

Graphical abstract
5-ASA induces membranous retention of E-cadherin through remodeling of N-glycans. Stabilization of E-cadherin at cell, contacts results in increased intercellular adhesion restricting tumor progression or restoring epithelial integrity in IBD.
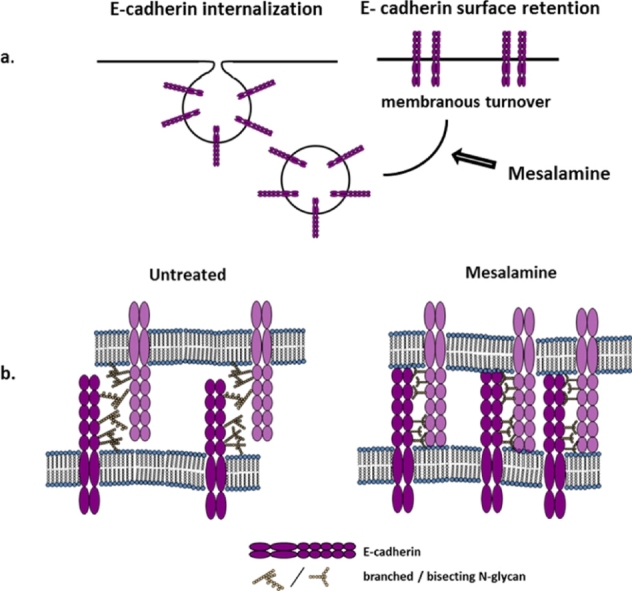
Abbreviations: CRC, colorectal cancer; IBD, inflammatory bowel diseases; UC, ulcerative colitis; GnT-III, N-acetylglucosaminyltransferase-III; AJ, adherens junction; PAK1, p21 activated kinase 1
Keywords: E-cadherin, GnT-III, 5-ASA, Glycosylation, Ulcerative colitis
Abstract
Genome wide association studies have implicated intestinal barrier function genes in the pathogenesis of ulcerative colitis. One of such loci CDH1, encoding E-cadherin, a transmembrane glycoprotein with known tumor suppressor functions, is also linked to the susceptibility to colorectal cancer. Loss of membranous E-cadherin expression is common in both colitis and cancer. We have recently demonstrated that mesalamine (5-ASA); the anti-inflammatory drug used to treat ulcerative colitis, induces membranous expression of E-cadherin and increases intercellular adhesion. Using colorectal cancer epithelial cells with aberrant E-cadherin expression, we investigated the mechanism underlying such an effect of 5-ASA. Post-translational modification of E-cadherin glycosylation was analyzed by biotin/streptavidin detection of sialylated glycoproteins. GnT-III (N-acetylglucosaminyltransferase III) expression was assessed by qRT-PCR, Western blot and immunofluorescence. GnT-III activity was analyzed by reactivity with E-4/L-4-PHA. Expression, localization and interaction of E-cadherin and β-catenin were analyzed by Western blot, immunocytochemistry and RNA interference. 5-ASA activity modulated E-cadherin glycosylation and increased both mRNA and protein levels of GnT-III and its activity as detected by increased E4-lectin reactivity. Intestinal APCMin polyps in mice showed low expression of GnT-III and 5-ASA was effective in increasing its expression. The data demonstrated that remodeling of glycans by GnT-III mediated bisect glycosylation, contributes to the membranous retention of E-cadherin by 5-ASA; facilitating intercellular adhesion. Induction of membranous expression of E-cadherin by 5-ASA is a novel mechanism for mucosal healing in colitis that might impede tumor progression by modulation of GnT-III expression.
1. Introduction
Ulcerative colitis (UC) is a form of inflammatory bowel disease (IBD) causing chronic inflammation of the colon. 5-ASA, the primary anti-inflammatory therapy in UC, induces mucosal healing in the majority of patients, although the underlying molecular mechanism is incompletely understood [1]. Chronic gut inflammation in UC disrupts the intestinal architecture as a consequence of altered function of molecules involved in epithelial integrity. Several transmembrane proteins are involved in the establishment and maintenance of the intercellular adhesion. Adherens junctions (AJs) are composed of multiprotein complexes, which are uniformly distributed along the plasma membrane and provide adhesive contact for intercellular adhesion. Establishment of polarized epithelia requires cell to cell contact that induces formation of AJs followed by assembly of tight junctions [2]. E-cadherin, a member of the cadherin family of transmembrane molecules, plays a dynamic role in maintaining the homeostasis of intestinal epithelium. Membranous expression of E-cadherin triggers assembly of AJs through its adhesive interaction with armadillo family members and cytoskeletal adapter proteins [3]. Abnormalities in epithelial architecture through disruption of AJs and impaired expression of E-cadherin are implicated in chronic gut inflammation and colorectal cancer (CRC). Recently, a genome-wide association study of UC identified CDH1 locus (encoding E-cadherin) which was also identified as one of the susceptibility loci for CRC [4], [5]. These observations provide a link for the role of E-cadherin in both UC and CRC. Membranous expression of E-cadherin is lost during wound healing, and epithelial-mesenchymal transition of cancer cells [6], [7]. Muise et al. demonstrated that IBD patients with the disease-associated single nucleotide polymorphisms in CDH1, have increased cytoplasmic accumulation of E-cadherin, likely as a result of a processing defect [8]. We recently reported that 5-ASA results in the membranous translocation of E-cadherin and concomitantly increases intercellular adhesion [9] indicating a molecular basis of its action in the restoration of epithelial barrier function in IBD patients.
Several studies suggest that molecular organization, stability and adhesive functions of E-cadherin can be modulated by remodeling of glycan structure [10]. It has been demonstrated previously that asparagine residues that undergo N-glycosylation are located in the ectodomain of the E-cadherin molecule [11]. These potential glycosylation sites have been implicated in the modulation of its function in pathophysiological conditions. Within the N-glycan biosynthetic pathway, GnT-III (N-acetylglucosaminyltransferase-III) introduces bisecting GlcNAc (N-acetylglucosamine) and suppresses processing of branched or complex glycans catalyzed by GnT-V. Bisecting glycans on E-cadherin delay its turnover on the membrane and increases cell adhesion [12], [13]. Using an epithelial cell line, Vagin et al. demonstrated that silencing GnT-III facilitates branching and complexity of N-glycans that was associated with increased permeability of the cell monolayer [14]. Moreover, the structure of N-glycans modulated by GnT-III on Na/K-ATPase, the cell adhesion protein investigated in this study, was associated with establishment of tight epithelium. GnT-III inhibited cytoplasmic and nuclear translocation of β-catenin from the membrane and transforming growth factor-β1 (TGF-β1) downregulated E-cadherin and GnT-III during epithelial-mesenchymal transition [15]. This confers a tumor suppressor role on GnT-III, as overexpression of this enzyme is associated with the inhibition of epithelial-mesenchymal transition and metastasis [16]. Altogether, these studies suggest that activity of glycosyltransferases is critical in pathophysiology of the disease and highlight them as attractive therapeutic targets. Nevertheless, the effect of N-glycosylation in gut epithelium is not well characterized. Considering the significance of membranous E-cadherin expression, which is critical for its role in epithelial integrity and cancer progression, we examined the underlying mechanism of membranous turnover of E-cadherin by 5-ASA and its effect on E-cadherin glycosylation, using colorectal cancer cell lines.
2. Materials and methods
2.1. Cell lines and reagents
Human colorectal carcinoma cell lines HCT116 and HT29 (obtained from ATCC) were grown in IMDM (Gibco/Invitrogen, Lofer, Austria) containing 10% fetal bovine serum (FBS; Biochrom, Berlin, Germany). 5-ASA (>99.9% pure; a generous gift from Shire Inc., Eysins, Switzerland) was dissolved in the culture medium at 20 mM or 40 mM final concentration (pH adjusted to 7.2 with NaOH).
2.2. Immunoprecipitation and labeling of sialylated glycoproteins on E-cadherin
HCT116 or HT29 cells were treated with 20 mM or 40 mM 5-ASA (for 24 h or left untreated. For immunoprecipitation, cell lysates were incubated with E cadherin antibody or control IgG, followed by protein G plus agarose beads (Santa Cruz). The beads were washed and either boiled directly in SDS sample buffer (for IP Western blot) or subsequently processed for glycoprotein labeling. Biotin labeling of sialylated glycoproteins was carried out as described [17], with minor modifications. Briefly, IP-beads were subjected to mild periodate oxidation with 1 mM NaIO4 at 4 °C, 20 min to generate a free aldehyde on sialic acids. Beads were washed with PBS followed by an aniline-catalyzed oxime ligation (PAL) with 100 μM aminooxy-biotin and 10 mM aniline in PBS (pH 6.7) for 90 min at 4 °C, to introduce a biotin tag. After washing with PBS, beads were boiled in SDS gel sample buffer and proteins were separated by SDS-PAGE. E-cadherin immunoblots were visualized by the Odyssey Infrared Imaging System (LI-COR Biosciences). Biotin labeling for sialylated glycoproteins was detected by a Streptavidin Alexa Fluor 488 conjugate on a Typhoon 9200 image scanner (Amersham Biosciences) or with IRDye 800CW Streptavidin (LiCor). Endoglycosidase H treatment of E_cadherin (IP) was performed according to manufacturer's protocol (New England Biolabs). One unit of Endo-H was used (1 h, 37 ̊C).
2.3. Cell adhesion assay
Cell adhesion assay was performed as described previously [9]. pCMV5Bmyc plasmid, with wt E-cadherin or E-cadherin N-glycosylation variant V13 (lacking site 1 in EC4 and site 3 in EC5) was a kind gift from Maria A. Kukuruzinska (Boston University, MA). CHO cells were transfected with the pCMV5Bmyc vector using Effectene (Qiagen). Cells were treated with 5-ASA (20 mM) for 24 h, washed in PBS, counted and an equal number of cells were plated in 24-well plates allowing them to attach to the surface. After 30 min of incubation, each plate was washed with PBS until no floating cells remained and then replaced with the fresh medium and MTT reagent. After 4 h, the medium was removed and the remaining precipitates were dissolved in DMSO/ethanol mixture (50/50 v/v) and absorbance was measured at 570 nm with an ELISA plate reader. The experiment was repeated two times, and for each dish, four wells were scored.
2.4. RNA interference
Transfections were performed using Mammalian Epithelial Cells Amaxa Basic Nucleofector Kit (Lonza, Basel, Switzerland) and cells were electroporated according to manufacturer's instructions. CTNNB1 FlexiTube siRNA was purchased from Qiagen (Cambridge, MA) and prepared based on the manufactures protocol. Control siRNA-A: sc-37007 (Santa Cruz Biotechnology, CA) was also prepared based on the manufacturer's instructions. 30 nM control siRNA and 30 nM CTNNB1 siRNA were utilized in all transfection experiments.
2.5. Immunofluorescence microscopy
Cells were fixed in methanol and immunostaining was performed using antibodies against β-catenin (clone 15B8; eBioscience), E-cadherin (Clone 36; BD Transduction Laboratories), GnT-III (F-20, Santa Cruz Biotechnology) PHA-L4-FITC and PHA-E4-FITC (amsbio). For protein visualization AlexaFluor 488 and 568 antibodies (Invitrogen) were used. Nuclear staining was performed using Vectashield with DAPI (Vector laboratories) for mounting. Images were scanned at 400× magnification on a LSM 700 (Zeiss) or Olympus BX 51). Digital images were processed with Zeiss LSM Browser.
2.6. Cell fractionation, Western blotting and antibodies
For cytosolic and membrane fractions cells were grown in 15 cm dishes and were collected with 400 μl cold hypotonic buffer (10 mM Tris–HCl pH 7.5, 0.2 mM MgCl2, with protease and phosphatase inhibitors) with a cell scraper. The extract was dounce homogenized and kept on ice for 30 min. The extract was spun at 15,000g for 45 min at 4 °C, and the supernatant was collected as the cytosolic fraction. The pellet was washed twice in hypotonic buffer and then resuspended by vortexing in 50 μl lysis buffer (150 mM NaCl, 20 mM Tris–HCl pH 7.5, 1% Triton X-100, and proteinase inhibitors). The extract was vortexed for 10 s every 10 min and kept on ice for 30 min. Afterwards, the supernatant was collected as the membrane fraction. Both fractions were incubated with Laemmli sample buffer containing 10% β-mercaptoethanol at 95 °C for 10 min and then analyzed by Western blot. Protein concentrations were measured by Bradford assay (Bio-Rad). Proteins were separated by SDS-PAGE and immunoblotted onto a PVDF membrane. Primary antibodies used were as follows: monoclonal antibody, anti-E-cadherin (clone 36 BD Transduction Laboratories), anti-β-catenin (clone 14/BD Transduction Laboratories), alpha-tubulin (cell signaling), GnT-III (Santa Cruz). The protein bands were visualized with anti-rabbit or anti-mouse antibodies coupled to horseradish peroxidase using the ECL kit (Amersham) or with IRDye coupled antibodies (either or both mouse/rabbit) and scanned on Odyssey imager (LI-COR Biotechnology).
2.7. GnT-III mRNA expression
Cells were treated with 20 mM 5-ASA for 24 h. Total RNA was extracted with TRIzol (Invitrogen). 1 μg of RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qRT-PCR was performed using the Fast SYBR Green Master Mix (Applied Biosystems) and run on the 7500 Fast Real-Time PCR System (Applied Biosystems). GnT-III (MGAT3) QuantiTect Primer Assay (QT01004381) was obtained from Qiagen. The reference housekeeping gene used was 36B4 that was found not to be affected by 5-ASA treatment.
2.8. Animals and experimental procedure
The animal experiment was performed as reported earlier [9]. Briefly, 4–6 week old heterozygous female and male C57BL/6J-ApcMin/+ mice (Jackson Laboratories) were fed with either 2500 mg/kg 5-ASA (A3537, Sigma-Aldrich) mixed into the chow or with a control diet (C1000, Altromin). The 5-ASA dose corresponds to the intake of 3 g/day in humans [18]. After 12 weeks the mice were euthanized, the whole gut dissected and coiled up to a Swiss roll. The intestine was fixed in neutral buffered formalin (10%) for 24 h and embedded in paraffin.
2.9. Immunohistochemistry
Immunohistochemistry analysis was done on paraffin-embedded mouse intestine. Serial tissue sections (4 μm) Swiss rolls were stained for GnT-III (F-20; sc-27287 Santa Cruz) using standard procedures. Briefly, slides were dried, de-waxed in xylol and rehydrated using a decreasing alcohol series. After blocking of endogenous peroxidase with 15% H2O2 in methanol, antigen retrieval was performed in 10 mM citrate buffer, pH 6. Subsequently slides were blocked in 2% horse serum, 3% BSA in TRIS buffer. GnT-III antibody was incubated 4 °C (overnight), followed by biotinylated anti-goat antibody and avidin-biotin-HRP complex. Staining was visualized using DAB and nuclear counterstaining was performed using hematoxylin. Slides were dehydrated and embedded in Histofluid.
2.10. Statistical analysis
The data was analyzed by Student's t-test. P-values less than 0.05 were considered statistically significant.
3. Results
3.1. 5-ASA mediated membranous turnover of E-cadherin is independent of β-catenin
An interaction between E-cadherin and β-catenin is one of the mechanisms of E-cadherin exocytosis on the cell membrane at AJs [19]. To test whether 5-ASA utilizes this mechanism, we examined the effect of silencing β-catenin on membranous expression of E-cadherin. HT29 cells (which are mutated in APC, have activated β-catenin signaling, and express low membranous E-cadherin) were transfected with control (con-siRNA) or siRNA for β-catenin (siβ-catenin) in the presence or absence of 5-ASA. In con-siRNA cells, 5-ASA treatment enhanced both E-cadherin and β-catenin localization at AJs (Fig. 1a). Moreover, there was an increased membranous co-localization of E-cadherin and β-catenin in con-siRNA cells (merged picture), confirming enhanced interaction of these molecules upon 5-ASA treatment. Interestingly, in siβ-catenin transfected cells, 5-ASA still induced membranous expression of E-cadherin. This observation indicated that the 5-ASA mediated membranous turnover of E-cadherin was independent of its interaction with β-catenin. Western blot was performed to confirm the findings of immunocytochemistry (Fig. 1b). Inhibition of β-catenin did not alter the 5-ASA-induced membranous increase in E-cadherin. Interestingly, the transmembrane protein Na/K. ATPase that was used to verify the purity of the membrane fraction also changed with E-cadherin expression. However, this effect might be expected with an increase in membranous E-cadherin as Na/K. ATPase is an essential component of the junctional complex and contributes to adhesion function of E-cadherin and epithelial polarity [14], [20]. We concluded that 5-ASA interferes with an alternative pathway involved in E-cadherin trafficking, which is independent of β-catenin.
Fig. 1.

Effect of silencing β-catenin on mesalamine (5-ASA) induced membranous translocation of E-cadherin. (a) HT29 cells were transfected with control and siβ-catenin RNA. Cells were grown on coverslips and treated with 5-ASA (20 mM) for 24 h. Immunostaining was performed using antibodies against β-catenin and E-cadherin. 5-ASA induced membranous translocation of both proteins. An increased interaction of β-catenin and E-cadherin was observed upon 5-ASA treatment in cells transfected with control RNA (merged image). Membranous turnover of E-cadherin was unaffected upon 5-ASA treatment in siβ-catenin transfected cells. Image magnification 400× (b) Western blot analysis of E-cadherin and β-catenin in total cell lysate (RIPA) and cellular fractions (membrane or cytoplasm) in siβ-catenin transfected cells. Anti-tubulin was used as loading control and Na–K ATPase was used to verify the purity of membranous fraction.
3.2. 5-ASA modulates E-cadherin glycosylation
Post-translational modification of E-cadherin by phosphorylation and glycosylation is implicated in the surface localization and binding with catenins and other partners at AJs [13]. We hypothesized that 5-ASA affects the structural modification of E-cadherin by remodeling of glycans. To test this, we examined E-cadherin glycosylation in 5-ASA treated cells. Biotin labeling of sialylated E-cadherin immunoprecipitates, followed by streptavidin detection revealed enhanced glycosylation of E-cadherin upon 5-ASA treatment (Fig. 2a). Both of the cell lines examined showed an increase in E-cadherin glycosylation as observed by high molecular weight bands. Enzymatic de-glycosylation by Endo-H resulted in the removal of glycosylated E-cadherin (Fig. 2b and c). The appearance of the E-cadherin glycosylated band and its disappearance upon Endo-H treatment indicates that 5-ASA induced modification of E-cadherin N-glycosylation could not be complex N-glycans, as these are resistant to Endo-H.
Fig. 2.

5-ASA modulates E-cadherin glycosylation. (a) Flow chart for the glycosylation assay performed on immunoprecipitated E-cadherin from HCT116 and HT29. (b) As indicated, Western blot was performed either on E-cadherin IP (±5-ASA) or on E-cadherin IP subjected to biotinylation (±5-ASA). Treatment increased glycosylation of E-cadherin (lane 6′) compared to untreated controls (lane 4′) which was detected as multiple high molecular weight bands. The experiment was performed again in the presence or absence of glycosidase Endo-H (right panel). Treatment with Endo-H resulted in the removal of 5-ASA induced glycosylated band (*) and a shift in E-cadherin band (*). (c) E-cadherin glycosylation assay with HT29 cells. *Indicate appearance of glycosylated band upon 5-ASA treatment (lane 2′) or shift in E-cadherin band upon glycosidase treatment. (d) Cell adhesion assay performed with CHO cells transfected with either wild type E-cadherin or N-glycosylation variant V-13 constructs. 5-ASA (20 mM; 24 h) increased cell adhesion in the cells with wt-E-cadherin but not in the cells transfected with E-cadherin V13 mutant. All N-glycosylation sites are intact in the wt-E-cadherin ectodomains (EC 4 and 5) while V13 mutant lacks Asn-404 and Asn-483.
It is known that E-cadherin can be modified by O-glycosylation and N-glycosylation. The former prevents its transport to the cell membrane [21], and N-glycosylation is the prominent modification which varies in its composition. Moreover, N-glycans at E-cadherin ectodomain were found to affect E-cadherin mediated cell adhesion [13]. To verify the role of N-glycosylation in 5-ASA mediated cell adhesion, we utilized an E-cadherin N-glycosylation mutant V13. It was demonstrated that CHO cells (that do not express endogenous E-cadherin), can organize E-cadherin-mediated AJs when transfected with E-cadherin [10]. CHO cells were transfected with E-cadherin wild-type or the V13 N-glycosylation mutant. Wild-type (wt) E-cadherin transfectants displayed increased adherence upon 5-ASA treatment (Fig. 2d), whereas no such effect was observed in V13-mutant transfected CHO cells. These observations demonstrated that 5-ASA modulates E-cadherin N-glycosylation, which affects cell adhesion. Using ECIS (electric cell-substrate impedance sensing), we have previously demonstrated that 5-ASA treatment affects intercellular adhesion. Moreover, 5-ASA activity resulted in membranous recruitment of AJ proteins [9]. Hence, we concluded that the increase in intercellular adhesion upon 5-ASA treatment is associated with remodeling of E-cadherin N-glycosylation.
3.3. GnT-III expression is upregulated by 5-ASA
It was clear that 5-ASA activity restored membranous expression of E-cadherin but it was independent of β-catenin. However, N-glycosylation was critical for increased cell adhesion. We speculated if modulation of N-glycosylation by 5-ASA contributes to the membranous retention of E-cadherin resulting in the stability of AJs.
Complexities of glycans that modify E-cadherin differ in proliferating cells where adhesion is inhibited compared to cells forming tight AJs restricting motility [10]. It has been shown that GnT-III modifies E-cadherin prolonging its membranous turnover and overexpression of this enzyme is responsible for enhanced adhesion [7]. We reasoned that 5-ASA-mediated modulation of E-cadherin glycosylation might promote AJ formation and hence, increased intercellular adhesion. We examined the expression levels of GnT-III upon 5-ASA treatment. Since addition of glycans on E-cadherin can also take place in the junctional complex at the cell membrane, we also examined the membrane fractions of these cells. 5-ASA induced a marked increase in GnT-III protein in HCT116 cells but only in the membranous fraction of HT29 cells (Fig. 3a). When assessing the effect of 5-ASA on the relative expression of GnT-III mRNA (MGAT3), an increase in MGAT3 transcripts was observed in both cell lines examined, the effect being more pronounced in HCT116 (Fig. 3b). Overall, the two cell lines examined differed in the expression of GnT-III. HT-29 cells expressed membranous GnT-III suggesting its activity at AJs. However, HCT116 showed a relatively higher increase in both MGAT3 mRNA and total protein levels upon 5-ASA treatment. To further validate these findings, immunofluorescence for GnT-III was performed in the presence or absence of 5-ASA. The cellular localization of GnT-III was not limited to cytoplasm. Although, the glycosyltransfereases are localized in Golgi complex, the protein was also present at the membrane in the CRC cells examined. Consistent with Western blot data, 5-ASA treatment resulted in an overall increase in GnT-III in HCT116. In HT29 cells 5-ASA modulated its localization at cell-to cell contacts (Fig. 3c). Altogether, this data supported the notion that 5-ASA modulates GnT-III expression in CRC cells.
Fig. 3.

5-ASA increases GnT-III expression. (a) Western blot with anti-GnT-III antibody upon 5-ASA (5-ASA; 20 mM) treatment of the cells in the whole cell extract (RIPA) or membranous fractions. α-tubulin was used as loading control (b) validation of increased transcription of GnT-III by qRT-PCR in 5-ASA treated (20 mM;24 h) cell lines. *p value = 0.018 (c) immunofluorescent detection for GnT-III expression in HCT116 and HT29 cells. Arrows indicate cell contacts. Image magnification 400×.
3.4. 5-ASA treatment inhibits l-PHA reactivity and increases bisecting glycans identified by E-PHA
5-ASA treatment modified N-glycosylation of E-cadherin with an increase in GnT-III expression in CRC cells. To further evaluate the products of GnT-III (bisecting GlcNAc) we performed lectin staining that distinguishes the products of GnT-III and GnT-V. The bisecting glycans are preferentially recognized by E4-PHA while L4-PHA recognizes branched GlcNAc (product of GnT-V). In untreated cells, L4-PHA reactivity was higher compared to E4-PHA indicating higher GnT-V activity. 5-ASA treatment resulted in marked inhibition of L4-PHA reactivity and increased glycans recognized by E4-PHA (Fig. 4) confirming the increase in GnT-III activity. This effect was more pronounced in HCT116 cells consistent with modulation of GnT-III expression by 5-ASA.
Fig. 4.

Detection of bisecting or branched GlcNAc structures. E-PHA and L-PHA lectin reactivity was assessed by immunofluorescence. In untreated CRC cells, branched GlcNAc (L4-PHA) were predominant suggesting higher activity of GnT-V. 5-ASA treatment inhibited GnT-V activity and an increased expression of bisecting GlcNAc (E4-PHA) structures was observed confirming increase in GnT-III enzymatic activity. Image magnification 400×.
The inhibition of L4-PHA reactivity upon 5-ASA treatment indicated inhibition of GnT-V activity. While E4-PHA reactivity demonstrated increased activity of GnT-III in 5-ASA treated cells. These results were in accordance with our previous observation that 5-ASA mediated cell adhesion was more pronounced in HCT116 compared to HT29 [9]. We concluded that increased expression of GnT-III by 5-ASA modifies glycan structures on E-cadherin, facilitates its membranous retention and promotes intercellular adhesion.
3.5. 5-ASA induced GnT-III expression in vivo
We further utilized tissue sections from our experiments in the APCMin mouse model of intestinal tumorigenesis, to examine the effect of 5-ASA activity on GnT-III expression in vivo [9]. APCMin mice develop spontaneous intestinal polyps due to mutation in the APC gene [22]. Our data and others have shown that 5-ASA reduced the number of polyps in these mice and restored membranous E-cadherin which showed aberrant expression in untreated polyps [9], [23], [24]. Immunohistochemistry was performed on intestinal sections from the mice kept on a 5-ASA diet as well as the untreated control group. GnT-III was moderately expressed in the cytoplasm of normal mucosa and APCMin polyps showed low expression of GnT-III (Fig. 5). 5-ASA treatment increased Gnt-III expression, both in the polyps and in the normal mucosa. The surrounding tissue also stained positive for GnT-III suggesting its role in the modification of mucus glycoproteins and/or staining for secreted protein. This data provided evidence that GnT-III expression and therefore, complexity of glycans, is modulated by 5-ASA activity.
Fig. 5.

5-ASA modulates GnT-III expression in vivo. Immunohistochemistry was performed on tissue sections from APCMin mice. Small intestinal APCMin polyp showed low expression of GnT-III while surrounding normal mucosa stained GnT-III positive with moderate expression (indicated by arrow). 5-ASA activity increased expression of GnT-III in mucosal lining. Image magnification 40×. Higher magnification (100×) shows that more cells stained GnT-III positive in the polyp from mice with 5-ASA in their diet as indicated by arrows. The figure represents polyps from the ileum.
4. Discussion
We have previously shown that 5-ASA activity is involved in E-cadherin/β-catenin-mediated intercellular adhesion through induction of membranous E-cadherin. While examining the mechanism of membranous E-cadherin expression by 5-ASA, the current study demonstrated that the 5-ASA modulates E-cadherin N-glycosylation. The increase in cell adhesion is dependent on N-glycosylation of E-cadherin as cells expressing the V13 N-glycosylation mutant, did not show this effect upon 5-ASA treatment. The data showed that N-glycosylation is modulated by 5-ASA through re-modeling of glycan structures that contribute to membranous retention of E-cadherin. 5-ASA-induced membranous translocation of E-cadherin was independent of β-catenin. Albeit, 5-ASA activity was found to be associated with upregulation of glycosyltransferase GnT-III, that stabilizes membranous E-cadherin. The increase in GnT-III activity was associated with decreased L4-PHA reactivity; indicating suppression of GnT-V activity and a concomitant increase in the bisect glycans recognizing E4-PHA. The increase in GnT-III expression by 5-ASA was also observed in APCMin polyps. This study provides a mechanistic explanation that GnT-III mediated modulation of bisect N-glycosylation on E-cadherin might be fostering its membranous retention, thereby promoting intercellular cell adhesion by 5-ASA. Remodeling of N-glycans by 5-ASA is a novel mechanism of its action in intestinal homeostasis.
Functional impairment of E-cadherin during tumor progression is attributed to transcriptional repression, promoter methylation along with altered post-translational modifications including glycosylation [7], [25]. Little is known regarding the role of N-glycans, the most prominent modification on E-cadherin, in the assembly and stability of AJs. Quantitative and qualitative changes in E-cadherin N-glycosylation were found to affect the molecular organization of AJs [10]. Bisecting glycans were shown to increase E-cadherin-mediated cell adhesion and reduce integrin-mediated cell spreading and migration [13]. These studies emphasize the significance of N-glycosylation and N-acetylglucosaminyltransferases in the impairment of E-cadherin functions in diseases such as IBD and cancer. We did not utilize an approach to silence GnT-III and validate the effect of 5-ASA on membranous translocation of E-cadherin. However, another study demonstrated that depletion of GnT-III resulted in membranous de-localization of E-cadherin without affecting its mRNA or protein levels [26]. The E-cadherin-catenin complex itself regulates GnT-III enzymatic activity [27] and our data supports this notion. The membranous increase of GnT-III upon 5-ASA treatment suggests its enzymatic activity in E-cadherin junctional complex and the likely cause of membranous retention of E-cadherin.
In a study from Jamal et al. [28], a hypo-glycosylated E-cadherin V13 mutant promoted the formation of stable AJs compared to wild-type. Using the same E-cadherin variant, however, we observed that 5-ASA was ineffective in increasing cell adhesion when the cells were transfected with the V13 mutant. Current data primarily suggest a role of GnT-III upregulation and glycan remodeling by 5-ASA; therefore, the possibility that the V13 residue is critical in GnT-III-induced alteration in glycan structure cannot be ruled out.
Upregulation of GnT-III expression and bisect glycosylation by 5-ASA might be attributed to inhibition of Wnt/β-catenin signaling. 5-ASA inhibits β-catenin nuclear localization and transcriptional activity. We have recently shown that 5-ASA activity is mediated by inhibition of serine–threonine kinase PAK1 and inhibition of PAK1 increased membranous localization of E-cadherin and β-catenin. PAK1 phosphorylates β-catenin and contributes to Wnt/β-catenin signaling. On other hand, activation of the Wnt/β-catenin pathway inhibited GnT-III expression [29]. Nevertheless, 5-ASA is an anti-inflammatory drug and modulates several pathways besides Wnt/β-catenin signaling. TGF-beta signaling induces GnT-V and inhibits Gnt-III; while 5-ASA acts as an antagonist for TGF-beta signaling [30]. Taken together, our data demonstrates that pharmacological inhibition of PAK1 and upregulation of Gnt-III by 5-ASA can reverse the impairment of E-cadherin membranous expression in CRC cells.
Our in vivo data in an APCMin mouse model gives a novel insight about GnT-III expression in intestinal polyposis. Though 5-ASA is not effective in lowering tumor incidence in this model, it does have an impact in lowering tumor multiplicity [23]. Upregulation of a metastasis suppressor protein like GnT-III, supports the beneficial effects of 5-ASA in restricting tumor burden. Knowledge about protein glycosylation in the development of intestinal disease is lacking. Further investigations are needed to understand the role of glycosyltransferses in the maintenance of gut physiology.
E-cadherin is critical in the establishment and maintenance of cell adhesion; it regulates intercellular contacts via several mechanisms. Reduced cell adhesiveness is associated with an increase in mucosal permeability. This study provides a mechanistic link for 5-ASA activity in improving intestinal barrier function through modulation of N-glycosylation. We propose that 5-ASA activity contributes in the interplay of an increased GnT-III expression and turnover of E-cadherin in the complex with β-catenin at AJs, thereby promoting intercellular adhesion. It is likely that 5-ASA might be interfering with E-cadherin endocytosis in CRC cells examined. Modulation of N-glycosylation associated with an upregulation of GnT-III is a novel mechanism of 5-ASA's activity, which can have implications in restoration of epithelial integrity in UC and in impeding tumor progression in colorectal carcinogenesis. 5-ASA might be effective in maintaining epithelial barrier function in other conditions leading to enhanced intestinal permeability such as NSAID colitis, graft-versus-host disease, multiple trauma and sepsis.
Grant support
The financial support by the Federal Ministry of Economy, Family and Youth and the National Foundation for Research, Technology and Development is gratefully acknowledged. This study was supported in part by the Austrian Science Fund (P24121 to CG).
Disclosures
CG has research collaboration with Shire Pharmaceuticals and received research support, lecturing or consulting honoraria from Ferring, Giuliani Tillot's and Dr Falk Pharma.
Acknowledgments
The pCMV5Bmyc plasmid with wt E-cadherin and N-glycosylation variant V13 (lacking site 1 in EC4 and site 3 in EC5) was a kind gift from Maria A. Kukuruzinska (Boston University, MA). Confocal images were taken in cooperation with the Imaging core facility, MUW, Vienna.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Romkens T.E., Kampschreur M.T., Drenth J.P., van Oijen M.G., de Jong D.J. High mucosal healing rates in 5-ASA-treated ulcerative colitis patients: results of a meta-analysis of clinical trials. Inflamm Bowel Dis. 2012;18:2190–2198. doi: 10.1002/ibd.22939. [DOI] [PubMed] [Google Scholar]
- 2.Farquhar M.G., Palade G.E. Junctional complexes in various epithelia. J Cell Biol. 1963;17:375–412. doi: 10.1083/jcb.17.2.375. 375-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Green K.J., Getsios S., Troyanovsky S., Godsel L.M. Intercellular junction assembly, dynamics, and homeostasis. Cold Spring Harb Perspect Biol. 2010;2:a000125. doi: 10.1101/cshperspect.a000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrett J.C., Lee J.C., Lees C.W., Prescott N.J., Anderson C.A., Phillips A. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009;41:1330–1334. doi: 10.1038/ng.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Houlston R.S., Webb E., Broderick P., Pittman A.M., Di Bernardo M.C., Lubbe S. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet. 2008;40:1426–1435. doi: 10.1038/ng.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmalhofer O., Brabletz S., Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metast Rev. 2009;28:151–166. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 7.Paredes J., Figueiredo J., Albergaria A., Oliveira P., Carvalho J., Ribeiro A.S. Epithelial E- and P-cadherins: role and clinical significance in cancer. Biochim Biophys Acta. 2012;1826:297–311. doi: 10.1016/j.bbcan.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Muise A.M., Walters T.D., Glowacka W.K., Griffiths A.M., Ngan B.Y., Lan H. Polymorphisms in E-cadherin (CDH1) result in a mis-localised cytoplasmic protein that is associated with Crohn's disease. Gut. 2009;58:1121–1127. doi: 10.1136/gut.2008.175117. [DOI] [PubMed] [Google Scholar]
- 9.Khare V., Lyakhovich A., Dammann K., Lang M., Borgmann M., Tichy B. Mesalamine modulates intercellular adhesion through inhibition of p-21 activated kinase-1. Biochem Pharmacol. 2013;85:234–244. doi: 10.1016/j.bcp.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liwosz A., Lei T., Kukuruzinska M.A. N-glycosylation affects the molecular organization and stability of E-cadherin junctions. J Biol Chem. 2006;281:23138–23149. doi: 10.1074/jbc.M512621200. [DOI] [PubMed] [Google Scholar]
- 11.Kornfeld R., Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. 631-664. [DOI] [PubMed] [Google Scholar]
- 12.Pinho S.S., Oliveira P., Cabral J., Carvalho S., Huntsman D., Gartner F. Loss and recovery of Mgat3 and GnT-III Mediated E-cadherin N-glycosylation is a mechanism involved in epithelial–mesenchymal–epithelial transitions. PLoS One. 2012;7:e33191. doi: 10.1371/journal.pone.0033191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Y., Sato Y., Isaji T., Fukuda T., Matsumoto A., Miyoshi E. Branched N-glycans regulate the biological functions of integrins and cadherins. FEBS J. 2008;275:1939–1948. doi: 10.1111/j.1742-4658.2008.06346.x. [DOI] [PubMed] [Google Scholar]
- 14.Vagin O., Tokhtaeva E., Yakubov I., Shevchenko E., Sachs G. Inverse correlation between the extent of N-glycan branching and intercellular adhesion in epithelia. Contribution of the Na, K—ATPase beta1 subunit. J Biol Chem. 2008;283:2192–2202. doi: 10.1074/jbc.M704713200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu Q., Isaji T., Lu Y., Gu W., Kondo M., Fukuda T. Roles of N-acetylglucosaminyltransferase III in epithelial-to-mesenchymal transition induced by transforming growth factor β1 (TGF-β1) in epithelial cell lines. J Biol Chem. 2012;287:16563–16574. doi: 10.1074/jbc.M111.262154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshimura M., Nishikawa A., Ihara Y., Taniguchi S., Taniguchi N. Suppression of lung metastasis of B16 mouse melanoma by N-acetylglucosaminyltransferase III gene transfection. PNAS. 1995;92:8754–8758. doi: 10.1073/pnas.92.19.8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeng Y., Ramya T.N., Dirksen A., Dawson P.E., Paulson J.C. High-efficiency labeling of sialylated glycoproteins on living cells. NatMethods. 2009;6:207–209. doi: 10.1038/nmeth.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reagan-Shaw S., Nihal M., Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y.T., Stewart D.B., Nelson W.J. Coupling assembly of the E-cadherin/beta-catenin complex to efficient endoplasmic reticulum exit and basal-lateral membrane targeting of E-cadherin in polarized MDCK cells. J Cell Biol. 1999;144:687–699. doi: 10.1083/jcb.144.4.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McNeill H., Ozawa M., Kemler R., Nelson W.J. Novel function of the cell adhesion molecule uvomorulin as an inducer of cell surface polarity. Cell. 1990;62:309–316. doi: 10.1016/0092-8674(90)90368-o. [DOI] [PubMed] [Google Scholar]
- 21.Zhu W., Leber B., Andrews D.W. Cytoplasmic O-glycosylation prevents cell surface transport of E-cadherin during apoptosis. EMBO J. 2001;20:5999–6007. doi: 10.1093/emboj/20.21.5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moser A.R., Pitot H.C., Dove W.F. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 23.Brown J.B., Lee G., Managlia E., Grimm G.R., Dirisina R., Goretsky T. Mesalamine inhibits epithelial beta-catenin activation in chronic ulcerative colitis. Gastroenterology. 2010;138:595–605. doi: 10.1053/j.gastro.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munding J., Ziebarth W., Pox C.P., Ladigan S., Reiser M., Huppe D. The influence of 5-aminosalicylic acid on the progression of colorectal adenomas via the beta-catenin signaling pathway. Carcinogenesis. 2012;33:637–643. doi: 10.1093/carcin/bgr306. [DOI] [PubMed] [Google Scholar]
- 25.Onder T.T., Gupta P.B., Mani S.A., Yang J., Lander E.S., Weinberg R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–3654. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- 26.Pinho S.S., Reis C.A., Paredes J., Magalhães A.M., Ferreira A.C., Figueiredo J. The role of N-acetylglucosaminyltransferase III and V in the post-transcriptional modifications of E-cadherin. Hum Mol Genet. 2009;18:2599–2608. doi: 10.1093/hmg/ddp194. [DOI] [PubMed] [Google Scholar]
- 27.Akama R., Sato Y., Kariya Y., Isaji T., Fukuda T., Lu L. N-acetylglucosaminyltransferase III expression is regulated by cell–cell adhesion via the E-cadherin–catenin–actin complex. Proteomics. 2008;8:3221–3228. doi: 10.1002/pmic.200800038. [DOI] [PubMed] [Google Scholar]
- 28.Jamal B.T., Nita-Lazar M., Gao Z., Amin B., Walker J., Kukuruzinska M.A. N-glycosylation status of E-cadherin controls cytoskeletal dynamics through the organization of distinct beta-catenin- and gamma-catenin-containing AJs. Cell Health Cytoskelet. 2009;2009:67–80. doi: 10.2147/chc.s5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Q., Akama R., Isaji T., Lu Y., Hashimoto H., Kariya Y. Wnt/beta-catenin signaling down-regulates N-acetylglucosaminyltransferase III expression: the implications of two mutually exclusive pathways for regulation. J Biol Chem. 2011;286:4310–4318. doi: 10.1074/jbc.M110.182576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koelink P.J., Hawinkels L.J., Wiercinska E., Sier C.F., ten Dijke P., Lamers C.B. 5-Aminosalicylic acid inhibits TGF-beta1 signalling in colorectal cancer cells. Cancer Lett. 2010;287:82–90. doi: 10.1016/j.canlet.2009.05.033. [DOI] [PubMed] [Google Scholar]