize with each other in the nucleus of cells transfected with both of the plasmids. D, Appearance of endogenous HSF1 and SIRT1 was analyzed in rat CGNs and HEK293T cells using HSF1 and SIRT1 antibodies. Immunocytochemistry was used to detect endogenous HSF1 (red) and SIRT1 (green). DAPI was used as a nuclear stain. HSF1 and SIRT1 were found to colocalize with each other endogenously in the nucleus. E, Schematic of wild-type SIRT1-Flag and SIRT1 deletion constructs used in our study (adapted from Kim et al., 2008). F, HEK293T cells were transfected with HSF1-Myc and SIRT1-Flag or with HSF1-Myc and 10 deletion constructs of SIRT1, which contains 60–80 sequential amino acid deletions. SIRT1 or the deletion constructs were immunoprecipitated using Flag antibody or IgG antibody. The immunoprecipitates and WCL were analyzed by Western blotting using the Myc and Flag antibodies. HSF1 did not interact with deletion constructs spanning regions 214–541 aa (SIRT1-Δ4-Δ7) and 610–696 aa (SIRT1-Δ9) of SIRT1. SIRT1-Flag and HSF1-Myc lysates pulled down with IgG were used as negative control. G, Rat CGNs were cotransfected with GFP and pLKO.1 or HSF1 shRNA2 or with SIRT1 and shRNA2 in a ratio of 1:2 for 48 h and then treated with HK or LK medium. Viability of neurons was quantified after 24 h using DAPI staining. The survival rates were normalized to GFP- and pLKO.1-cotransfected cultures treated with HK. Data represent the mean ± SD from three independent experiments. **p ≤ 0.01 compared with neurons cotransfected with GFP and pLKO.1 and treated with HK.
We examined the interaction of HSF1 with SIRT1 mutants in which regions of 60–80 residues were deleted (Fig. 12E). As shown in Figure 12F, interaction with HSF1 was sharply reduced with deletion constructs SIRT1-Δ4 to SIRT1-Δ7 and with SIRT1-Δ9. The catalytic domain of SIRT1 spans the region between 236 and 490, portions of which are deleted in SIRT1-Δ4 to SIRT1-Δ7 (Fig. 12D). It was recently demonstrated that a 25 aa region between residues 631–655 of SIRT1 was obligatory for its catalytic activity (Kang et al., 2011). This region, designated previously by Kang et al. (2011) as ESA (essential for SIRT1 activity), is within the region deleted in SIRT1-Δ9. These results suggest that the interaction of HSF1 with SIRT1 is dependent on the catalytic activity of SIRT1.
We previously reported that SIRT1-mediated neuroprotection does not require its catalytic activity and is not reduced by pharmacological agents that inhibit it. However, neuroprotection by SIRT1 was suppressed by inhibitors against classical HDACs such as TSA (J.A.P. and S.R.D., unpublished observations). Neuroprotection by HSF1 is also inhibited by TSA (Fig. 10). Pharmacological inhibitors against other signaling molecules described in Figure 10, which do not inhibit HSF1-mediated protection, also have no effect on neuroprotection by SIRT1 (J.A.P. and S.R.D., unpublished observations). Nicotinamide, a nonspecific sirtuin inhibitor, or Sirtinol, a specific inhibitor for SIRT1, did not inhibit the HSF1 mediated neuroprotection (Fig. 10). Our studies have also shown that SIRT1 neuroprotection is itself not reduced by the use of these two inhibitors (Pfister et al., 2008). These results, along with the finding that the two proteins interact, raise the possibility that HSF1 and SIRT1 cooperate to protect neurons. In accordance with this idea, knock-down of HSF1 expression blocks SIRT1-mediated neuroprotection (Fig. 12G).
Neuroprotection by HSF1 against proteinopathic degeneration is also mediated by the noncanonical mechanism
Polyglutamine expansion of huntingtin (Htt) causes Huntington's disease (HD), a genetic neurodegenerative disorder (Zuccato et al., 2010). Overexpression of mutant Htt (Htt-138) with an expanded polyglutamine tract in cultured cells is the best-studied and most used model of proteinopathic toxicity. Although in the brains of HD patients, death occurs selectively in neurons of the striatum, in cell culture systems, the overexpression of mutant Htt has toxic effects on a variety of neuronal and non-neuronal cell types, which is associated with its aggregation (Zuccato et al., 2010). We recently showed that mutant Htt promotes death of CGNs (Bardai and D'Mello, 2011; Bardai et al., 2013). To determine whether HSF1 can protect CGNs against mutant Htt neurotoxicity, we coexpressed the two proteins. As shown in Figure 13A, overexpression of HSF1 completely blocked mutant Htt toxicity both in HK and LK medium. Very surprisingly given the conclusions of previous studies (Orosz et al., 1996; Farkas et al., 1998; Lu et al., 2008; Lu et al., 2009), trimerization-deficient HSF1 was as protective as the wild-type protein. Similarly, other HSF1 constructs with mutations affecting the HSP-related transcriptional activity were also protective (Fig. 13B). Although it is possible that the canonical HSP-dependent pathway also protects against proteinopathic neurodegeneration, our results suggest that protection can be mediated independently of this pathway in CGNs.
Figure 13.

HSF1 promotes protection against mutant HTT-mediated toxicity. A, Rat CGNs transfected with GFP, HSF1-Flag, Q138 (mut HTT-GFP), or HSF1-Flag and Q138 for 8 h and then switched to HK or LK medium for 24 h. Viability of transfected cells was quantified by DAPI staining. The survival rates were normalized to GFP-transfected cultures treated with HK. Data represent the mean ± SD from three independent experiments. **p ≤ 0.01 compared with neurons transfected with GFP and treated with LK; **p ≤ 0.01 compared with neurons transfected with Q138 and treated with HK. B, Rat CGNs transfected with GFP, Q138, Q138, and HSF1-Flag or Q138 and the HSF1 deletion constructs in a 1:2 ratio were treated with HK or LK medium for 24 h. Viability of transfected cells was quantified by DAPI staining. The survival rates were normalized to GFP-transfected cultures treated with HK. Data represent the mean ± SD from three independent experiments. **p ≤ 0.01 compared with neurons transfected with GFP, HttQ138, or HSF1 and treated with HK.
Discussion
Because the overexpression of HSPs protect against neurodegeneration induced by the accumulation of misfolded proteins in different disease models, they have been considered as a therapeutic target. Because HSP production is under the control of HSF1, the effect of pharmacological HSF1 activation or its overexpression has been tested in tissue culture and in in vivo models of proteinopathic neurodegenerative disease (Nollen et al., 2004; Fujimoto et al., 2005; Kraemer et al., 2006; Wang et al., 2009; Hayashida et al., 2010; Liangliang et al., 2010; Zhang et al., 2011; Kondo et al., 2013). These investigations showed that HSF1 is neuroprotective, leading to the belief that its beneficial effect is mediated by the stimulation of HSP production (Fujimoto et al., 2005; Neef et al., 2010; Neef et al., 2011).
Studies on HSF1 as a neuroprotective protein have been limited to models of proteinopathic degeneration. Whether HSF1 can protect neurons in response to nonproteotoxic stimuli has not been investigated directly. Also not fully explored is the issue of whether HSP stimulation is the only mechanism by which HSF1 can protect neurons. We show that depletion of HSF1 from otherwise healthy neurons is sufficient to promote death, indicating that an elevated level of HSF1 is necessary to maintain neuronal survival. Overexpressed HSF1 can protect neurons even when death is not due to the accumulation of protein aggregates. We find that both under nonproteotoxic conditions and in response to proteinopathic stress, HSF1 protects neurons through a noncanonical, HSP-independent mechanism. Evidence for this includes full neuroprotection by a mutant form of HSF1 that completely lacks the trimerization domain and is therefore incapable of oligomerization. Trimerization is required for the binding of HSF1 to HSE sequences within the promoters of HSP genes. Furthermore, other mutant constructs lacking activation domains essential for heat-shock-related stimulation of HSF transcription or with mutations in key residues, such as S230, are also fully neuroprotective. Finally, pharmacological stimulation of HSP production has no protective effect, at least in nonproteotoxic conditions.
Evidence for the existence of an HSP-independent pathway for the cytoprotective effect of HSF1 has been emerging recently in models in which cell death is induced by heat shock. For example, it has been shown that HSF3, which is incapable of activating the heat shock response, protects MEF cells from heat-shock-induced death just as efficiently as HSF1 (Fujimoto et al., 2010). Similarly, chicken HSF1 can protect against heat shock fully, even though it has only a marginal stimulatory effect on HSP expression. Even in models of neurodegenerative disease, there is evidence for HSP-independent protective mechanisms. In particular, a recent publication reported that protection by HSF1 against polyQ-Htt toxicity involves the transcriptional stimulation of NFATc2 expression (Hayashida et al., 2010). We have not been able to detect NFATc2 expression in CGNs. Although overexpression of the related NFAT3 protein protects CGNs against LK, we find that its expression does not correlate with the expression pattern of HSF1. Another study reported that HSF1 overexpression protects against α-synuclein toxicity in neuroblastoma cells even though the expression of key HSPs necessary for a heat-shock response was not increased (Liangliang et al., 2010). It is noteworthy that of the ∼400 genes identified as inhibitors of aggregation-associated neurodegenerative disease in genome-wide Caenorhabditis elegans RNAi screens, only a small number encode HSPs or other types of chaperones (Nollen et al., 2004; Kraemer et al., 2006; van Ham et al., 2008; Wang et al., 2009). Other genome-wide screens have also identified large numbers of non-HSP genes that are regulated by HSF1 in the absence or presence of heat shock (Trinklein et al., 2004; Page et al., 2006).
HSF1 activity and function is generally regulated by posttranslational modifications. In most systems, the intracellular level of HSF1 is tightly regulated and does not generally change. However, in neurons primed to die through either LK or HCA treatment, HSF1 mRNA and protein levels are reduced well before signs of degeneration are evident, suggesting that reduction in HSF1 expression is a critical event in neurodegeneration. A recent study found that when mice deficient in HSF1 were crossed with spinal and bulbar muscular atrophy mice, disease progression was exacerbated and pathology was found in areas not normally affected (Kondo et al., 2013). Also consistent with a crucial role for HSF1 in maintaining neuronal survival are the observations from two separate groups that mice lacking HSF1 display neurodegeneration and astrogliosis (Santos and Saraiva, 2004; Homma et al., 2007). Intriguingly, another group failed to detect neuronal loss or any other neuropathological abnormalities (Steele et al., 2008). This may be related to differences in genetic background of the mice used in these studies. Other members of the HSF family may also compensate for the survival-promoting function of HSF1. HSF2 is known to cooperate with HSF1 in the heat shock response (He et al., 2003; Ostling et al., 2007) and mice lacking HSF2 display brain abnormalities including thinning of the cerebral cortex (Kallio et al., 2002; Wang et al., 2003). HSF3 can also protect cells against heat-shock-induced death through an HSP-independent mechanism (Fujimoto et al., 2010).
How does trimerization-deficient HSF1 act? Our results show that HSF1 requires its DNA-binding domain for neuroprotection. Because binding at HSEs requires trimeric HSF1, it is possible that for neuroprotection HSF1 binds non-HSE sequences, perhaps as a monomer and in conjunction with other transcription factors. Transcriptional activation through binding at non-HSE sites has also been proposed by Hahn et al. (2004), who reported that more than half of the HSF target promoters identified in yeast through a ChIP-microarray approach lacked a consensus HSE. In addition to chaperone proteins, this study identified genes encoding proteins with a variety of other functions, including protein trafficking, cell signaling, maintenance of cell integrity, and energy generation as putative HSF1 targets (Hahn et al., 2004).
We have found that inhibition of a variety of neuroprotective signaling pathways and molecules, including the MEK-ERK and PI-3K-Akt pathways, CaMK, CKII, and PKA, have no effect on HSF1-mediated protection. Although more work is needed, we provide evidence consistent with the possibility that HSF1 cooperates with SIRT1 to protect neurons. This cooperation is likely mediated through a direct interaction between HSF1 and SIRT1 in the nucleus of neurons. SIRT1 has been shown previously to protect neurons in both cell culture and in in vivo models of neurodegenerative disease (Qin et al., 2006; Kim et al., 2007; Pfister et al., 2008; Tang and Chua, 2008; Jeong et al., 2012; Jiang et al., 2011). Interestingly, we have found previously that the catalytic activity of SIRT1 is not needed for neuroprotection by SIRT1 (Pfister et al., 2008). We have also observed recently (unpublished observation) that SIRT1 neuroprotection is blocked by inhibition of classical HDACs. Consistent with HSF1 and SIRT1 acting cooperatively, classical HDAC inhibition also blocks neuroprotection by HSF1.
In summary, we describe here three novel findings with regard to HSF1. First, we find that HSF1 is necessary for the survival of otherwise healthy neurons and can protect neurons against nonproteotoxic death when overexpressed. Second, although HSF1 activity is generally regulated posttranslationally, HSF1 expression is reduced in neurons induced to die. Although further investigations are needed to determine whether expression of HSF1 is reduced in disease states, approaches that prevent the downregulation of HSF1 or promote increased production could have value in the treatment of neurodegenerative conditions. Third, and most importantly, although the protective effect of HSF1 against proteotoxic stress is generally believed to be mediated through the transcriptional stimulation of HSP production, we provide evidence indicating that both under nonproteotoxic conditions and in response to proteotoxic stress, HSF1 can protect neurons through an HSP-independent mechanism. Indeed, mutant forms that lack domains necessary for trimerization or the transcriptional stimulation of HSPs protect neurons just as effectively as the wild-type protein.
Footnotes
This work was supported by the National Institute of Neurological Diseases and Stroke, National Institutes of Health (Grant NS40408 to S.R.D.).
References
- Aridon P, Geraci F, Turturici G, D'Amelio M, Savettieri G, Sconzo G. Protective role of heat shock proteins in Parkinson's disease. Neurodegener Dis. 2011;8:155–168. doi: 10.1159/000321548. [DOI] [PubMed] [Google Scholar]
- Auluck PK, Bonini NM. Pharmacological prevention of parkinson disease in Drosophila. Nat Med. 2002;8:1185–1186. doi: 10.1038/nm1102-1185. [DOI] [PubMed] [Google Scholar]
- Bardai FH, D'Mello SR. Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3beta. J Neurosci. 2011;31:1746–1751. doi: 10.1523/JNEUROSCI.5704-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardai FH, Price V, Zaayman M, Wang L, D'Mello SR. Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J Biol Chem. 2012;287:35444–35453. doi: 10.1074/jbc.M112.394544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardai FH, Verma P, Smith C, Rawat V, Wang L, D'Mello SR. Disassociation of histone deacetylase-3 from normal huntingtin underlies mutant huntingtin neurotoxicity. J Neurosci. 2013;33:11833–11838. doi: 10.1523/JNEUROSCI.5831-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batulan Z, Taylor DM, Aarons RJ, Minotti S, Doroudchi MM, Nalbantoglu J, Durham HD. Induction of multiple heat shock proteins and neuroprotection in a primary culture model of familial amyotrophic lateral sclerosis. Neurobiol Dis. 2006;24:213–225. doi: 10.1016/j.nbd.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Benedito AB, Lehtinen M, Massol R, Lopes UG, Kirchhausen T, Rao A, Bonni A. The transcription factor NFAT3 mediates neuronal survival. J Biol Chem. 2005;280:2818–2825. doi: 10.1074/jbc.M408741200. [DOI] [PubMed] [Google Scholar]
- Björk JK, Sistonen L. Regulation of the members of the mammalian heat shock factor family. FEBS J. 2010;277:4126–4139. doi: 10.1111/j.1742-4658.2010.07828.x. [DOI] [PubMed] [Google Scholar]
- Bonini NM. Chaperoning brain degeneration. Proc Natl Acad Sci U S A. 2002;99:16407–16411. doi: 10.1073/pnas.152330499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodezt K, D'Mello SR. Decreased expression of the metabotropic glutamate receptor-4 gene is associated with neuronal apoptosis. J Neurosci Res. 1998;53:531–541. doi: 10.1002/(SICI)1097-4547(19980901)53:5<531::AID-JNR3>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Cao L, Cao DX, Su XW, Chen LJ, Liu AL, Jian WJ, Wu YP, Qiu PX, Yan GM. Activation of PI3-K/Akt pathway for thermal preconditioning to protect cultured cerebellar granule neurons against low potassium-induced apoptosis. Acta Pharmacol Sin. 2007;28:173–179. doi: 10.1111/j.1745-7254.2007.00504.x. [DOI] [PubMed] [Google Scholar]
- Chen LJ, Su XW, Qiu PX, Huang YJ, Yan GM. Thermal preconditioning protected cerebellar granule neurons of rats by modulating HSP70 expression. Acta Pharmacol Sin. 2004;25:458–461. [PubMed] [Google Scholar]
- Dastidar SG, Narayanan S, Stifani S, D'Mello SR. Transducin-like enhancer of split-1 (TLE1) combines with forkhead box protein G1 (FoxG1) to promote neuronal survival. J Biol Chem. 2012;287:14749–14759. doi: 10.1074/jbc.M111.328336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng WG, Zhu Y, Montero A, Wu KK. Quantitative analysis of binding of transcription factor complex to biotinylated DNA probe by a streptavidin-agarose pulldown assay. Anal Biochem. 2003;323:12–18. doi: 10.1016/j.ab.2003.08.007. [DOI] [PubMed] [Google Scholar]
- D'Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: Inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci U S A. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas T, Kutskova YA, Zimarino V. Intramolecular repression of mouse heat shock factor 1. Mol Cell Biol. 1998;18:906–918. doi: 10.1128/mcb.18.2.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikake N, Nagai Y, Popiel HA, Okamoto Y, Yamaguchi M, Toda T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem. 2008;283:26188–26197. doi: 10.1074/jbc.M710521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Nakai A. The heat shock factor family and adaptation to proteotoxic stress. FEBS J. 2010;277:4112–4125. doi: 10.1111/j.1742-4658.2010.07827.x. [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Takaki E, Hayashi T, Kitaura Y, Tanaka Y, Inouye S, Nakai A. Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J Biol Chem. 2005;280:34908–34916. doi: 10.1074/jbc.M506288200. [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Hayashida N, Katoh T, Oshima K, Shinkawa T, Prakasam R, Tan K, Inouye S, Takii R, Nakai A. A novel mouse HSF3 has the potential to activate nonclassical heat-shock genes during heat shock. Mol Biol Cell. 2010;21:106–116. doi: 10.1091/mbc.E09-07-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci. 1995;15:1172–1179. doi: 10.1523/JNEUROSCI.15-02-01172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn JS, Hu Z, Thiele DJ, Iyer VR. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol Cell Biol. 2004;24:5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskew-Layton RE, Payappilly JB, Smirnova NA, Ma TC, Chan KK, Murphy TH, Guo H, Langley B, Sultana R, Butterfield DA, Santagata S, Alldred MJ, Gazaryan IG, Bell GW, Ginsberg SD, Ratan RR. Controlled enzymatic production of astrocytic hydrogen peroxide protects neurons from oxidative stress via an Nrf2-independent pathway. Proc Natl Acad Sci U S A. 2010;107:17385–17390. doi: 10.1073/pnas.1003996107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashida N, Inouye S, Fujimoto M, Tanaka Y, Izu H, Takaki E, Ichikawa H, Rho J, Nakai A. A novel HSF1-mediated death pathway that is suppressed by heat shock proteins. EMBO J. 2006;25:4773–4783. doi: 10.1038/sj.emboj.7601370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashida N, Fujimoto M, Tan K, Prakasam R, Shinkawa T, Li L, Ichikawa H, Takii R, Nakai A. Heat shock factor 1 ameliorates proteotoxicity in cooperation with the transcription factor NFAT. EMBO J. 2010;29:3459–3469. doi: 10.1038/emboj.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Soncin F, Grammatikakis N, Li Y, Siganou A, Gong J, Brown SA, Kingston RE, Calderwood SK. Elevated expression of heat shock factor (HSF) 2A stimulates HSF1-induced transcription during stress. J Biol Chem. 2003;278:35465–35475. doi: 10.1074/jbc.M304663200. [DOI] [PubMed] [Google Scholar]
- Holmberg CI, Hietakangas V, Mikhailov A, Rantanen JO, Kallio M, Meinander A, Hellman J, Morrice N, MacKintosh C, Morimoto RI, Eriksson JE, Sistonen L. Phosphorylation of serine 230 promotes inducible transcriptional activity of heat shock factor 1. EMBO J. 2001;20:3800–3810. doi: 10.1093/emboj/20.14.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma S, Jin X, Wang G, Tu N, Min J, Yanasak N, Mivechi NF. Demyelination, astrogliosis, and accumulation of ubiquitinated proteins, hallmarks of CNS disease in hsf1-deficient mice. J Neurosci. 2007;27:7974–7986. doi: 10.1523/JNEUROSCI.0006-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inouye S, Fujimoto M, Nakamura T, Takaki E, Hayashida N, Hai T, Nakai A. Heat shock transcription factor 1 opens chromatin structure of interleukin-6 promoter to facilitate binding of an activator or a repressor. J Biol Chem. 2007;282:33210–33217. doi: 10.1074/jbc.M704471200. [DOI] [PubMed] [Google Scholar]
- Jain MK, Fujita KP, Hsieh CM, Endege WO, Sibinga NE, Yet SF, Kashiki S, Lee WS, Perrella MA, Haber E, Lee ME. Molecular cloning and characterization of SmLIM, a developmentally regulated LIM protein preferentially expressed in aortic smooth muscle cells. J Biol Chem. 1996;271:10194–10199. doi: 10.1074/jbc.271.17.10194. [DOI] [PubMed] [Google Scholar]
- Jain MK, Kashiki S, Hsieh CM, Layne MD, Yet SF, Sibinga NE, Chin MT, Feinberg MW, Woo I, Maas RL, Haber E, Lee ME. Embryonic expression suggests an important role for CRP2/SmLIM in the developing cardiovascular system. Circ Res. 1998;83:980–985. doi: 10.1161/01.RES.83.10.980. [DOI] [PubMed] [Google Scholar]
- Jeong H, Cohen DE, Cui L, Supinski A, Savas JN, Mazzulli JR, Yates JR, 3rd, Bordone L, Guarente L, Krainc D. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat Med. 2012;18:159–165. doi: 10.1038/nm.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Wang J, Fu J, Du L, Jeong H, West T, Xiang L, Peng Q, Hou Z, Cai H, Seredenina T, Arbez N, Zhu S, Sommers K, Qian J, Zhang J, Mori S, Yang XW, Tamashiro KL, Aja S, Moran TH, Luthi-Carter R, Martin B, Maudsley S, Mattson MP, Cichewicz RH, Ross CA, Holtzman DM, Krainc D, Duan W. Neuroprotective role of Sirt1 in mammalian models of huntington's disease through activation of multiple Sirt1 targets. Nat Med. 2011;18:153–158. doi: 10.1038/nm.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio M, Chang Y, Manuel M, Alastalo TP, Rallu M, Gitton Y, Pirkkala L, Loones MT, Paslaru L, Larney S, Hiard S, Morange M, Sistonen L, Mezger V. Brain abnormalities, defective meiotic chromosome synapsis and female subfertility in HSF2 null mice. EMBO J. 2002;21:2591–2601. doi: 10.1093/emboj/21.11.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Suh JY, Jung YS, Jung JW, Kim MK, Chung JH. Peptide switch is essential for Sirt1 deacetylase activity. Mol Cell. 2011;44:203–213. doi: 10.1016/j.molcel.2011.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Sang C, Adachi H, Minamiyama M, Waza M, Tanaka F, Doyu M, Sobue G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A. 2005;102:16801–16806. doi: 10.1073/pnas.0506249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10:402–405. doi: 10.1038/nm1021. [DOI] [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH. SIRT1 deacetylase protects against neurodegeneration in models for alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JA, Kim Y, Kwon BM, Han DC. The natural compound cantharidin induces cancer cell death through inhibition of heat shock protein 70 (HSP70) and bcl-2-associated athanogene domain 3 (BAG3) expression by blocking heat shock factor 1 (HSF1) binding to promoters. J Biol Chem. 2013;288:28713–28726. doi: 10.1074/jbc.M113.488346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- Kodeboyina S, Balamurugan P, Liu L, Pace BS. cJun modulates ggamma-globin gene expression via an upstream cAMP response element. Blood Cells Mol Dis. 2010;44:7–15. doi: 10.1016/j.bcmd.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo N, Katsuno M, Adachi H, Minamiyama M, Doi H, Matsumoto S, Miyazaki Y, Iida M, Tohnai G, Nakatsuji H, Ishigaki S, Fujioka Y, Watanabe H, Tanaka F, Nakai A, Sobue G. Heat shock factor-1 influences pathological lesion distribution of polyglutamine-induced neurodegeneration. Nat Commun. 2013;4:1405. doi: 10.1038/ncomms2417. [DOI] [PubMed] [Google Scholar]
- Koulich E, Nguyen T, Johnson K, Giardina C, D'mello S. NF-κB is involved in the survival of cerebellar granule neurons: association of Iκβ phosphorylation with cell survival. J Neurochem. 2001;76:1188–1198. doi: 10.1046/j.1471-4159.2001.00134.x. [DOI] [PubMed] [Google Scholar]
- Kraemer BC, Burgess JK, Chen JH, Thomas JH, Schellenberg GD. Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum Mol Genet. 2006;15:1483–1496. doi: 10.1093/hmg/ddl067. [DOI] [PubMed] [Google Scholar]
- Langley B, D'Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, Yang L, Ko B, Fisher M, Cho S, Beal MF, Ratan RR. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J Neurosci. 2008;28:163–176. doi: 10.1523/JNEUROSCI.3200-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liangliang X, Yonghui H, Shunmei E, Shoufang G, Wei Z, Jiangying Z. Dominant-positive HSF1 decreases alpha-synuclein level and alpha-synuclein-induced toxicity. Mol Biol Rep. 2010;37:1875–1881. doi: 10.1007/s11033-009-9623-2. [DOI] [PubMed] [Google Scholar]
- Lindquist S. The heat-shock response. Annu Rev Biochem. 1986;55:1151–1191. doi: 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- Lowenstein DH, Chan PH, Miles MF. The stress protein response in cultured neurons: Characterization and evidence for a protective role in excitotoxicity. Neuron. 1991;7:1053–1060. doi: 10.1016/0896-6273(91)90349-5. [DOI] [PubMed] [Google Scholar]
- Lu M, Kim HE, Li CR, Kim S, Kwak IJ, Lee YJ, Kim SS, Moon JY, Kim CH, Kim DK, Kang HS, Park JS. Two distinct disulfide bonds formed in human heat shock transcription factor 1 act in opposition to regulate its DNA binding activity. Biochemistry. 2008;47:6007–6015. doi: 10.1021/bi702185u. [DOI] [PubMed] [Google Scholar]
- Lu M, Lee YJ, Park SM, Kang HS, Kang SW, Kim S, Park JS. Aromatic-participant interactions are essential for disulfide-bond-based trimerization in human heat shock transcription factor 1. Biochemistry. 2009;48:3795–3797. doi: 10.1021/bi802255c. [DOI] [PubMed] [Google Scholar]
- Luo W, Sun W, Taldone T, Rodina A, Chiosis G. Heat shock protein 90 in neurodegenerative diseases. Mol Neurodegener. 2010;5 doi: 10.1186/1750-1326-5-24. 24–1326-5–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao RQ, Fontana J, Fulton D, Lin MI, Harrison KD, Sessa WC. Dominant-negative Hsp90 reduces VEGF-stimulated nitric oxide release and migration in endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:105–111. doi: 10.1161/ATVBAHA.107.155499. [DOI] [PubMed] [Google Scholar]
- Morimoto RI. Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Fujikake N, Popiel HA, Wada K. Induction of molecular chaperones as a therapeutic strategy for the polyglutamine diseases. Curr Pharm Biotechnol. 2010;11:188–197. doi: 10.2174/138920110790909650. [DOI] [PubMed] [Google Scholar]
- Neef DW, Turski ML, Thiele DJ. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 2010;8:e1000291. doi: 10.1371/journal.pbio.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef DW, Jaeger AM, Thiele DJ. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat Rev Drug Discov. 2011;10:930–944. doi: 10.1038/nrd3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nollen EA, Garcia SM, van Haaften G, Kim S, Chavez A, Morimoto RI, Plasterk RH. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci U S A. 2004;101:6403–6408. doi: 10.1073/pnas.0307697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orosz A, Wisniewski J, Wu C. Regulation of Drosophila heat shock factor trimerization: Global sequence requirements and independence of nuclear localization. Mol Cell Biol. 1996;16:7018–7030. doi: 10.1128/mcb.16.12.7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostling P, Björk JK, Roos-Mattjus P, Mezger V, Sistonen L. Heat shock factor 2 (HSF2) contributes to inducible expression of hsp genes through interplay with HSF1. J Biol Chem. 2007;282:7077–7086. doi: 10.1074/jbc.M607556200. [DOI] [PubMed] [Google Scholar]
- Page TJ, Sikder D, Yang L, Pluta L, Wolfinger RD, Kodadek T, Thomas RS. Genome-wide analysis of human HSF1 signaling reveals a transcriptional program linked to cellular adaptation and survival. Mol Biosyst. 2006;2:627–639. doi: 10.1039/b606129j. [DOI] [PubMed] [Google Scholar]
- Pfister JA, Ma C, Morrison BE, D'Mello SR. Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS One. 2008;3:e4090. doi: 10.1371/journal.pone.0004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, Puigserver P, Sadoshima J, Deng H, Pedrini S, Gandy S, Sauve AA, Pasinetti GM. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rordorf G, Koroshetz WJ, Bonventre JV. Heat shock protects cultured neurons from glutamate toxicity. Neuron. 1991;7:1043–1051. doi: 10.1016/0896-6273(91)90348-4. [DOI] [PubMed] [Google Scholar]
- Rossi A, Coccia M, Trotta E, Angelini M, Santoro MG. Regulation of cyclooxygenase-2 expression by heat: A novel aspect of heat shock factor 1 function in human cells. PLoS One. 2012;7:e31304. doi: 10.1371/journal.pone.0031304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos SD, Saraiva MJ. Enlarged ventricles, astrogliosis and neurodegeneration in heat shock factor 1 null mouse brain. Neuroscience. 2004;126:657–663. doi: 10.1016/j.neuroscience.2004.03.023. [DOI] [PubMed] [Google Scholar]
- Steele AD, Hutter G, Jackson WS, Heppner FL, Borkowski AW, King OD, Raymond GJ, Aguzzi A, Lindquist S. Heat shock factor 1 regulates lifespan as distinct from disease onset in prion disease. Proc Natl Acad Sci U S A. 2008;105:13626–13631. doi: 10.1073/pnas.0806319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang BL, Chua CE. SIRT1 and neuronal diseases. Mol Aspects Med. 2008;29:187–200. doi: 10.1016/j.mam.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Trinklein ND, Murray JI, Hartman SJ, Botstein D, Myers RM. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol Biol Cell. 2004;15:1254–1261. doi: 10.1091/mbc.E03-10-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turturici G, Sconzo G, Geraci F. Hsp70 and its molecular role in nervous system diseases. Biochem Res Int. 2011;2011:618127. doi: 10.1155/2011/618127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ham TJ, Thijssen KL, Breitling R, Hofstra RM, Plasterk RH, Nollen EA. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008;4:e1000027. doi: 10.1371/journal.pgen.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Zhang J, Moskophidis D, Mivechi NF. Targeted disruption of the heat shock transcription factor (hsf)-2 gene results in increased embryonic lethality, neuronal defects, and reduced spermatogenesis. Genesis. 2003;36:48–61. doi: 10.1002/gene.10200. [DOI] [PubMed] [Google Scholar]
- Wang J, Farr GW, Hall DH, Li F, Furtak K, Dreier L, Horwich AL. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 2009;5:e1000350. doi: 10.1371/journal.pgen.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med. 2005;11:1088–1095. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- Westerheide SD, Anckar J, Stevens SM, Jr, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin A, Koulich E, Mohamed S, Liu L, D'Mello SR. Apoptosis in cerebellar granule neurons is associated with reduced interaction between CREB-binding protein and NF-kappaB. J Neurochem. 2003;84:397–408. doi: 10.1046/j.1471-4159.2003.01540.x. [DOI] [PubMed] [Google Scholar]
- Zhang T, Mullane PC, Periz G, Wang J. TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum Mol Genet. 2011;20:1952–1965. doi: 10.1093/hmg/ddr076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in huntington's disease. Physiol Rev. 2010;90:905–981. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]