

Abstract
Ischemic stroke is a debilitating clinical disorder that affects millions of people, yet lacks effective neuroprotective treatments. Fish oil is known to exert beneficial effects against cerebral ischemia. However, the underlying protective mechanisms are not fully understood. The present study tests the hypothesis that omega-3 polyunsaturated fatty acids (n-3 PUFAs) attenuate ischemic neuronal injury by activating nuclear factor E2-related factor 2 (Nrf2) and upregulating heme oxygenase-1 (HO-1) in both in vitro and in vivo models. We observed that pretreatment of rat primary neurons with docosahexaenoic acid (DHA) significantly reduced neuronal death following oxygen-glucose deprivation. This protection was associated with increased Nrf2 activation and HO-1 upregulation. Inhibition of HO-1 activity with tin protoporphyrin IX attenuated the protective effects of DHA. Further studies showed that 4-hydroxy-2E-hexenal (4-HHE), an end-product of peroxidation of n-3 PUFAs, was a more potent Nrf2 inducer than 4-hydroxy-2E-nonenal derived from n-6 PUFAs. In an in vivo setting, transgenic mice overexpressing fatty acid metabolism-1, an enzyme that converts n-6 PUFAs to n-3 PUFAs, were remarkably resistant to focal cerebral ischemia compared with their wild-type littermates. Regular mice fed with a fish oil-enhanced diet also demonstrated significant resistance to ischemia compared with mice fed with a regular diet. As expected, the protection was associated with HO-1 upregulation, Nrf2 activation, and 4-HHE generation. Together, our data demonstrate that n-3 PUFAs are highly effective in protecting the brain, and that the protective mechanisms involve Nrf2 activation and HO-1 upregulation by 4-HHE. Further investigation of n-3 PUFA neuroprotective mechanisms may accelerate the development of stroke therapies.
Introduction
Ischemic stroke is a debilitating clinical disorder that affects millions of people, yet lacks effective neuroprotective treatments. Large-scale studies have shown that consumption of oily fish reduces the incidence of stroke events and associated mortality (Skerrett and Hennekens, 2003; He et al., 2004; Turunen et al., 2008). Recent reports have also demonstrated that omega-3 polyunsaturated fatty acids (n-3 PUFAs) are beneficial against brain ischemia (Black et al., 1979; Marcheselli et al., 2003; Akbar et al., 2005; Belayev et al., 2005; Zhang et al., 2010). Although the neuroprotective mechanisms underlying n-3 PUFAs are not fully understood, some have suggested that n-3 PUFAs exert protection primarily via anti-inflammatory effects (Bazan, 2005; Zhang et al., 2010; Lalancette-Hébert et al., 2011). Inflammatory processes typically involve interactions between multiple cell types. However, in studies using pure cultures of single cell types, such as neurons (Akbar et al., 2005), astrocytes (Sergeeva et al., 2005), or endothelial cells (Majkova et al., 2011), the protective properties of n-3 PUFAs are also evident in vitro. These studies all suggest that n-3 PUFAs may also directly protect individual cell types in the absence of anti-inflammatory intercellular interactions. However, the nature of these self-protective mechanisms is not well understood.
Cytoprotection against oxidative stress, toxicity, and carcinogens is primarily mediated by phase 2 enzymes, including heme oxygenase 1 (HO-1; Dinkova-Kostova and Talalay, 2008; Zhang et al., 2013). The expression of phase 2 enzymes is regulated by nuclear factor E2-related factor 2 (Nrf2), a transcription factor. Under basal conditions, Nrf2 is silenced by its partner protein, Kelch-like ECH-associated protein 1 (Keap1; Itoh et al., 1999; Kobayashi et al., 2004). Keap1 is a cysteine-rich cytosolic protein that functions as an adaptor, linking Nrf2 and a Cullin3-related E3 ligase, thereby facilitating proteasomal degradation of Nrf2 (Kobayashi et al., 2004). Cysteine is nucleophilic and highly reactive with electrophiles via a Michael-type reaction. Under stressful conditions, Nrf2 inducers react with Keap1 cysteines, leading to disruption of its association with Nrf2. Once freed from proteasomal degradation, Nrf2 rapidly undergoes nuclear translocation, initiating expression of phase 2 enzymes (Wakabayashi et al., 2004). However, it is still not known whether Nrf2 and HO-1 contribute to the neuroprotection afforded by n-3 PUFAs.
The present study tested the hypotheses that n-3 PUFAs protect against ischemic brain injury via oxidation to 4-hydroxy-2E-hexenal (4-HHE), which in turn upregulates HO-1 expression by acting as a potent activator of the Nrf2 pathway. These hypotheses were based on the following observations: (1) the concentration of n-3 PUFAs is high in the nervous system and n-3 PUFAs are more susceptible to oxidation than either n-6 or n-9 PUFAs (Guichardant et al., 2004; Valentine and Valentine, 2004), (2) the rate constant of 4-HHE during its reaction with the cysteine in glutathione is ∼1.5-fold higher than that of 4-hydroxy-2E-nonenal (4-HNE; Esterbauer et al., 1975, 1991), and (3) 4-HHE has a higher electrophilicity index than 4-HNE (Table 1).
Table 1.
Comparison between n-6 and n-3 PUFA lipid peroxidation productsa

aFour-HNE and 4-HHE are lipid electrophiles derived from n-6 and n-3 PUFAs, respectively. Both are α,β-unsaturated carbonyl aldehydes and their reactive carbons are marked with stars. However, the electrophilicity index (ώ) of 4-HHE (n-3) is higher than that of 4-HNE (n-6). Consequently, 4-HHE (n-3) has a higher rate constant (k1) than 4-HNE (n-6) during the reaction with the cysteine in glutathione.
bElectrophilicity index was calculated using Spartan 104 software (Wavefunction).
cRate constants were measured and reported by Esterbauer et al. (1975).
Materials and Methods
Creation of fatty acid metabolism-1 gene transgenic mice and lipid profile analysis.
All animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and Stroke Treatment and Academic Roundtable (STAIR) guidelines. Transgenic fatty acid metabolism-1 (fat-1) gene mice were generated as described previously (Kang et al., 2004). Heterozygous fat-1 male mice were bred with C57BL/6 female mice to produce offspring. Mouse tail DNA was extracted for genotyping. The specific primers used were as follows: (F) 5′-CGGTTTCTGCGATGGATCCCAC-3′ and (R) 5′-CCGGTGAAAACGCAGAAGTTGTTG-3′. Amplification of a 631 bp band confirmed the fat-1 genotype. Fat-1 transgenic mice and wild-type littermates were fed with a standard laboratory rodent diet (Prolab 5P76 Isopro 3000, PMI Nutrition). Lipid extraction and fatty acid analysis of forebrains were performed as described previously (Zhang et al., 2010). Forebrain samples were harvested and fast-frozen and then dried into powder in a vacuum freeze dryer until analysis. The fatty acids were extracted as described previously (Zhang et al., 2010). Brain fatty acid methyl ester peak identification was performed by comparison to the peak retention times of a 30-component methyl ester standard (Sigma-Aldrich). The concentration of each fatty acid was determined by calculating areas of the peaks. All values are expressed as mole percentage.
Fish oil treatment and murine model of transient focal ischemia.
Male C57BL/6 mice (The Jackson Laboratory) were randomly divided into three groups: sham, control (regular diet), and experimental [fish oil (FO) diet]. Control mice were fed with a regular laboratory rodent diet (Prolab 5P76 Isopro 3000, PMI Nutrition) containing low concentrations of n-3 PUFAs (0.34%). For the experimental group, 5% FO (w/w) was added to the regular diet, which increased the n-3 PUFA [docosahexaenoic acid (DHA) plus eicosapentaenoic acid (EPA)] content from 0.34 to 1.5%, and decreased the n-6:n-3 PUFA ratio from 5:1 to 1:1 (Zhang et al., 2010). Mice were fed for 6 weeks before fatty acid analysis and ischemia were performed. Fat-1 transgenic mice and C57BL/6 mice in all groups were anesthetized with 1.5% isoflurane in a 30% O2/70% N2O mixture under spontaneous breathing. Transient focal cerebral ischemia was induced with standard, previously published procedures (Zhang et al., 2012). Rectal temperature was maintained at 37.0 ± 0.5°C with a temperature-regulated heating pad throughout the surgery, and mean arterial blood pressure was monitored with a tail cuff. The respective diets for the mice were continued after ischemia until the animals were killed.
As dictated by STAIR guidelines, regional cerebral blood flow (rCBF) was evaluated before, during, and after the surgery. Animals underwent middle cerebral artery occlusion (MCAO) for 60 min and then reperfusion for the indicated durations. Quantitative rCBF measurement of wild-type and fat-1 mice was also performed at 15 min after the onset of MCAO by [14C]-iodoantipyrine (IAP) autoradiography (Sawada et al., 2000; Stetler et al., 2008), or by laser Doppler flowmetry. Mice were excluded from the study if their rCBF did not fall below 20% of baseline or if they failed to exhibit neurological deficits after anesthesia recovery. For the HO-1 inhibition study, 30 μg of tin protoporphyrin IX (Sn-PPIX; Frontier Scientific), a potent competitive inhibitor of heme oxidation, was injected intracerebroventricularly 3 h after MCAO in 5 μl of vehicle (PBS; Zhang et al., 2012).
Evaluation of ischemic outcomes.
Neurobehavioral and histological studies were performed to evaluate ischemic outcomes by a blinded observer. For the short-term studies (48 h), neurologic dysfunction was scored by a blinded observer at 48 h after MCAO using the five-point method (Zhang et al., 2010), with 0 being the least dysfunction and 4 the worst dysfunction. Mice were then killed and brains removed and sliced into 1-mm-thick coronal sections. Sections were then stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) to determine infarct volume (Zhang et al., 2007). For the long-term studies (21 d), the corner test and rotarod test (IITC Life Science) were performed up to 7 d after MCAO. The Morris water maze test was performed from day 15 to day 21 following MCAO by a blinded observer to evaluate cognitive function (Stetler et al., 2008). Following this test, animals were killed by perfusion and the brain sections (5 μm) were stained with antimicrotubule-associated protein 2 (MAP2). The infarct was measured by a blinded observer using MCID software (MCID Image Analysis) and was expressed by the following equation: (viable area of contralateral hemisphere minus viable area of ipsilateral hemisphere)/viable area of contralateral hemisphere. The viable or healthy area in this equation was defined as the MAP2 immunoreactive zone.
Primary neuronal cultures and treatment.
Primary cultures of cortical neurons were dissected from embryonic day 17 fetal rats and maintained in Neurobasal A medium supplemented with B27 (Invitrogen), as previously described (Zhang et al., 2007). Experiments were performed 10 d after seeding. Oxygen glucose deprivation (OGD) was used to mimic in vitro ischemia with standard, previously published procedures (Zhang et al., 2007). Cortical neurons pretreated with EPA (10, 20, 40, 100 μm) or DHA (10, 20, 40, 100 μm) for 24 h were subjected to OGD for 60 min, and then returned to normal culture media supplied with the same concentration of DHA or EPA. To examine the impact of Nrf2 on n-3 PUFA-mediated protection against OGD, select neuronal cultures were infected with lentiviral particles bearing rat-specific Nrf2 or scramble shRNAs (Santa Cruz Biotechnology) for 3 d before DHA treatment and OGD. To examine the role of HO-1 in neuroprotection, select cultures were treated with 10 μm Sn-PPIX or vehicle 3 h after OGD. To detect the effects of electrophiles on HO-1 expression and cell viability, neurons were incubated with 4-HNE (5, 10, 20 μm) or 4-HHE (5, 10, 20 μm) for the indicated durations.
Cell death and viability.
Twenty-four hours after OGD, cell viability and death were quantitatively evaluated by Hoechst 33258 nuclear staining (Invitrogen), the live/dead cell viability assay (Invitrogen) or the lactate dehydrogenase (LDH) release assay. For Hoechst staining, the percentage of cells showing nuclear condensation was quantified and expressed as a function of total neurons (cell death percentage). The live/dead cell viability assay was performed according to the manufacturer's instructions (Invitrogen). In this assay, red dots (fluorescent ethidium homodimer-1) represent dying neurons with compromised membranes and green dots (fluorescent membrane-permeant calcein AM) represent live cells. The numbers of red dots were expressed as a function of red plus green dots and are presented as cell death percentage. For cell counting, three random fields were captured per well by a blinded observer. Four to six wells per condition per experiment were selected, and the experiments were repeated on three independent occasions. Extracellular LDH released from damaged cells was measured by an LDH detection kit (Pointe Scientific). All data were expressed as percentage of LDH release compared with the control group.
Western blots and Western slot blots.
Primary neurons or brain tissues were harvested at the indicated time points after ischemia or after 4-HHE and 4-HNE treatments (n = 4 per experimental condition). Whole-cell lysates were prepared as described previously (Wang et al., 2008). Nuclear and cytoplasmic fractions were extracted using an NE-PER nuclear and cytoplasmic extraction kit (Thermo Scientific) according to the manufacturer's instructions. Protein concentrations were determined by the bicinchoninic protein assay kit (Bio-Rad). Equal amounts of protein samples were loaded and probed with antibodies recognizing HO-1 (1:3000; Enzo Life Science), Nrf2 (1:1000; Enzo Life Science), Keap1 (1:1000; Abcam), β-actin (1:8000; Millipore), or histone (1:1000; Cell Signaling Technology). After incubating with secondary antibodies for 1 h, the membranes were incubated with LumiGLO solution (KPL). MCID imaging software was used for gel analysis by a blinded observer. For the Western slot blot, proteins were extracted from the brain tissues, and equivalent amounts of proteins were loaded and transferred to polyvinylidene difluoride membranes using a Bio-Dot Microfiltration apparatus (Bio-Rad). Anti-4-HHE (1:500; JaICA) or anti-4-HNE (1:1000, R&D) was used as the primary antibody. The remaining procedures were the same as for the Western blots.
Immunohistochemistry.
At the indicated time points, primary neurons were fixed with 4% paraformaldehyde for 10 min. Alternating sections from each experimental condition were exposed to all solutions except primary antibodies to verify lack of nonspecific staining. Mice were killed at 2 (for HO-1 immunolabeling) or 21 d (for MAP2 immunolabeling) after MCAO. DAPI was used as the nuclear counterstain in HO-1 stained sections. Following fixation, sections were cut at 5 μm thickness in the coronal plane. HO-1 (1:300; Enzo Life Science) or MAP2 (1:200; Santa Cruz Biotechnology) antibodies were applied and visualized with Cy3-conjugated secondary antibodies. The MAP2-positive area in the ipsilateral versus contralateral hemisphere was determined by a blinded observer using the MCID system and was used to represent tissue loss, as outlined above.
Nrf2 nuclear translocation assay.
EGFP-tagged human Nrf2 (pcDNA3-EGFP-C4-Nrf2, Addgene) was inserted into the lentiviral transfer vector FSW under the control of neuron-specific synapsin I promoter (Furukawa and Xiong, 2005; Stetler et al., 2008; Zhang et al., 2012). To detect whether lipid electrophiles induce nuclear translocation, neuronal cultures were infected for 3 d with lenti-Nrf2, and then treated with 4-HNE or 4-HHE (5 μm) for 1 h. The cultures were then counterstained with DAPI and photographed.
Statistical analysis.
Results are presented as mean ± SD, unless indicated otherwise. The difference between means was assessed by the Student's t test (single comparisons) or by ANOVA and post hoc Scheffe's tests (for multiple comparisons). A p value ≤0.05 was considered statistically significant.
Results
n-3 PUFAs alleviate acute brain injury induced by focal ischemia in fat-1 mice
To test our primary hypothesis that n-3 PUFAs protect the brain against ischemic injury, we generated transgenic mice carrying a fat-1 gene. Fat-1 was originally identified in Caenorhabditis elegans and encodes an n-3 desaturase that adds an extra double bond to n-6 fatty acids, thereby converting n-6 PUFAs into their corresponding n-3 PUFAs (Ge et al., 2002; Kang et al., 2004). The fat-1 cDNA was synthesized and constructed into a pST180 vector driven by a chicken β-actin promoter and a cytomegalovirus enhancer. Fat-1 heterozygotes were interbred with wild-type C57/B6 mice to generate fat-1 transgenic mice. Both fat-1 mice and their wild-type littermates were fed a regular diet rich in n-6 PUFA (n-6:n-3 ratio, 5:1; ProLab IsoPro RMH 3000, PMI).
Fatty acid analysis of forebrains was performed using gas chromatography. As shown in Figure 1A, fat-1 mice exhibited an increase in overall n-3/n-6 PUFA ratio. Individually, the contents of all major n-3 PUFAs were increased (Fig. 1A), including α-linoleic acid (ALA; 18:3 n-3), EPA (20:5 n-3), and DHA (22:6 n-3). These results indicate our success in generating fat-1 transgenic mice with altered brain fatty acid profiles.
Figure 1.
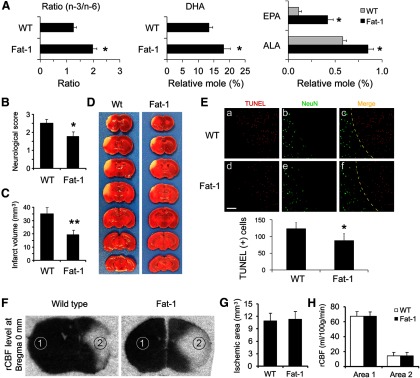
n-3 PUFA protects the brain against transient focal ischemia in mice. A, Fatty acid analysis of forebrains from wild-type (WT) and fat-1 transgenic mice. Compared with WT mice, fat-1 mice showed an increased total n-3/n-6 ratio (left), increased levels of DHA (middle), and increased levels of ALA and EPA. Data are presented as mean ± SD (n = 6; *p ≤ 0.01 vs WT). Focal brain ischemia was then induced by 60 min MCAO followed by 48 h reperfusion. Neurological function was examined at 48 h after ischemia. B, Fat-1 mice showed improved neurological function recovery after ischemia. C, Fat-1 mice had smaller infarct volumes compared with WT mice. Data are presented as mean ± SD (n = 9 per group, *p ≤ 0.05 and **p ≤ 0.01 vs WT by t test). D, Representative photographs of TTC-stained brain sections showing reduced infarct sizes in fat-1 mice. E, Representative microphotographs and counts of TUNEL-positive cells in the infarct areas 48 h after MCAO in WT and fat-1 mice, expressed as the number of cells per field of view (200×) in randomly selected regions. Scale bar, 100 μm. Yellow dotted lines in C and F indicate the boundary between infarct (upper right) and peri-infarct (lower left) zones (n = 6 per group; *p ≤ 0.05 vs WT littermates). F, Representative autoradiographs of C14-IAP at bregma 0 mm, showing rCBF changes in mouse brains during MCAO. Dark region represents normal blood flow, while light area represents reduced blood flow. Area 1, contralateral hemisphere; area 2, ischemic inner boundary; area 3, ischemic core. G, WT and fat-1 mice demonstrated similar ischemic areas during MCAO. Data are presented as mean ± SD, analyzed with t test (n = 6 per group). H, WT and fat-1 mice demonstrated similar levels of CBF reduction during MCAO. Data are presented as mean ± SD, and analyzed with ANOVA and post hoc tests.
We next determined whether n-3 PUFAs protect the brain against ischemia induced by 60 min MCAO and followed by 48 h reperfusion. As shown in Figure 1B, fat-1 mice exhibited less severe neurological deficits than their wild-type littermates. Furthermore, TTC staining showed that fat-1 mice also developed a significantly smaller infarct than wild-type mice (Fig. 1C,D). TUNEL-positive cells were significantly decreased in fat-1 mice compared with their wild-type littermates (Fig. 1E), suggesting a neuroprotective effect of n-3 PUFAs against ischemia. This protection was not dependent on rCBF changes, as wild-type and fat-1 mice demonstrated equivalent rCBF changes (Fig. 1F–H), which were measured by IAP autoradiography 15 min after the onset of MCAO (Sawada et al., 2000; Stetler et al., 2008). These data strongly indicate that n-3 PUFAs alleviate acute brain injury induced by focal ischemia in mice.
FO supplementation provides long-term functional and histological protection against brain ischemia in mice
Experiments using transgenic animals, such as fat-1 mice usually generate clear-cut and precise results; however, those results may not readily translate to humans. One practical way to increase n-3 PUFAs in humans is through ingestion of oily fish or FO capsules. Dietary supplementation can be mimicked in animal models to determine whether oral n-3 PUFAs also provide neuroprotection, especially over the long term.
To investigate whether dietary n-3 PUFAs confer similar protection as fat-1 transgene expression, we fed C57BL/6 mice with either a regular diet or FO-enriched food for 6 weeks, and analyzed their brain lipid profiles. As shown in Figure 2A, the overall n-3/n-6 ratio was significantly increased by FO dietary supplementation. DHA (Fig. 2B), ALA, and EPA levels (Fig. 2C) were also increased after feeding with FO, indicating that dietary supplementation is effective in increasing n-3 PUFA components in the brain. In a separate cohort of similarly fed animals, we subjected mice to 60 min MCAO and evaluated their sensorimotor function in the first or third week, followed by tissue loss analysis (Fig. 2D). Laser Doppler flowmetry was used to measure rCBF changes, which showed no significant differences between these two groups (data not shown). As shown in Figure 2E,F, intact mice in both the control and FO groups displayed no significant difference in sensorimotor function. One week following stroke, however, FO-fed mice demonstrated improved performance in the corner tests (Fig. 2E) and rotarod tests (Fig. 2F). We further evaluated cognitive function with the Morris water maze in another set of mice in the third week after ischemia. As shown in Figure 2G, FO-fed mice showed improved cognitive function, as indicated by reduced latency to find the submerged platform. At the end of the 21 d recovery period, MAP2 staining was used to determine the long-term protective effect of FO treatment. As expected from the behavioral assays, FO-fed mice also showed less tissue loss than control mice (Fig. 2H). Collectively, these data support our hypothesis that n-3 PUFAs provide potent and long-term neuroprotection against brain ischemia.
Figure 2.

FO treatment confers long-term neuroprotection against stroke in mice. Mice were fed with either a regular diet or an FO-enriched diet for 6 weeks. Their brains were collected and the lipid profiles of forebrains were analyzed. A–C, The overall n-3/n-6 ratio (A), DHA content (B), and ALA and EPA contents (C) were calculated, showing increased n-3 PUFA contents in the brains (n = 3, *p ≤ 0.05 vs regular diet mice). D, A diagram showing the timeline of FO feeding and the time points for MCAO and various assessments. E, F, The corner test (E) and rotarod test (F) show that FO-fed mice recovered sensorimotor function better than control mice (n = 6–8, *p ≤ 0.05 vs regular diet mice). G, Cognitive function in the Morris water maze during the third week after MCAO is presented as latency to escape the water bath. FO-fed mice recovered better than control mice (n = 5–6, *p ≤ 0.05 vs regular diet-fed mice). H, Representative photographs of MAP2-stained coronal sections and quantitative analyses of tissue loss at day 21 after MCAO (n = 6; data are presented as mean ± SD, *p ≤ 0.05 vs control).
n-3 PUFAs reduce ischemic neuronal injury induced by OGD in vitro
n-3 PUFAs are well established to reduce inflammatory cascades. Thus, their protective effects in vivo might involve interactions between multiple cell types, such as microglial-neuronal communication. Alternatively, n-3 PUFAs could also act directly on neurons in the absence of anti-inflammatory effects. To test the latter hypothesis, we determined whether n-3 PUFAs enhanced self-defense in pure primary cortical neuron cultures. The major long-chain n-3 PUFAs in cellular membranes are 18-carbon ALA, 20-carbon EPA, and 22-carbon DHA. Among them, DHA is the most abundant. We pretreated the neuronal cultures (purity, >97%) with DHA or EPA for 24 h and then challenged the cultures for 60 min with OGD, an in vitro model of ischemia. Twenty-four hours later, neuronal death was assessed with LDH release and cell counts using a live/dead viability/toxicity kit. Both DHA (Fig. 3A) and EPA (Fig. 3B) reduced LDH release in a concentration-dependent manner between 10 and 100 μm, indicating a neuroprotective effect. To extend these findings, we stained primary neurons with the live/dead assay. As shown in Figure 3C,D, both DHA and EPA reduced the percentage of dead neurons after OGD. It should be noted that higher concentrations of n-3 PUFAs (>250 μm) did not elicit neuroprotection and might be cytotoxic (data not shown), supporting previous reports (Chan and Fishman, 1978; Belayev et al., 2005). Collectively, these results indicate that n-3 PUFAs increase neuronal resistance against ischemic injury induced by OGD, and that this neuroprotection can occur in the absence of anti-inflammatory interactions between multiple cell types.
Figure 3.
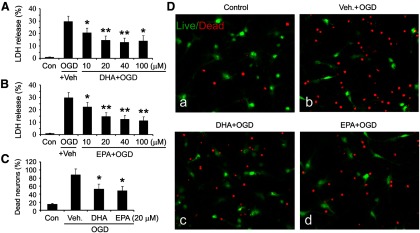
n-3 PUFA pretreatment reduces neuronal death after in vitro ischemia. A, B, DHA-treated (A) or EPA-treated (B) neuronal cultures were challenged with 60 min OGD; 24 h later, LDH release was measured. Data are presented as mean ± SE from three independent experiments, analyzed with ANOVA and post hoc tests (*p ≤ 0.05 and **p ≤ 0.01 vs OGD group). C, Neuronal cultures were treated with DHA or EPA, followed by OGD. Cultures were stained with a live/dead viability/toxicity kit 24 h later, and live and dead neurons were counted. Data are presented as the percentage of dead neurons as a function of total neuron number, and analyzed by χ2 tests (*p ≤ 0.05 vs OGD group). D, Representative micrographs of primary neuronal cultures 24 h after n-3 PUFA treatment and OGD. The cultures were stained with a live/dead kit; healthy neurons appear green and the condensed nuclei of dead neurons appear red.
HO-1 and Nrf2 are essential in DHA-mediated neuroprotection against ischemic neuronal injury
We next investigated the cytoprotective mechanisms of n-3 PUFAs against ischemic neuronal injury. Cellular self-defense is mediated by two types of mechanisms (Dinkova-Kostova and Talalay, 2008). The first line of defense consists of a group of small-molecule antioxidants. These are pre-existing, short-lived, and killed in the process of antioxidative activity. Therefore, small molecules need to be continuously resupplied or regenerated after use. Well-known examples include glutathione and vitamin E. In contrast, the second line of defense is offered by a group of enzymes that are minimally expressed under normal conditions but can be induced by cellular stress. Once produced, however, they are effective for longer periods, as they operate catalytically without being consumed. Examples include HO-1, glutathione-S-transferase, γ-glutamylcysteine synthetase, and NAD(P)H:quinone oxidoreductase 1 (Dinkova-Kostova and Talalay, 2008; Zhang et al., 2013). Among these enzymes, HO-1 is particularly noteworthy not only because it degrades toxic heme but also because it generates multiple neuroprotective molecules, including the antioxidant biliverdin (Deguchi et al., 2008) and carbon monoxide (Imuta et al., 2007).
We next tested the hypothesis that n-3 PUFAs facilitate the HO-1 response to ischemic injury. Neuronal cultures were pretreated with vehicle, DHA, or EPA (20 or 40 μm) for 24 h and then subjected to OGD. The neurons were harvested at 1, 6, or 24 h after OGD and total proteins were extracted for Western blot analysis. As shown in Figure 4A, HO-1 was expressed at trace levels in control cells and at 1 h after OGD. However, HO-1 was significantly increased by 6 h after OGD and peaked at 24 h. Notably, DHA or EPA alone did not upregulate HO-1 (Lanes 5 and 9), but only facilitated the HO-1 response to ischemia. DHA or EPA treatment augmented HO-1 expression at 6 and 24 h after OGD (Fig. 4A). To determine the maximally effective DHA concentration, we treated neuronal cultures with a range of concentrations of DHA for 24 h. Half of the cultures were then challenged with OGD. As shown in Figure 4B, n-3 PUFAs themselves did not upregulate HO-1 expression, but enhanced HO-1 expression after OGD in a concentration-dependent manner. Immunofluorescent staining confirmed that DHA enhanced HO-1 expression after OGD in neuronal cultures (Fig. 4C), suggesting that HO-1 may mediate the neuroprotective effects of n-3 PUFAs. To verify the role of HO-1 and Nrf2 in neuroprotection, we knocked down Nrf2 or inhibited HO-1 with Sn-PPIX, a specific inhibitor of HO-1 activity. As shown in Figure 4D,E, both Sn-PPIX and Nrf2 shRNA significantly blocked the neuroprotective effect of DHA. Finally, we also showed that Nrf2 knockdown significantly reduced neuronal HO-1 levels after DHA and OGD treatments (Fig. 4F), verifying that Nrf2 mediates the rise in HO-1 in this model. Together, these results strongly support the hypothesis that HO-1 and Nrf2 are essential in n-3 PUFA-mediated neuroprotection in vitro.
Figure 4.

HO-1 mediates the neuroprotective effects of n-3 PUFAs in vitro. Primary neuronal cultures were treated with n-3 PUFAs and harvested for Western blotting or fixed for Hoechst staining. A, Time course of HO-1 expression. Representative Western blots and semiquantitative analyses of HO-1 levels after n-3 PUFA and OGD treatments (*p ≤ 0.05 and **p ≤ 0.01 vs control and vehicle-treated OGD groups at the same time points; #p ≤ 0.05 and ##p ≤ 0.01 vs control group. B, Dose–response data related to HO-1 expression. Representative Western blots and semiquantitative analyses of HO-1 levels after n-3 PUFA treatment, with or without OGD (**p ≤ 0.01 vs control, ##p ≤ 0.01 vs control and OGD groups). C, Representative micrographs of HO-1 immunofluorescence in neuronal cultures. HO-1 was immunostained red with Cy3 and nuclei were stained blue with Hoechst. D, E, Quantitative analysis (D) and representative micrographs (E) showed that knockdown of Nrf2 or inhibition of HO-1 activity with 10 μm Sn-PPIX diminishes the neuroprotective effect of n-3 PUFAs (*p ≤ 0.05 vs control group, #p ≤ 0.05 vs OGD group). F, Nrf2 knockdown reduces neuronal HO-1 levels. Primary neurons were infected with Lenti-scramble or Lenti-Nrf2 shRNAs for 3 d, followed by DHA and OGD treatments. HO-1 levels were detected 6 h after OGD using Western blot (p ≤ 0.05 vs control group, and #p ≤ 0.05 vs lentiviral groups).
DHA activates the Nrf2 pathway after OGD
Nrf2 is the major transcription factor responsible for the upregulation of HO-1 and other phase 2 enzymes. To investigate whether n-3 PUFAs enhance the nuclear translocation of Nrf2, we treated neuronal cultures with either vehicle or DHA followed by OGD, and then extracted the nuclear fraction. Western blots showed that there was only a trace level of Nrf2 in nuclei under normal conditions. However, nuclear Nrf2 increased at 6 h after OGD and subsided by 24 h (Fig. 5A,B). In DHA-treated groups, Nrf2 nuclear translocation was significantly increased, especially at 6 and 24 h after OGD (Fig. 5A,B). The cytosolic levels of Nrf2 and of Keap1, the binding partner of Nrf2, remained stable. These data suggest that n-3 PUFAs enhance the nuclear translocation of Nrf2 after ischemia. We then performed an electrophoretic mobility shift assay to detect whether DHA increases the DNA binding activity of Nrf2 after OGD in neurons. As shown in Figure 5C,D, DHA treatment significantly increased the antioxidant response element binding activity of Nrf2 at 6 and 24 h after OGD compared with vehicle groups. The binding was specific, as the competition assay showed that adding cold probes inhibited the DNA binding of Nrf2.
Figure 5.

DHA enhances the nuclear translocation and DNA binding activity of Nrf2 in neurons. Primary neuronal cultures were treated with vehicle or DHA followed by OGD. A, B, Representative Western blots (A) and semiquantitative analyses (B) of Nrf2 levels, showing increased nuclear Nrf2 in DHA-treated groups (*p ≤ 0.05 vs control and vehicle-treated OGD groups at the same time points; #p ≤ 0.05 vs control groups, n = 3 per condition). C, D, Representative electrophoretic mobility shift assay (C) and semiquantitative analyses (D) of DNA binding of Nrf2 (*p ≤ 0.05 vs control and vehicle-treated OGD groups at the same time points; #p ≤ 0.05 vs control groups; and &p ≤ 0.01 vs DHA-treated OGD at 24 h; n = 3 per condition). The specificity of DNA binding activity was determined by a competition assay, whereas threefold (C1) or 50-fold (C2) excess of cold probe was added as a specific competitor.
HO-1 plays a critical role in n-3 PUFA-mediated neuroprotection against MCAO in mice
To investigate whether HO-1 also plays a similar protective role in vivo, we fed mice with standard or FO-enriched diets for 6 weeks, subjected them to 60 min MCAO, and harvested the MCA territory at 4, 24, or 72 h after ischemia for Western blotting. As shown in Figure 6A, HO-1 was expressed at trace levels in the sham-operated group but began to rise 4 h after ischemia, peaking at 72 h. As expected, FO treatment significantly augmented HO-1 expression. Immunohistochemistry revealed that HO-1 was barely detectable in the sham group (data not shown). One day after ischemia (Fig. 6B, top), however, HO-1 was moderately expressed in neurons and Iba1-positive cells in the ischemic core, and was barely expressed in astrocytes. In the peri-infarct regions, HO-1was also weakly expressed in neurons and Iba1-positive cells, and barely expressed in astrocytes. At day 3 (Fig. 6B, bottom), HO-1 was strongly and exclusively expressed in Iba1-postive cells in the core, indicating a probable macrophage origin. In the peri-infarct regions, HO-1 was barely expressed in neurons, mildly expressed in microglia, and strongly expressed in some but not all astrocytes. FO increased HO-1 levels but did not change its pattern or cellular distribution after MCAO. To detect whether HO-1 contributes to the neuroprotective effects of FO treatment, we subjected mice to MCAO and injected 30 μg/5 μl of Sn-PPIX into the cerebral ventricles 3 h after ischemia and evaluated ischemic outcomes 24 h later. As shown in Figure 6C, FO-fed mice exhibited improved neurological functions and reductions in infarct volume compared with control mice (Fig. 6D,E). Furthermore, Sn-PPIX partially blocked the beneficial effects of FO on ischemia. Together, these data show that n-3 PUFAs boost HO-1 expression after ischemia, and that this rise in HO-1 partially mediates the neuroprotective effects of n-3 PUFAs against ischemic brain injury in vivo.
Figure 6.
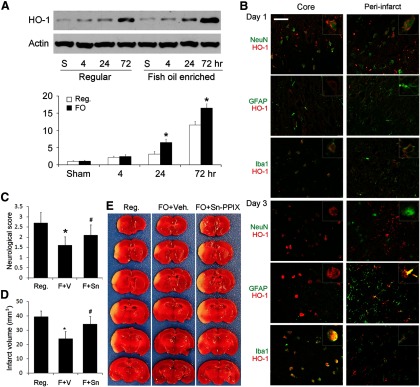
FO protects the brain against focal ischemia via HO-1. Mice were fed with a regular diet (Reg.) or FO-enhanced diet for 6 weeks followed by 60 min MCAO. A, Representative Western blots and semiquantitative analyses of HO-1 levels in mouse brain after ischemia, showing increased HO-1 levels in the FO group (n = 3, *p ≤ 0.05 vs sham and regular diet at the same time points). B, Representative microphotographs of HO-1 expression and cellular distribution in the ischemic core and surrounding regions at 1 or 3 d after ischemia. HO-1 was stained red and cellular markers were stained green. Scale bar, 50 μm. Insets show high-power images of representative cells. C–E, Neurological score (C), infarct volumes (D), and representative TTC staining (E) at 48 h after MCAO, showing that FO treatment reduced neurological dysfunction and infarct volumes and that protection was partially blocked by Sn-PPIX, a HO-1 activity inhibitor (n = 6, *p ≤ 0.05 vs Reg., #p ≤ 0.05 vs FO mice injected with vehicle).
To determine whether postischemic treatment with n-3 PUFAs can also upregulate HO-1 and offer neuroprotection in wild-type mice, we injected DHA (50 μmol/kg) subcutaneously at the onset of reperfusion, and harvested the brain tissues for Western blots. As showed in Figure 7A,B, HO-1 levels in the DHA group were higher than in the vehicle group. In another set of mice, we performed neurological examinations and TTC staining 48 h after MCAO. As expected, DHA treatment significantly reduced neurological dysfunctions (Fig. 7C) and infarct volume (Fig. 7D,E) compared with the vehicle group. Intracerebroventricular injection of Sn-PPIX partially blocked the protective effects of DHA (Fig. 7C–E). Together, these data indicate that postischemic DHA treatment also alleviates acute brain injury induced by focal ischemia in mice, and that the protective mechanisms also involve HO-1 upregulation.
Figure 7.

Postischemic DHA treatment protects the brain against focal ischemia. MCAO was induced in C57BL/6 mice and DHA was subcutaneously injected at the onset of reperfusion. A, B, Representative Western blots (A) and semiquantitative analyses (B) of HO-1 levels in mouse brain after ischemia, showing increased HO-1 levels in the DNA group (n = 3, *p ≤ 0.05 vs sham and vehicle group at the same time points). S, Sham-operated; V, vehicle; D, DHA. C–E, Neurological score (C), infarct volumes (D), and representative TTC staining (E) at 48 h after MCAO, showing that DHA treatment reduced neurological dysfunction and infarct volumes and that the protection was partially blocked by Sn-PPIX (n = 8, *p ≤ 0.05 vs vehicle, #p ≤ 0.05 vs DHA group).
4-HHE is a strong inducer of the Nrf2 pathway
Although the above findings show that n-3 PUFAs enhance Nrf2 activation and HO-1 upregulation after brain ischemia, the specific compounds responsible for HO-1 induction are unknown. According to Holtzclaw et al. (2004), Nrf2 inducers can be classified into 10 categories based on their chemical structures; among these, the only category that can be generated endogenously are the Michael reaction acceptors, also known as electrophiles. Endogenous electrophiles belong to a group of chemicals known as α,β-unsaturated carbonyl compounds, which are primarily lipid peroxidation products (Satoh et al., 2006; LoPachin et al., 2009b). Known Nrf2 inducers include 4-HNE from n-6 PUFA (Pryor and Porter, 1990; Esterbauer et al., 1991) and 4-HHE from n-3 PUFA (Guichardant et al., 2004; Long and Picklo, 2010). Both are highly reactive with cysteine thiol groups. Electrophiles derived from n-3 and n-6 PUFA differ in their anti-inflammatory and antioxidative effects, with n-3 PUFA derivatives having preferable effects (Bagga et al., 2003; van Beelen et al., 2006; Schmitz and Ecker, 2008). We hypothesized that endogenous lipid electrophiles are generated via lipid peroxidation in the brain after ischemia and that n-3 PUFA-derived 4-HHE is a stronger Nrf2 inducer than n-6 PUFA-derived 4-HNE. To test this hypothesis, we first calculated the orbital energies and eletrophilicity index of 4-HNE and 4-HHE using Spartan ‘08 software (Wavefunction), and found that 4-HHE has a higher eletrophilicity index than 4-HNE (Table 1). To test whether 4-HHE induces stronger HO-1 expression than 4-HNE, we treated primary neurons with 4-HHE and 4-HNE for 0.5 to 2 h, harvested the cells, and extracted both nuclear and cytosolic proteins. As shown by Western blot in Figure 8A,B, HO-1 induction by 4-HHE was more rapid and robust than that induced by 4-HNE. Four-HHE also increased the nuclear translocation of Nrf2 more than 4-HNE (Fig. 8A,C,D), indicating that 4-HHE is a more potent inducer of Nrf2 activation. To further test whether 4-HHE offers neuroprotection against OGD, we incubated cultured neurons with either 4-HHE or 4-HNE for 2 h, and then challenged the neurons with OGD on the following day. Hoechst staining (Fig. 8E) and cell counts (Fig. 8F) showed that 4-HHE treatment significantly reduced the number of dead neurons after OGD compared with the vehicle and 4-HNE groups, suggesting that 4-HHE mediates the protective effects of n-3 PUFAs.
Figure 8.

Four-HHE is a potent Nrf2 activator. Primary cultures were incubated with 4-HHE (n-3) or 4-HNE (n-6) at a concentration of 10 μm for the indicated duration. A, Representative Western blots of cytosolic HO-1 and nuclear Nrf2. B, C, Quantitative analyses of HO-1 (B) and Nrf2 (C) levels indicated that lipid electrophiles can directly induce Nrf2 nuclear translocation and HO-1 expression, and that 4-HHE (n-3) is more potent than 4-HNE (n-6) in inducing Nrf2 activation. Data are presented as mean ± SE, and analyzed with ANOVA and post hoc tests (*p ≤ 0.05 vs 4-HNE-treated group at the same time point). D, Representative microphotographs of Nrf2-EGFP transfected neuronal cultures, showing that 4-HHE was a stronger inducer of Nrf2 than 4-HNE. Primary neurons were infected with lenti-Nrf2-EGFP for 3 d followed by treatment with 4-HHE and 4-HNE (10 μm) for 1 h. Cells were then fixed and counterstained with DAPI (blue). E, F, Representative Hoechst staining (E) and dead cell counts (F), showing that 4-HHE pretreatment reduced neuronal death. Primary neurons were treated with vehicle, 4-HNE, and 4-HHE (10 μm) for 2 h. After overnight recovery, neurons were challenged with OGD for 60 min and then stained with Hoechst (p ≤ 0.05 vs control, vehicle-treated, and 4-HNE-treated OGD groups.
Lipid electrophiles (4-HHE and 4-HNE) are generated in mouse brains after MCAO
We then investigated whether lipid electrophiles (4-HNE and 4-HHE) are produced in the brain and whether FO treatment leads to increased generation of 4-HHE. As endogenous electrophiles readily form adducts with nucleophiles, they rarely exist as free forms but commonly as adducted forms. We therefore measured the levels of total proteins that were modified by either 4-HNE or 4-HHE using Western slot blotting. Six weeks after ingestion of regular or FO-enhanced diets, mice were subjected to MCAO. Brain tissues were harvested 4 or 24 h after ischemia and protein samples were subjected to slot blot. As shown in Figure 9, only trace levels of 4-HNE-modified and 4-HHE-modified proteins were detected in the sham group. Following stroke, the levels of 4-HNE-modified and 4-HHE-modified proteins were significantly increased, especially at 24 h after MCAO, indicating that both 4-HNE and 4-HHE are both generated after cerebral ischemia. Compared with animals on a regular diet, FO treatment significantly augmented the rise in 4-HHE-modified proteins after ischemia (Fig. 9). These results suggest that FO enhances 4-HHE generation in mouse brain after brain ischemia.
Figure 9.

FO treatment increases 4-HHE levels in mouse brains after MCAO. Mice were fed with a regular diet or an FO-enhanced diet for 6 weeks followed by 60 min MCAO. At the indicated time points, brain tissues were harvested and subjected to Western slot blots. A, B, Representative slot blots (A) and semiquantitative analyses (B) of the levels of 4-HHE modified proteins, showing increased 4-HHE levels in FO-treated groups (n = 3; **p ≤ 0.01 and *p ≤ 0.05 vs control and regular diet groups at the same time points). C, D, Representative slot blots (C) and semiquantitative analyses (D) of the levels of 4-HHE modified proteins, showing decreased 4-HNE levels in FO-treated groups (n = 3; **p ≤ 0.01 and *p ≤ 0.05 vs control and regular diet groups at the same time points).
Discussion
The present study demonstrates that n-3 PUFAs elicit robust and prolonged neuroprotection against ischemic brain injury in fat-1 transgenic mice or in C57/B6 mice fed with an FO-enhanced diet. DHA treatment increases Nrf2 nuclear translocation and HO-1 expression, and thus preserves neuronal viability after OGD. Importantly, inhibition of HO-1 with Sn-PPIX significantly attenuates neuroprotection in vitro and in vivo, strongly suggesting a mechanistic role for this enzyme. Nrf2 activation and HO-1 upregulation are mediated by 4-HHE, an n-3 PUFA oxidative product. These findings support our hypothesis that n-3 PUFAs protect the brain against ischemic injury by oxidizing to 4-HHE, activating Nrf2, and upregulating HO-1 expression (Fig. 10).
Figure 10.

Potential mechanism of n-3 PUFA-mediated neuroprotection. 1, Under physiological conditions, Nrf2 is physically “locked” with Keap1, which leads to proteasomal degradation of Nrf2 via the Cul3 (E3)-dependent pathway. 2, Under oxidative stress conditions, such as brain ischemia and reperfusion, lipid oxidation produces α,β-unsaturated carbonyl electrophiles. n-3 PUFA-derived 4-HHE is more powerful than n-6 PUFA-derived 4-HNE in electrophilicity. 3, Four-HHE covalently reacts with cysteine residues of Keap1, which causes conformational changes in Keap1, setting Nrf2 free. 4, Nrf2 then accumulates, translocates into the nucleus, and binds the antioxidant response element (ARE) of phase 2 genes, leading to upregulation of HO-1 and other enzymes. 5, HO-1 protects the brain against ischemic damage by breaking down heme into biliverdin (antioxidative) and carbon monoxide (CO; anti-inflammatory), reducing calcium overload and positive feedback on HO-1 expression. C, Cysteines; Cul3, cullin3; PLA2, phospholipase A2; Ub, ubiquitin.
Neuroprotective effects of n-3 PUFAs against ischemic brain injury have been shown in several stroke models (Black et al., 1979; Marcheselli et al., 2003; Akbar et al., 2005; Belayev et al., 2005, 2009; Moreira et al., 2010; Zhang et al., 2010). These studies focused on the acute phase of ischemia assessed from 24 h to 7 d postinjury in focal ischemic stroke models. We have expanded our study to 21 d postinjury. Our data therefore demonstrate that oral administration of FO for 6 weeks before ischemia improved outcomes for the long term. Demonstrating the long-term efficacy of FO is critical for the translation of our findings to human studies (Eady et al., 2012). It is also important to note here that oral intake of commercially available FO is safe, inexpensive, and convenient.
The protective mechanisms of n-3 PUFAs are not fully understood, although anti-inflammatory effects and Akt activation have been reported to be contributing factors. For example, n-3 PUFAs suppress microglial activation, reduce proinflammatory cytokine release, and inhibit neutrophil infiltration after stroke (Mayer et al., 2002; Marcheselli et al., 2003; Belayev et al., 2005; Musiek et al., 2008; Zhang et al., 2010). It has also been reported that n-3 PUFAs increased the phosphorylation of Akt after ischemia by regulating phosphatidylserine (Akbar et al., 2005) or neuroprotectin D1 (Eady et al., 2012). However, these factors may not comprise the entire extent of protective mechanisms because DHA is still protective in single cell-type cultures (Akbar et al., 2005; Begum et al., 2012) and the involvement of Akt in n-3 PUFA-mediated protection after ischemia is controversial (Moreira et al., 2010). Thus, it was not clear before our studies whether n-3 PUFAs participate in direct cellular protection. To fully understand the protective mechanisms, it is first necessary to recognize basic characteristics of n-3 PUFAs and the key differences between n-3 PUFAs and n-6 PUFAs, as discussed below.
The dominant human dietary fatty acids are n-6, not n-3, PUFAs (Schmitz and Ecker, 2008). The only structural difference between them is that there is an extra carbon–carbon double bond between the third and fourth carbons of n-3 PUFAs. However, this small difference confers two distinct features to n-3 PUFAs. First, it makes n-3 PUFAs shorter and the cell membranes thinner, thereby increasing membrane fluidity and flexibility (Eldho et al., 2003; Schmitz and Ecker, 2008). Fluidity and flexibility are essential for highly curved or coiled membranes, such as those in the synapses of neurons and rhodopsin disks of the retina, which contain ∼35 and 50% DHA, respectively (Salem et al., 2001; Das, 2006), making the CNS rich in n-3 PUFAs. The second feature of n-3 PUFAs is their increased susceptibility to free radical attack (Roberts et al., 1998; Fam et al., 2002; Gao et al., 2007; Yin et al., 2007; Brooks et al., 2008), because the unsaturated double-carbon bonds in PUFAs are extremely vulnerable and n-3 PUFAs have one more double-carbon bond than n-6 PUFAs (Valentine and Valentine, 2004). For example, 3.4-fold more oxidized product was generated from DHA compared with n-6 arachidonic acid in a cell-free assay (Roberts et al., 1998).
Once oxidized, PUFAs undergo enzymatic or nonenzymatic chain reactions, eventually generating end products of lipid peroxidation over a series of intermediate products. For example, the specific end product of n-3 PUFAs is 4-HHE, whereas the corresponding n-6 PUFA derivative is 4-HNE (Table 1). Four-HHE and 4-HNE are both aldehydes, relatively stable, and high in concentration compared with intermediates of lipid peroxidation; therefore, they may exert more biological effects than their intermediates. Their biological effects are determined by their electrophilicities and concentrations. Reactive oxygen species (ROS) and their lipid peroxidation products are classically considered to be detrimental; however, recent studies suggest that both also play important roles in cellular signaling cascades (Yang et al., 2003; Levonen et al., 2004; Lopachin et al., 2009a). This is especially true to the electrophiles (Satoh and Lipton, 2007; Uchida, 2007; Rudolph and Freeman, 2009; Bindoli and Rigobello, 2013). FO or n-3 PUFAs may enhance this redox signaling because n-3 PUFAs are more susceptible to ROS and 4-HHE is a stronger electrophile than 4-HNE. In support of this notion, our data also indicate that 4-HHE is a potent inducer of Nrf2 activation in neurons, and that more 4-HHE is generated in FO-fed mice. Thus 4-HHE may mediate the neuroprotective effects of n-3 PUFAs against ischemic brain injury. In addition to neuronal protection, protective effects of n-3 PUFAs have also been reported in other cell types, such as adipocytes (Kusunoki et al., 2013), liver cells (van Beelen et al., 2006), and mammary cells (Gao et al., 2007). These protective effects are also thought to occur via oxidation products, subsequent Nrf2 activation, and phase 2 enzyme upregulation, suggesting that these mechanisms commonly underlie n-3 PUFA-mediated protection.
The neuroprotective effects of Nrf2 (Shih et al., 2005; Shah et al., 2007) and HO-1 (Panahian et al., 1999; Shah et al., 2011) against ischemia are well recognized. Nrf2 has a short half-life and exists at low basal levels under normal conditions because it is degraded after binding Keap1 (Itoh et al., 1999). Keap1 is rich in cysteine, and its Cys-151 and Cys-275 play important roles in degrading Nrf2 (Kobayashi et al., 2004; Zhang et al., 2013). A recent study shows that 4-HNE interacts with Cys-151 of Keap1 via Michael addition (McMahon et al., 2010), leading to Nrf2 activation. Four-HHE may function in a similar manner, though direct evidence for this is not yet available. As a target gene of Nrf2, HO-1 is also protective against brain ischemia, as HO-1 knock-out in mice exacerbates infarcts (Shah et al., 2011), whereas HO-1 overexpression reduces infarcts (Panahian et al., 1999; Zhang et al., 2012). In supporting previous reports (Nimura et al., 1996; Fu et al., 2006), we also observed moderate HO-1 expression in neurons early after ischemia in the core and penumbra. In addition to degrading heme, this neuronal HO-1 may also protect by suppressing superoxide production and maintaining nitric oxide bioavailability (Chao et al., 2013). The strongest HO-1 signal was observed on day 3 in macrophages in the ischemic core and in astrocytes in the peri-infarct zone. Their role in neuroprotection is not clear. However, it is possible that Nrf2 and HO-1 contribute to glial self-defense, as well as to the clearance of cellular debris (Manoonkitiwongsa et al., 2001), the neuroplasticity of macrophages (Michael-Titus and Priestley, 2013), and the generation of glial cell line-derived neurotrophic factor and glutathione for astrocytes (Shin et al., 2012). All of these features may contribute to long-term protection observed in the present study. Further investigation will clarify their precise roles in neuroprotection.
In summary, our results reveal a novel neuroprotective mechanism underlying n-3 PUFA-mediated protection in the brain (Fig. 10). The present study is the first demonstration of the mechanistic involvement of 4-HHE and HO-1 in n-3 PUFA-mediated neuroprotection in the ischemic brain. Thus, further research into the activation of the Nrf2 pathway by n-3 PUFAs is justified. Our study raises several novel questions. For example, the precise amount of 4-HHE generated in the n-3 PUFA-treated ischemic mouse brain is not known. It should also be noted that Nrf2/HO-1 may not be the only mechanism through which n-3 PUFAs and lipid electrophiles protect cells, as lipid electrophiles may also activate (Sethi et al., 2002) or inhibit additional signaling pathways (Zhang et al., 2010; Shearn et al., 2011). This hypothesis is also consistent with our findings that HO-1 inhibition only partially attenuates n-3 PUFA-mediated protection in vivo. Therefore, n-3 PUFAs may elicit long-term protection through additional pathways other than Nrf2/HO-1. Further clarification of these issues may help develop new dietary strategies for stroke treatment.
Footnotes
This work was supported by grants from the National Institutes of Health/National Institute of Neurological Disorders and Stroke (NS36736, NS43802, and NS45048 to J.C.), the American Heart Association (10SDG2560122 to F.Z.), the Natural Science Foundation of China (81228008 to J.C.; 81271276 to F.Z.; 81271275 and 81070947 to B.S.; 81020108021, 81171149, and 81371306 to Y.G.), the Doctoral Fund of Ministry of Education of China (20120071110042 to J.C.), and the Natural Science Foundation of Shandong (Y2007C014 to B.S.). We thank Dr. Richard M. LoPachin (Albert Einstein College of Medicine) for calculating the electrophilicity indexes of 4-HNE and 4-HHE. We also thank Carol Culver for editorial assistance and Pat Strickler for secretarial support.
The authors declare no competing financial interests.
References
- Akbar M, Calderon F, Wen Z, Kim HY. Docosahexaenoic acid: a positive modulator of Akt signaling in neuronal survival. Proc Natl Acad Sci U S A. 2005;102:10858–10863. doi: 10.1073/pnas.0502903102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagga D, Wang L, Farias-Eisner R, Glaspy JA, Reddy ST. Differential effects of prostaglandin derived from omega-6 and omega-3 polyunsaturated fatty acids on COX-2 expression and IL-6 secretion. Proc Natl Acad Sci U S A. 2003;100:1751–1756. doi: 10.1073/pnas.0334211100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan NG. Neuroprotectin D1 (NPD1): a DHA-derived mediator that protects brain and retina against cell injury-induced oxidative stress. Brain Pathol. 2005;15:159–166. doi: 10.1111/j.1750-3639.2005.tb00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum G, Kintner D, Liu Y, Cramer SW, Sun D. DHA inhibits ER Ca2+ release and ER stress in astrocytes following in vitro ischemia. J Neurochem. 2012;120:622–630. doi: 10.1111/j.1471-4159.2011.07606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belayev L, Marcheselli VL, Khoutorova L, Rodriguez de Turco EB, Busto R, Ginsberg MD, Bazan NG. Docosahexaenoic acid complexed to albumin elicits high-grade ischemic neuroprotection. Stroke. 2005;36:118–123. doi: 10.1161/01.STR.0000149620.74770.2e. [DOI] [PubMed] [Google Scholar]
- Belayev L, Khoutorova L, Atkins KD, Bazan NG. Robust docosahexaenoic acid-mediated neuroprotection in a rat model of transient, focal cerebral ischemia. Stroke. 2009;40:3121–3126. doi: 10.1161/STROKEAHA.109.555979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindoli A, Rigobello MP. Principles in redox signaling: from chemistry to functional significance. Antioxid Redox Signal. 2013;18:1557–1593. doi: 10.1089/ars.2012.4655. [DOI] [PubMed] [Google Scholar]
- Black KL, Culp B, Madison D, Randall OS, Lands WE. The protective effects of dietary fish oil on focal cerebral infarction. Prostaglandins Med. 1979;3:257–268. doi: 10.1016/0161-4630(79)90067-3. [DOI] [PubMed] [Google Scholar]
- Brooks JD, Milne GL, Yin H, Sanchez SC, Porter NA, Morrow JD. Formation of highly reactive cyclopentenone isoprostane compounds (A3/J3-isoprostanes) in vivo from eicosapentaenoic acid. J Biol Chem. 2008;283:12043–12055. doi: 10.1074/jbc.M800122200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PH, Fishman RA. Brain edema: induction in cortical slices by polyunsaturated fatty acids. Science. 1978;201:358–360. doi: 10.1126/science.663662. [DOI] [PubMed] [Google Scholar]
- Chao XD, Ma YH, Luo P, Cao L, Lau WB, Zhao BC, Han F, Liu W, Ning WD, Su N, Zhang L, Zhu J, Fei Z, Qu Y. Up-regulation of heme oxygenase-1 attenuates brain damage after cerebral ischemia via simultaneous inhibition of superoxide production and preservation of NO bioavailability. Exp Neurol. 2013;239:163–169. doi: 10.1016/j.expneurol.2012.09.020. [DOI] [PubMed] [Google Scholar]
- Das UN. Essential fatty acids: biochemistry, physiology and pathology. Biotechnol J. 2006;1:420–439. doi: 10.1002/biot.200600012. [DOI] [PubMed] [Google Scholar]
- Deguchi K, Hayashi T, Nagotani S, Sehara Y, Zhang H, Tsuchiya A, Ohta Y, Tomiyama K, Morimoto N, Miyazaki M, Huh NH, Nakao A, Kamiya T, Abe K. Reduction of cerebral infarction in rats by biliverdin associated with amelioration of oxidative stress. Brain Res. 2008;1188:1–8. doi: 10.1016/j.brainres.2007.07.104. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Talalay P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res 52 [Suppl] 2008;1:S128–S138. doi: 10.1002/mnfr.200700195. [DOI] [PubMed] [Google Scholar]
- Eady TN, Belayev L, Khoutorova L, Atkins KD, Zhang C, Bazan NG. Docosahexaenoic acid signaling modulates cell survival in experimental ischemic stroke penumbra and initiates long-term repair in young and aged rats. PLoS One. 2012;7:e46151. doi: 10.1371/journal.pone.0046151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldho NV, Feller SE, Tristram-Nagle S, Polozov IV, Gawrisch K. Polyunsaturated docosahexaenoic vs docosapentaenoic acid-differences in lipid matrix properties from the loss of one double bond. J Am Chem Soc. 2003;125:6409–6421. doi: 10.1021/ja029029o. [DOI] [PubMed] [Google Scholar]
- Esterbauer H, Zollner H, Scholz N. Reaction of glutathione with conjugated carbonyls. Z Naturforsch C. 1975;30:466–473. doi: 10.1515/znc-1975-7-808. [DOI] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- Fam SS, Murphey LJ, Terry ES, Zackert WE, Chen Y, Gao L, Pandalai S, Milne GL, Roberts LJ, Porter NA, Montine TJ, Morrow JD. Formation of highly reactive A-ring and J-ring isoprostane-like compounds (A4/J4-neuroprostanes) in vivo from docosahexaenoic acid. J Biol Chem. 2002;277:36076–36084. doi: 10.1074/jbc.M205638200. [DOI] [PubMed] [Google Scholar]
- Fernandes MX, Castanho MA, de la Torre JG. Brownian dynamics simulation of the unsaturated lipidic molecules oleic and docosahexaenoic acid confined in a cellular membrane. Biochim Biophys Acta. 2002;1565:29–35. doi: 10.1016/S0005-2736(02)00501-1. [DOI] [PubMed] [Google Scholar]
- Fu R, Zhao ZQ, Zhao HY, Zhao JS, Zhu XL. Expression of heme oxygenase-1 protein and messenger RNA in permanent cerebral ischemia in rats. Neurol Res. 2006;28:38–45. doi: 10.1179/016164106X91852. [DOI] [PubMed] [Google Scholar]
- Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Wang J, Sekhar KR, Yin H, Yared NF, Schneider SN, Sasi S, Dalton TP, Anderson ME, Chan JY, Morrow JD, Freeman ML. Novel n-3 fatty acid oxidation products activate Nrf2 by destabilizing the association between Keap1 and Cullin3. J Biol Chem. 2007;282:2529–2537. doi: 10.1074/jbc.M607622200. [DOI] [PubMed] [Google Scholar]
- Ge Y, Wang X, Chen Z, Landman N, Lo EH, Kang JX. Gene transfer of the Caenorhabditis elegans n-3 fatty acid desaturase inhibits neuronal apoptosis. J Neurochem. 2002;82:1360–1366. doi: 10.1046/j.1471-4159.2002.01077.x. [DOI] [PubMed] [Google Scholar]
- Guichardant M, Chantegrel B, Deshayes C, Doutheau A, Moliere P, Lagarde M. Specific markers of lipid peroxidation issued from n-3 and n-6 fatty acids. Biochem Soc Trans. 2004;32:139–140. doi: 10.1042/BST0320139. [DOI] [PubMed] [Google Scholar]
- He K, Song Y, Daviglus ML, Liu K, Van Horn L, Dyer AR, Goldbourt U, Greenland P. Fish consumption and incidence of stroke: a meta-analysis of cohort studies. Stroke. 2004;35:1538–1542. doi: 10.1161/01.STR.0000130856.31468.47. [DOI] [PubMed] [Google Scholar]
- Holtzclaw WD, Dinkova-Kostova AT, Talalay P. Protection against electrophile and oxidative stress by induction of phase 2 genes: the quest for the elusive sensor that responds to inducers. Adv Enzyme Regul. 2004;44:335–367. doi: 10.1016/j.advenzreg.2003.11.013. [DOI] [PubMed] [Google Scholar]
- Imuta N, Hori O, Kitao Y, Tabata Y, Yoshimoto T, Matsuyama T, Ogawa S. Hypoxia-mediated induction of heme oxygenase type I and carbon monoxide release from astrocytes protects nearby cerebral neurons from hypoxia-mediated apoptosis. Antioxid Redox Signal. 2007;9:543–552. doi: 10.1089/ars.2006.1519. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JX, Wang J, Wu L, Kang ZB. Transgenic mice: fat-1 mice convert n-6 to n-3 fatty acids. Nature. 2004;427:504. doi: 10.1038/427504a. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusunoki C, Yang L, Yoshizaki T, Nakagawa F, Ishikado A, Kondo M, Morino K, Sekine O, Ugi S, Nishio Y, Kashiwagi A, Maegawa H. Omega-3 polyunsaturated fatty acid has an anti-oxidant effect via the Nrf-2/HO-1 pathway in 3T3–L1 adipocytes. Biochem Biophys Res Commun. 2013;430:225–230. doi: 10.1016/j.bbrc.2012.10.115. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Julien C, Cordeau P, Bohacek I, Weng YC, Calon F, Kriz J. Accumulation of dietary docosahexaenoic acid in the brain attenuates acute immune response and development of postischemic neuronal damage. Stroke. 2011;42:2903–2909. doi: 10.1161/STROKEAHA.111.620856. [DOI] [PubMed] [Google Scholar]
- Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD, Darley-Usmar VM. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochemical J. 2004;378:373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long EK, Picklo MJ., Sr Trans-4-hydroxy-2-hexenal, a product of n-3 fatty acid peroxidation: make some room HNE. Free Radic Biol Med. 2010;49:1–8. doi: 10.1016/j.freeradbiomed.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Lopachin RM, Geohagen BC, Gavin T. Synaptosomal toxicity and nucleophilic targets of 4-hydroxy-2-nonenal. Toxicol Sci. 2009a;107:171–181. doi: 10.1093/toxsci/kfn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T, Petersen DR, Barber DS. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chem Res Toxicol. 2009b;22:1499–1508. doi: 10.1021/tx900147g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majkova Z, Layne J, Sunkara M, Morris AJ, Toborek M, Hennig B. Omega-3 fatty acid oxidation products prevent vascular endothelial cell activation by coplanar polychlorinated biphenyls. Toxicol Appl Pharmacol. 2011;251:41–49. doi: 10.1016/j.taap.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoonkitiwongsa PS, Jackson-Friedman C, McMillan PJ, Schultz RL, Lyden PD. Angiogenesis after stroke is correlated with increased numbers of macrophages: the clean-up hypothesis. J Cereb Blood Flow Metab. 2001;21:1223–1231. doi: 10.1097/00004647-200110000-00011. [DOI] [PubMed] [Google Scholar]
- Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, Musto A, Hardy M, Gimenez JM, Chiang N, Serhan CN, Bazan NG. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem. 2003;278:43807–43817. doi: 10.1074/jbc.M305841200. [DOI] [PubMed] [Google Scholar]
- Mayer K, Merfels M, Muhly-Reinholz M, Gokorsch S, Rosseau S, Lohmeyer J, Schwarzer N, Krüll M, Suttorp N, Grimminger F, Seeger W. Omega-3 Fatty acids suppress monocyte adhesion to human endothelial cells: role of endothelial PAF generation. Am J Physiol Heart Circ Physiol. 2002;283:H811–H818. doi: 10.1152/ajpheart.00235.2002. [DOI] [PubMed] [Google Scholar]
- McMahon M, Lamont DJ, Beattie KA, Hayes JD. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc Natl Acad Sci U S A. 2010;107:18838–18843. doi: 10.1073/pnas.1007387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael-Titus AT, Priestley JV. Omega-3 fatty acids and traumatic neurological injury: from neuroprotection to neuroplasticity? Trends Neurosci. 2013;pii doi: 10.1016/j.tins.2013.10.005. S0166-2236(13)00198-7. [DOI] [PubMed] [Google Scholar]
- Moreira JD, Knorr L, Thomazi AP, Simão F, Battú C, Oses JP, Gottfried C, Wofchuk S, Salbego C, Souza DO, Perry ML, Vinadé L. Dietary omega-3 fatty acids attenuate cellular damage after a hippocampal ischemic insult in adult rats. J Nutr Biochem. 2010;21:351–356. doi: 10.1016/j.jnutbio.2009.01.013. [DOI] [PubMed] [Google Scholar]
- Musiek ES, Brooks JD, Joo M, Brunoldi E, Porta A, Zanoni G, Vidari G, Blackwell TS, Montine TJ, Milne GL, McLaughlin B, Morrow JD. Electrophilic cyclopentenone neuroprostanes are anti-inflammatory mediators formed from the peroxidation of the omega-3 polyunsaturated fatty acid docosahexaenoic acid. J Biol Chem. 2008;283:19927–19935. doi: 10.1074/jbc.M803625200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimura T, Weinstein PR, Massa SM, Panter S, Sharp FR. Heme oxygenase-1 (HO-1) protein induction in rat brain following focal ischemia. Brain Res Mol Brain Res. 1996;37:201–208. doi: 10.1016/0169-328X(95)00315-J. [DOI] [PubMed] [Google Scholar]
- Panahian N, Yoshiura M, Maines MD. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J Neurochem. 1999;72:1187–1203. doi: 10.1111/j.1471-4159.1999.721187.x. [DOI] [PubMed] [Google Scholar]
- Pryor WA, Porter NA. Suggested mechanisms for the production of 4-hydroxy-2-nonenal from the autoxidation of polyunsaturated fatty acids. Free Radic Biol Med. 1990;8:541–543. doi: 10.1016/0891-5849(90)90153-A. [DOI] [PubMed] [Google Scholar]
- Roberts LJ, 2nd, Montine TJ, Markesbery WR, Tapper AR, Hardy P, Chemtob S, Dettbarn WD, Morrow JD. Formation of isoprostane-like compounds (neuroprostanes) in vivo from docosahexaenoic acid. J Biol Chem. 1998;273:13605–13612. doi: 10.1074/jbc.273.22.13605. [DOI] [PubMed] [Google Scholar]
- Rudolph TK, Freeman BA. Transduction of redox signaling by electrophile-protein reactions. Sci Signal. 2009;2:re7, 1–13. doi: 10.1126/scisignal.290re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem N, Jr, Litman B, Kim HY, Gawrisch K. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids. 2001;36:945–959. doi: 10.1007/s11745-001-0805-6. [DOI] [PubMed] [Google Scholar]
- Satoh T, Lipton SA. Redox regulation of neuronal survival mediated by electrophilic compounds. Trends Neurosci. 2007;30:37–45. doi: 10.1016/j.tins.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Satoh T, Okamoto SI, Cui J, Watanabe Y, Furuta K, Suzuki M, Tohyama K, Lipton SA. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic [correction of electrophillic] phase II inducers. Proc Natl Acad Sci U S A. 2006;103:768–773. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada M, Alkayed NJ, Goto S, Crain BJ, Traystman RJ, Shaivitz A, Nelson RJ, Hurn PD. Estrogen receptor antagonist ICI182,780 exacerbates ischemic injury in female mouse. J Cereb Blood Flow Metab. 2000;20:112–118. doi: 10.1097/00004647-200001000-00015. [DOI] [PubMed] [Google Scholar]
- Schmitz G, Ecker J. The opposing effects of n-3 and n-6 fatty acids. Prog Lipid Res. 2008;47:147–155. doi: 10.1016/j.plipres.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Sergeeva M, Strokin M, Reiser G. Regulation of intracellular calcium levels by polyunsaturated fatty acids, arachidonic acid and docosahexaenoic acid, in astrocytes: possible involvement of phospholipase A2. Reprod Nutr Dev. 2005;45:633–646. doi: 10.1051/rnd:2005050. [DOI] [PubMed] [Google Scholar]
- Sethi S, Ziouzenkova O, Ni H, Wagner DD, Plutzky J, Mayadas TN. Oxidized omega-3 fatty acids in fish oil inhibit leukocyte-endothelial interactions through activation of PPARα. Blood. 2002;100:1340–1346. doi: 10.1182/blood-2002-01-0316. [DOI] [PubMed] [Google Scholar]
- Shah ZA, Li RC, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Doré S. Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience. 2007;147:53–59. doi: 10.1016/j.neuroscience.2007.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah ZA, Nada SE, Doré S. Heme oxygenase 1, beneficial role in permanent ischemic stroke and in Gingko biloba (EGb 761) neuroprotection. Neuroscience. 2011;180:248–255. doi: 10.1016/j.neuroscience.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn CT, Smathers RL, Stewart BJ, Fritz KS, Galligan JJ, Hail N, Jr, Petersen DR. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) inhibition by 4-hydroxynonenal leads to increased Akt activation in hepatocytes. Mol Pharmacol. 2011;79:941–952. doi: 10.1124/mol.110.069534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25:10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Kim SW, Jin Y, Kim ID, Lee JK. Ethyl pyruvate-mediated Nrf2 activation and hemeoxygenase 1 induction in astrocytes confer protective effects via autocrine and paracrine mechanisms. Neurochem Int. 2012;61:89–99. doi: 10.1016/j.neuint.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Skerrett PJ, Hennekens CH. Consumption of fish and fish oils and decreased risk of stroke. Prev Cardiol. 2003;6:38–41. doi: 10.1111/j.1520-037X.2003.00959.x. [DOI] [PubMed] [Google Scholar]
- Stetler RA, Cao G, Gao Y, Zhang F, Wang S, Weng Z, Vosler P, Zhang L, Signore A, Graham SH, Chen J. Hsp27 protects against ischemic brain injury via attenuation of a novel stress-response cascade upstream of mitochondrial cell death signaling. J Neurosci. 2008;28:13038–13055. doi: 10.1523/JNEUROSCI.4407-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen AW, Verkasalo PK, Kiviranta H, Pukkala E, Jula A, Männistö S, Räsänen R, Marniemi J, Vartiainen T. Mortality in a cohort with high fish consumption. Int J Epidemiol. 2008;37:1008–1017. doi: 10.1093/ije/dyn117. [DOI] [PubMed] [Google Scholar]
- Uchida K. Lipid peroxidation and redox-sensitive signaling pathways. Curr Atheroscler Rep. 2007;9:216–221. doi: 10.1007/s11883-007-0022-7. [DOI] [PubMed] [Google Scholar]
- Valentine RC, Valentine DL. Omega-3 fatty acids in cellular membranes: a unified concept. Prog Lipid Res. 2004;43:383–402. doi: 10.1016/j.plipres.2004.05.004. [DOI] [PubMed] [Google Scholar]
- van Beelen VA, Aarts JM, Reus A, Mooibroek H, Sijtsma L, Bosch D, Rietjens IM, Alink GM. Differential induction of electrophile-responsive element-regulated genes by n-3 and n-6 polyunsaturated fatty acids. FEBS Lett. 2006;580:4587–4590. doi: 10.1016/j.febslet.2006.07.028. [DOI] [PubMed] [Google Scholar]
- Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Xing Z, Vosler PS, Yin H, Li W, Zhang F, Signore AP, Stetler RA, Gao Y, Chen J. Cellular NAD replenishment confers marked neuroprotection against ischemic cell death: role of enhanced DNA repair. Stroke. 2008;39:2587–2595. doi: 10.1161/STROKEAHA.107.509158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Sharma R, Sharma A, Awasthi S, Awasthi YC. Lipid peroxidation and cell cycle signaling: 4-hydroxynonenal, a key molecule in stress mediated signaling. Acta Biochim Pol. 2003;50:319–336. [PubMed] [Google Scholar]
- Yin H, Brooks JD, Gao L, Porter NA, Morrow JD. Identification of novel autoxidation products of the omega-3 fatty acid eicosapentaenoic acid in vitro and in vivo. J Biol Chem. 2007;282:29890–29901. doi: 10.1074/jbc.M703108200. [DOI] [PubMed] [Google Scholar]
- Zhang F, Wang S, Signore AP, Chen J. Neuroprotective effects of leptin against ischemic injury induced by oxygen-glucose deprivation and transient cerebral ischemia. Stroke. 2007;38:2329–2336. doi: 10.1161/STROKEAHA.107.482786. [DOI] [PubMed] [Google Scholar]
- Zhang F, Wang S, Zhang M, Weng Z, Li P, Gan Y, Zhang L, Cao G, Gao Y, Leak RK, Sporn MB, Chen J. Pharmacological induction of heme oxygenase-1 by a triterpenoid protects neurons against ischemic injury. Stroke. 2012;43:1390–1397. doi: 10.1161/STROKEAHA.111.647420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, An C, Gao Y, Leak RK, Chen J, Zhang F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol. 2013;100:30–47. doi: 10.1016/j.pneurobio.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Hu X, Yang W, Gao Y, Chen J. Omega-3 polyunsaturated fatty acid supplementation confers long-term neuroprotection against neonatal hypoxic-ischemic brain injury through anti-inflammatory actions. Stroke. 2010;41:2341–2347. doi: 10.1161/STROKEAHA.110.586081. [DOI] [PMC free article] [PubMed] [Google Scholar]