

Abstract
The optimal combination of bevacizumab with cytotoxic or cytostatic drugs in recurrent glioblastoma is unknown. We performed a phase 2 trial of combined bevacizumab and fotemustine for patients with glioblastoma at first relapse after radiotherapy and temozolomide. The primary endpoint was 6-month progression-free survival (PFS), while secondary endpoints were overall survival (OS), response rate based on RANO criteria and toxicity. Fifty-four patients with recurrent GBM were enrolled. The authors observed a 6-month PFS rate of 42.6 % (95 % CI 29.3–55.2) and a median PFS of 5.2 months (95 % CI 3.8–6.6). The median OS was 9.1 months (95 % CI 7.3–10.3). Twenty-eight patients (52 %) had a radiographic response, and a significant neurological improvement with steroid reduction was observed in 25/42 symptomatic patients (60 %). MGMT promoter methylation was significantly associated with improved PFS in univariate analysis. Most unifocal tumors at baseline had a focal enhancing progression (76 %), while the diffuse non-enhancing progression accounted for 9.5 %. Response or survival were not associated with any pattern of progression. Survival after failure of treatment was short. Twelve out of 54 patients (22 %) discontinued fotemustine for grade 3/4 myelotoxicity, while 4/54 (7.4 %) discontinued bevacizumab. This study failed to demonstrate a superiority of the combination of bevacizumab and fotemustine over either bevacizumab or fotemustine alone as historical controls. Future studies should explore alternative regimens of combination of the two drugs.
Keywords: Bevacizumab, Fotemustine, Nitrosoureas, Glioblastoma, Recurrence
Introduction
Glioblastoma (GBM) is the most common malignant primary brain tumor, and the standard therapy involves maximal safe surgical resection, followed by radiotherapy with concomitant and adjuvant temozolomide (TMZ) [1, 2]. Despite optimal treatment, GBMs inevitably recur with a median survival of 15–18 months [3].
Treatment options at recurrence are of limited efficacy, and there is no accepted standard of care [4]. GBMs are highly vascularized tumors with elevated expression of vascular endothelial grow factor (VEGF), that drives endothelial cell proliferation and thus new blood vessels formation [5]. Recent studies with bevacizumab, a humanized monoclonal antibody against VEGF, alone or associated with chemotherapy or targeted drugs, have reported higher response rates and prolongation of median and 6-month progression-free survival compared to historical controls with non-bevacizumab treatments [6–8]. As a consequence US Food and Drug Administration (FDA) granted approval of single-agent bevacizumab in May 2009; conversely, the European Medical Agency (EMA) refused bevacizumab approval, mainly for the uncertain impact on overall survival and the lack of a non-bevacizumab control arm in the registration studies. Nitrosoureas are in Europe the standard salvage option in recurrent GBMs. Fotemustine is a chloroethylnitrosourea compound with elevated lipophilic properties that has shown some activity in recurrent GBMs [9–12]. The combination of bevacizumab plus fotemustine has been recently suggested as active and relatively safe in untreated metastatic melanoma patients [13]. No prospective studies are available on the combination of bevacizumab and fotemustine in recurrent GBMs.
Here we present the final results of a phase II Italian study that investigated the role of the combination of bevacizumab and fotemustine in GBMs at first relapse after standard radiotherapy and TMZ.
Materials and methods
Eligibility criteria
Inclusion criteria for the study were as follows: age ≥18 years; Karnofsky performance status (KPS) score ≥60; histological diagnosis of glioblastoma at original surgery or at reoperation; first progression after radiotherapy and concomitant/adjuvant temozolomide; measurable disease on enhanced MRI (≥1 cm) within 1 week prior to treatment; stable corticosteroid dose for ≥7 days before baseline MRI; adequate hematologic, hepatic and renal function: hematocrit >29 %; absolute neutrophil count (ANC) ≥1,000 μL; platelets count ≥100,000 μL; serum aspartate aminotranferase, bilirubin and creatinine <1.5 times normal At least 3 months between completion of radiotherapy and at least 1 month between reoperation and enrollment were required. All patients provided informed consent. Key exclusion criteria included: evidence of CNS hemorrhage on baseline MRI; concurrent therapeutic anticoagulation (LMWH allowed); uncontrolled hypertension; cardiac arrhythmias; history of congestive heart failure or stroke; active infection requiring intravenous antibiotics; urine protein : creatinine ratio >1; pregnancy or nursing; prior stereotactic radiosurgery or any other antiangiogenic agent. The protocol was approved by the Institutional Review Board.
Study design and treatment
This was a multicenter, single arm, open label, phase II study. The primary endpoint was 6-month progression-free survival (PFS-6), while secondary endpoints were overall survival (OS), response rate (RR) and toxicity.
The treatment consisted of an induction phase with bevacizumab at 10 mg/kg intravenously on day 1 and 15 and fotemustine at 75 mg/m2 intravenously on day 1 and day 8, followed after an interval of 3 weeks by a maintenance phase with bevacizumab at 10 mg/kg and fotemustine at 75 mg/m2 every 3 weeks until tumor progression, unacceptable toxicity or withdrawal of consent. Fotemustine doses were held for grade 3 or 4 non-hematological toxicity, grade 3 thrombocytopenia, grade 4 neutropenia, and fever associated with any grade of neutropenia until the event resolved to grade 1 or pretreatment values. Thereafter, doses of fotemustine were reduced by 25 %; chemotherapy doses were also reduced by 25 % for any related-event requiring >2 weeks to recover. Patients who required more that 3 chemotherapy dose reductions were allowed to remain on study and receive bevacizumab alone. Bevacizumab was discontinued for uncontrollable hypertension, grade 2 or greater hemorrhage, arterial thrombosis, severe proteinuria or congestive heart failure. Bevacizumab was held until other related grade 3 events resolved to grade ≤1. Dose reductions of bevacizumab were not allowed. Initiation of each cycle required: an ANC ≥ 1,500 μL; a platelet count ≥100.000 μL; aspartate aminotranferase, bilirubin and creatinine less than twice the institutional upper limit of normal; proteinuria grade ≤2 on urinanalysis; and resolution of any related grade ≥3 event to grade ≤1. A complete blood count and metabolic panel with urinanalysis were obtained every 4 weeks; blood pressure was checked before every infusion of bevacizumab.
Response evaluation
Study investigators determined response by neurological examination and contrast enhanced MRI (performed on a 1.5 Tesla scanner) after the induction phase and then after every other maintenance cycle. One Investigator (R.S.) reviewed all MRI examinations. Response was evaluated based on the recently published response assessment in neuro-oncology (RANO) criteria that require the evaluation of both the enhancing and non-enhancing (hyperintense in T2/FLAIR) components of the tumor [14].
MGMT promoter methylation analysis
Genomic DNA was isolated from paraffin sections of glioblastoma tissue, denaturated with sodium hydroxide in a volume of 35 μL and subjected to bisulfite treatment in a volume of 350 μL for 5 h at 55 °C and then purified. The methylation-specific PCR was performed in a two-step approach.
Statistical analysis
When this study was designed, the sole available study employing bevacizumab on recurrent malignant gliomas useful for comparison was that of Vredenburgh et al. [15]. To achieve 90 % power to detect an increase of 20 % in 6-month PFS (from 40 to 60 %) with 5 % type-one error we aimed to recruit 52 patients.
The characteristics of the patients were described using medians and interquartile ranges for the continuous variables. Percentage frequencies were used for the categorical variables.
PFS was defined as the time from the start of therapy to disease progression or death or last follow-up.
OS was defined as the time from the start of therapy to death or last contact if censored.
Age, gender, Karnofsky score, type of first surgery, second surgery, tumor extension, time from original diagnosis and MGMT status were categorized and analyzed as factors potentially influencing PFS, OS and response.
A Cox proportional hazard model was used to estimate crude and adjusted hazard ratios (HRs) and 95 % confidence intervals (95 % CIs) for a set of potential, pre defined, risk factors of progression of disease and mortality. We included in the multivariate analysis only those variables known in the literature as significant prognostic factors.
Statistical analyses were performed using Stata 11.2 (StataCorp LP, College Station, TX, USA).
Results
Patients characteristics
From May 15, 2007 to Dec 31, 2010, 54 patients with recurrent GBM were enrolled. Patients’ characteristics at study entry are summarized in Table 1. Twenty-four patients (44 %) had a Karnofsky Performance Status of ≥90 at the time of enrollment. Forty-six patients (85 %) had unifocal tumors, while 8 (15 %) multifocal tumors. Twenty patients (37 %) had MGMT unmethylated tumors while 18 (33 %) had MGMT methylated tumors; in the remaining 16 patients data were not available. Eleven out of 54 patients (20 %) were reoperated before inclusion into the study. The median time from original diagnosis to study enrollment was 11 months. Forty-one patients (76 %) were receiving dexamethasone with a median total daily dose of 4 mg (range 2–8). Twenty-nine patients (54 %) were on antiepileptic medication with non-EIAEDs.
Table 1.
Patients characteristics at study entry
| No. | % | |
|---|---|---|
| Gender | ||
| Female | 19 | 35.19 |
| Male | 35 | 64.81 |
| Age (years)a | 57.1 | [14.9] |
| Antiepiletic drugs | ||
| No | 25 | 46.3 |
| Yes | 29 | 53.7 |
| Steroids | ||
| No | 13 | 24.07 |
| Yes | 41 | 75.93 |
| Karnofsky score | ||
| Normal activity (90–100) | 24 | 44.44 |
| Normal activity with effort (80) | 22 | 40.74 |
| Cares for self, unable to carry on normal activity (60–70) | 8 | 14.81 |
| Type of first surgery | ||
| Biopsy | 5 | 9.26 |
| Partial resection | 19 | 35.19 |
| Subtotal/total resection | 30 | 55.56 |
| MGMT status | ||
| Unmethylated | 20 | 37.04 |
| Methylated | 18 | 33.33 |
| Not evaluable | 16 | 29.63 |
| Time from original diagnosis (months)a | 11 | [9.43] |
| Second surgery before treatment | ||
| No | 43 | 79.63 |
| Yes | 11 | 20.37 |
| Tumor extension | ||
| Unifocal | 46 | 85.19 |
| Multifocal | 8 | 14.81 |
| Total | 54 | 100 |
aMedian [iqr]
All patients completed the induction phase, and a median of 7 maintenance cycles were administered (range 1–72).
As for June 15, 2012 study therapy was discontinued due to PD in 50/54 (91 %) patients, while 3 patients were free of tumor progression and alive, and 1 patient died suddenly at home for an unknown reason. The median follow-up for all patients was 9.3 months (95 % CI 7.3–10.5).
Progression-free and overall survival
PFS rate at 6 months was 42.6 % (95 % CI 29.3–55.2) and the median PFS was 5.2 months (95 % CI 3.8–6.6) (Fig. 1a). MGMT status was associated with PFS, although not statistically significant in the adjusted analysis: the progression risk was halved in methylated patients (HR 0.48, 95 % CI 0.21–1.09). Gender, age, Karnofsky score, time from the original diagnosis, type of first surgery, second surgery and tumor extension did not influence PFS in both univariate and multivariate analysis (Table 2).
Fig. 1.
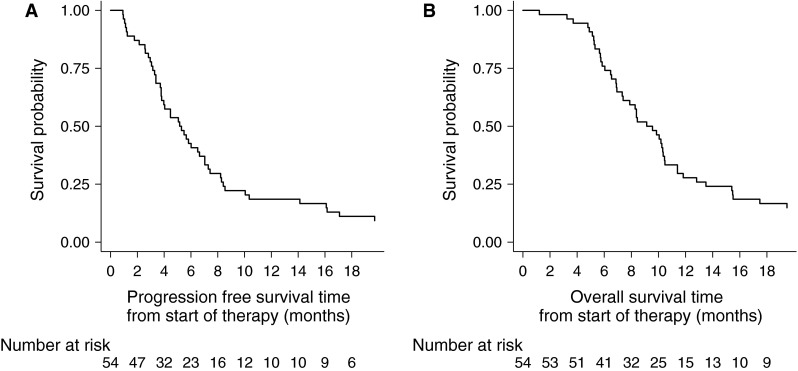
a Progression-free survival from study entry; b overall survival from study entry
Table 2.
Crude and adjusted hazard ratios (HR) of progression free survival
| Univariate effect | Multivariate effect | |||||
|---|---|---|---|---|---|---|
| HR | 95 % CI | p | HR | 95 % CI | p | |
| Gender | ||||||
| Female | 1 | 1 | ||||
| Male | 0.95 | [0.53,1.69] | 0.867 | 1.58 | [0.74,3.40] | 0.241 |
| Age (years) | 0.99 | [0.97,1.02] | 0.654 | 0.99 | [0.96,1.02] | 0.486 |
| Karnofsky score | ||||||
| Normal activity (90–100) | 1 | 1 | ||||
| Normal activity with effort (80) | 0.82 | [0.45,1.51] | 0.53 | 0.98 | [0.50,1.95] | 0.965 |
| Cares for self, unable to carry on normal activity (60–70) | 2.23 | [0.96,5.18] | 0.063 | 1.97 | [0.69,5.63] | 0.203 |
| Type of first surgery | ||||||
| Biopsy/partial | 1 | 1 | ||||
| Subtotal/total | 0.93 | [0.53,1.62] | 0.785 | 1 | [0.53,1.87] | 0.989 |
| MGMT status | ||||||
| Unmethylated | 1 | 1 | ||||
| Methylated | 0.50 | [0.25,0.98] | 0.045 | 0.48 | [0.21,1.09] | 0.078 |
| Not evaluable | 0.65 | [0.34,1.27] | 0.211 | 0.53 | [0.22,1.30] | 0.163 |
| Time from original diagnosis | ||||||
| <11 months | 1 | 1 | ||||
| ≥11 months | 1.43 | [0.81,2.51] | 0.218 | 1.64 | [0.81,3.34] | 0.168 |
| Tumor extension | ||||||
| Unifocal | 1 | |||||
| Multifocal | 1.21 | [0.56,2.61] | 0.621 | |||
| Second surgery | ||||||
| No | 1 | |||||
| Yes | 0.81 | [0.40,1.63] | 0.551 | |||
OS rate was 75.9 % (95 % CI 62.2–85.2) and 29.7 % (95 % CI 18.2–42.0) at 6 and 12 months, respectively, and the median OS was 9.1 months (95 % CI 7.3–10.3) (Fig. 1b).
The risk of death increased in patients with the worst performance status at the enrollment (HR = 3.45, 95 % CI 1.32–9.01). Gender, age, Karnofsky score, time from the original diagnosis, type of first surgery, second surgery, tumor extension and MGMT status did not influence OS in both univariate and multivariate analysis (Table 3).
Table 3.
Crude and adjusted hazard ratios (HR) of overall survival
| Univariate effect | Multivariate effect | |||||
|---|---|---|---|---|---|---|
| HR | 95 % CI | p | HR | 95 % CI | p | |
| Gender | ||||||
| Female | 1 | 1 | ||||
| Male | 0.93 | [0.52,1.65] | 0.793 | 1.35 | [0.64,2.88] | 0.431 |
| Age (years) | 0.99 | [0.96,1.02] | 0.54 | 0.98 | [0.95,1.02] | 0.363 |
| Karnofsky score | ||||||
| Normal activity (90–100) | 1 | 1 | ||||
| Normal activity with effort (80) | 0.78 | [0.43,1.43] | 0.423 | 0.77 | [0.39,1.51] | 0.443 |
| Cares for self, unable to carry on normal activity (60–70) | 3.43 | [1.47,7.99] | 0.004 | 3.45 | [1.32,9.01] | 0.011 |
| Type of first surgery | ||||||
| Biopsy/partial | 1 | 1 | ||||
| Subtotal/total | 0.99 | [0.57,1.74] | 0.983 | 0.95 | [0.52,1.74] | 0.874 |
| MGMT status | ||||||
| Unmethylated | 1 | 1 | ||||
| Methylated | 0.72 | [0.37,1.43] | 0.35 | 0.82 | [0.38,1.74] | 0.602 |
| Not evaluable | 0.98 | [0.50,1.90] | 0.942 | 0.98 | [0.45,2.13] | 0.95 |
| Time from original diagnosis | ||||||
| <11 months | 1 | 1 | ||||
| ≥11 months | 1.66 | [0.95,2.90] | 0.077 | 1.52 | [0.79,2.91] | 0.206 |
| Tumor extension | ||||||
| Unifocal | 1 | |||||
| Multifocal | 1.14 | [0.53,2.45] | 0.737 | |||
| Second surgery | ||||||
| No | 1 | |||||
| Yes | 0.96 | [0.46,1.98] | 0.902 | |||
The median time of OS after treatment failure was 3.45 months (95 % CI 2.20–4.53) (Fig. 2a).
Fig. 2.

a Whole-cohort: overall survival after treatment failure; b unifocal tumors at baseline: overall survival after treatment failure by patterns of progression
Response
The RR was as follows: CR in 2/54 patients (4 %), PR in 26/54 (48 %), SD in 20/54 (37 %) and PD in 6/54 (11 %). The overall response rate (CR + PR) was 52 %. The median time to maximum response was 4 weeks (range 4–12 weeks). Among the 41 patients who were on dexamethasone at study initiation, 32 (78 %) were able to taper, including 17 (41.5 %) who completely discontinued the treatment, while 9 patients (22 %) required a stable dexamethasone dose. Of the 13 patients who were not receiving dexamethasone at study enrollment, 2 (15 %), who had progression at first evaluation, required therapy with dexamethasone. Overall, a significant neurologic improvement was observed in 25/42 symptomatic patients (60 %). Response was not predicted by any clinical factor, including the MGMT status .
Patterns of failure
Patterns of tumor failure on MRI after study treatment were available in 50/54 patients (92.5 %). All 8 tumors, that were multifocal before treatment, had a multifocal progression.
Forty-two patients had unifocal tumors before treatment, and patterns of progression were as follows: local (enhancing) in 32/42 (76 %), distant (enhancing) in 3/42 (7 %), multifocal (local + distant enhancing) in 2/42 (4.5 %), diffuse (nonenhancing) in 4/42 (9.5 %) and isolated leptomeningeal spread in 1/42 (2 %). The pattern of tumor failure (local vs. non local) was not associated with the previous response to treatment. Overall survival after failure was not significantly different according to patterns of tumor progression (local vs. non local) (Fig. 2b).
Salvage chemotherapy after progression on bevacizumab + fotemustine
Following progression on bevacizumab + fotemustine, 21/50 patients (42 %) received salvage chemotherapy (18 PCV, 1 dose-dense temozolomide, and 2 high-dose tamoxifen) and in 1 of these patients bevacizumab was maintained beyond progression in association with chemotherapy. Twenty-nine out of 50 patients (58 %) received supportive care alone. Patients who received salvage chemotherapy had a median survival of 4.53 months (95 % CI 3.19–5.52), while patients who received supportive care alone had a median survival of 2.20 months (95 % CI 1.31–3.45).
Toxicity
Toxicity is summarized in Table 4. Most patients experienced grade 1 or 2 toxicities. Grade 3 toxicities were predominantly hematologic, including neutropenia in 7 patients (13 %) and thrombocytopenia in 5 patients (9 %). Other grade 3 toxicities included wound dehiscence in 3 patients (5.5 %), fatigue and deep venous thrombosis in 2 patients (4 %), and hypertension and hemorrhage (61) in 1 patient (1.8 %). Grade 4 toxicities included pulmonary embolism in 2 patients (4 %), and hypertension with reversible posterior encephalopathy, stroke, neutropenia and thrombocytopenia in 1 patient, respectively. One patient experienced grade 5 CNS hemorrhage. Twelve out of 54 patients (22 %) with persistent grade 3 or 4 neutropenia or thrombocytopenia discontinued fotemustine and 11 required dose modification. Four out of 54 patients (7.4 %) discontinued bevacizumab (1 stroke, 1 intratumoral hemorrhage, 1 GI perforation and 1 pulmonary embolism).
Table 4.
Toxicities
| Adverse event | Grade: no. of patients | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| Nonhematologic toxicity | |||||
| Hypertension | 2 | 6 | 1 | 1 | – |
| Infection | – | – | – | – | – |
| Wound dehiscence | – | – | 3 | – | – |
| Fatigue | 18 | 5 | 2 | – | – |
| Proteinuria | 7 | 10 | – | – | – |
| Rash | – | – | – | – | – |
| Hyperpigmentation | 2 | – | – | – | – |
| Nausea/vomiting | – | – | – | – | – |
| Hypophonia | 5 | – | – | – | – |
| GI perforation/fistula | – | – | – | – | – |
| Hemorrhage, CNS | 2 | – | – | – | 1 |
| Hemorrhage, GI | – | 1 | 1 | – | – |
| Epistaxis | 2 | – | – | – | – |
| Stroke | – | – | – | 1 | – |
| DVT | – | – | 2 | – | – |
| PE | – | – | – | 2 | – |
| Transaminase elevation | – | – | – | – | – |
| Hematologic toxicity | |||||
| Anemia | 2 | – | – | – | – |
| Neutropenia | 1 | 4 | 7 | 1 | – |
| Thrombocitopenia | 3 | 8 | 5 | 1 | – |
Discussion
This is the first phase II trial that has explored the combination of bevacizumab and fotemustine in patients with GBMs. A series of retrospective and prospective studies have evaluated the association of bevacizumab with miscellaneous other agents, including irinotecan, etoposide, temozolomide, carboplatin, cetuximab and erlotinib in patients with recurrent GBMs [15–25], and all have achieved outcomes that are not better than bevacizumab monotherapy [6–8, 26]. Overall, combination studies have shown PFS-6 of 19–50 % and median OS of 6–10.2 months compared with PFS-6 of 25–42.6 % and OS of 6.5–9.2 months with bevacizumab alone. Similar results were reported in terms of radiologic responses (20–57 % with drug combinations vs. 29–42 % with bevacizumab alone). In our study we observed a 6-month PFS of 42.6 %, a median PFS of 5.2 months, an OS of 9.1 months and a RR of 52 %: these results are not significantly superior over the best results reported with either bevacizumab alone or in combination. Unfortunately, in this trial we were not able to evaluate the quality of life over time with specific questionnaires.
We chose a nitrosourea to be combined with bevacizumab based on the rationale that bevacizumab might enhance the delivery of an active cytotoxic drug, and adequate safety would be expected with this regimen due to non-overlapping primary toxicities of each of the agents. Four phase II trials evaluated fotemustine in recurrent glioblastomas. Three studies [9–11] used the same induction/maintenance schedule of our study, and reported similar outcome results: PFS-6 of 20.9–52 %, PFS of 1.7–6.1 months and OS of 6–9.1 months. Another study [12], that used longer rest periods (2 weeks instead of 1 week) between doses during the induction phase, reported better results (PFS-6 of 61 %, mPFS of 6.7 months and OS of 11.1 months), and reduced grade 3 or 4 myelotoxicity. In our study 22 % of patients discontinued fotemustine due to grade 3 or 4 myelotoxicity after the induction phase: maybe we could not exploit entirely the potential synergistic effect of the combination. In an ongoing Dutch phase II randomized trial on recurrent GBMs (BELOB trial), comparing bevacizumab + lomustine versus either bevacizumab or lomustine alone, the initial dose of the nitrosourea was lowered due to a high grade 3 and 4 myelotoxicity, and the preliminary results in terms of survival are encouraging [27]. Another explanation for the lack of synergism of our combination is that in the maintenance phase we used bevacizumab at the dose of 10 mg/kg every 3 weeks instead of every 2 weeks: however, to date there is no evidence of a relationship between dose, schedule and response [28].
The role of MGMT as a prognostic or predictive marker in patients with recurrent GBM receiving alkylating chemotherapy (temozolomide, nitrosoureas) is debated. Some studies found longer PFS and OS in patients with MGMT promoter methylated tumors [29–31], whereas others did not [32–37]. Addeo et al. [12], who used fotemustine alone, observed a trend toward prolonged PFS-6 for methylated patients. In our study we found an association between MGMT promoter methylation and longer PFS that was statistically significant in univariate analysis only. In general, the lack of a correlation between MGMT promoter methylation and outcome in recurrent GBMs may be due to the small sample size of the studies, the poor outcome of GBM at relapse or an absence of a true association.
In the present study we analyzed the patterns of failure after treatment using the same categories employed in recent studies [38–40]. As nearly 15 % of our patients had bilateral multifocal disease (both enhancing and non-enhancing) at baseline, thus rendering difficult and unreliable a distinction between local and non local progression and between true tumor progression and radiation effects, we restricted the analysis to unifocal tumors at baseline. The majority of these patients did not have a shift in the patterns of failure, with a rate of local enhancing progression of 76 %, which is similar to that has been reported in the literature [23, 38, 41]. Conversely, the proportion of patients displaying a diffuse non-enhancing pattern of tumor progression (“gliomatosis-like”) was significantly lower (9.5 %) than that reported in previous studies [40–44], thus raising the possibility that fotemustine could have some anti-infiltrative properties.
Some clinical reports have suggested that the non-enhancing and multifocal patterns of tumor progression could be associated with a previous response [42] or poorer outcome [43, 44]. We did not observe any difference in terms of response or survival between patients who had local versus non-local progression, as already reported by Pope et al. (2011) in the BRAIN study.
Overall, our correlations have some limitations: the sample size was small, the physician that evaluated the MRI scans was not blind to clinical data, and as a single investigator analyzed the tumor patterns, interobserver reliability to confirm pattern assignments was not assessed.
Last, we confirm that the outcome following disease progression on bevacizumab-based regimen is poor [24, 45]. A longer survival was observed among patients receiving salvage chemotherapy, as it was more commonly offered to patients with younger age and higher Karnofsky score.
In conclusion, in this trial we were unable to demonstrate a superiority of the combination of bevacizumab and fotemustine over either bevacizumab or fotemustine alone as historical controls. Future studies should investigate whether a clinically meaningful benefit from the combination could emerge by employing lower doses of bevacizumab and/or more protracted schedules of fotemustine. More in general, the issue of the potential benefit of adding a nitrosourea to bevacizumab is still unresolved, and ultimately the ongoing EORTC 26101 phase III study will hopefully give an answer.
Conflict of interest
Dr. Soffietti served on an advisory board with Genentech-Roche and has received compensation. No other author has a financial disclosure.
Ethical standard
This study was performed complying with Italian laws.
References
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Grossman SA, Ye X, Piantadosi S, et al. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin Cancer Res. 2010;16:2443–2449. doi: 10.1158/1078-0432.CCR-09-3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma—are we there yet? Neuro Oncol. 2013;15:4–27. doi: 10.1093/neuonc/nos273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plate KH, Risau W. Angiogenesis in malignant gliomas. Glia. 1995;15:339–347. doi: 10.1002/glia.440150313. [DOI] [PubMed] [Google Scholar]
- 6.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 7.Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–745. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chamberlain MC, Johnston SK. Salvage therapy with single agent bevacizumab for recurrent glioblastoma. J Neurooncol. 2010;96:259–269. doi: 10.1007/s11060-009-9957-6. [DOI] [PubMed] [Google Scholar]
- 9.Scoccianti S, Detti B, Sardaro A, et al. Second-line chemotherapy with fotemustine in temozolomide-pretreated patients with relapsing glioblastoma: a single institution experience. Anticancer Drugs. 2008;19:613–620. doi: 10.1097/CAD.0b013e3283005075. [DOI] [PubMed] [Google Scholar]
- 10.Brandes AA, Tosoni A, Franceschi E, et al. Fotemustine as second-line treatment for recurrent or progressive glioblastoma after concomitant and/or adjuvant temozolomide: a phase II trial of Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO) Cancer Chemother Pharmacol. 2009;64:769–775. doi: 10.1007/s00280-009-0926-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fabrini MG, Silvano G, Lolli I, et al. A multi-institutional phase II study on second-line Fotemustine chemotherapy in recurrent glioblastoma. J Neurooncol. 2009;92:79–86. doi: 10.1007/s11060-008-9739-6. [DOI] [PubMed] [Google Scholar]
- 12.Addeo R, Caraglia M, De Santi MS, et al. A new schedule of fotemustine in temozolomide-pretreated patients with relapsing glioblastoma. J Neurooncol. 2011;102:417–424. doi: 10.1007/s11060-010-0329-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Del Vecchio M, Mortarini R, Canova S, et al. Bevacizumab plus fotemustine as first-line treatment in metastatic melanoma patients: clinical activity and modulation of angiogenesis and lymphangiogenesis factors. Clin Cancer Res. 2010;16:5862–5872. doi: 10.1158/1078-0432.CCR-10-2363. [DOI] [PubMed] [Google Scholar]
- 14.Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28:1963–1972. doi: 10.1200/JCO.2009.26.3541. [DOI] [PubMed] [Google Scholar]
- 15.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–1259. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 16.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25:4722–4729. doi: 10.1200/JCO.2007.12.2440. [DOI] [PubMed] [Google Scholar]
- 17.Gilbert MR, Wang M, Aldape KD, et al. (2009) RTOG 0625: a phase II study of bevacizumab with irinotecan in recurrent glioblastoma (GBM). J Clin Oncol. 27(15S) Abstract no 2011
- 18.Nghiemphu PL, Liu W, Lee Y, et al. Bevacizumab and chemotherapy for recurrent glioblastoma: a single-institution experience. Neurology. 2009;72:1217–1222. doi: 10.1212/01.wnl.0000345668.03039.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Francesconi AB, Dupre S, Matos M, et al. Carboplatin and etoposide combined with bevacizumab for the treatment of recurrent glioblastoma multiforme. J Clin Neurosci. 2010;17:970–974. doi: 10.1016/j.jocn.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 20.Hasselbalch B, Lassen U, Hansen S, et al. Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: a phase II trial. Neuro Oncol. 2010;12:508–516. doi: 10.1093/neuonc/nop063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sathornsumetee S, Desjardins A, Vredenburgh JJ, et al. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro Oncol. 2010;12:1300–1310. doi: 10.1093/neuonc/noq099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verhoeff JJ, Lavini C, van Linde ME, et al. Bevacizumab and dose-intense temozolomide in recurrent high-grade glioma. Ann Oncol. 2010;21:1723–1727. doi: 10.1093/annonc/mdp591. [DOI] [PubMed] [Google Scholar]
- 23.Desjardins A, Reardon DA, Coan A, et al. Bevacizumab and daily temozolomide for recurrent glioblastoma. Cancer. 2012;118:1302–1312. doi: 10.1002/cncr.26381. [DOI] [PubMed] [Google Scholar]
- 24.Reardon DA, Desjardins A, Peters KB, et al. Phase II study of carboplatin, irinotecan, and bevacizumab for bevacizumab naïve, recurrent glioblastoma. J Neurooncol. 2012;107:155–164. doi: 10.1007/s11060-011-0722-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Field KM, Simes J, Wheeler H, et al. (2013) A randomized phase II study of carboplatin and bevacizumab in recurrent glioblastoma multiforme (CABARET). J Clin Oncol 31(15S). Abstract no 2017
- 26.Raizer JJ, Grimm S, Chamberlain MC, et al. A phase 2 trial of single agent bevacizumab given in an every-3-week schedule for patients with recurrent high-grade gliomas. Cancer. 2010;116:5297–5305. doi: 10.1002/cncr.25462. [DOI] [PubMed] [Google Scholar]
- 27.Taal W, Oosterkamp HM, Walenkamp AME, Beerepoot LV, et al. (2013) A randomized phase II study of bevacizumab versus bevacizumab plus lomustine versus lomustinw single agent in recurrent glioblastoma: The Dutch BELOB study. J Clin Oncol 31(15S). Abstract no 2001
- 28.Wong ET, Gautam S, Malchow C, Lun M, Pan E, Brem S. Bevacizumab for recurrent glioblastoma multiforme: a meta-analysis. J Natl Compr Canc Netw. 2011;9:403–407. doi: 10.6004/jnccn.2011.0037. [DOI] [PubMed] [Google Scholar]
- 29.Metellus P, Coulibaly B, Nanni I, et al. Prognostic impact of O6-methylguanine-DNA methyltransferase silencing in patients with recurrent glioblastoma multiforme who undergo surgery and carmustine wafer implantation: a prospective patient cohort. Cancer. 2009;115:4783–4794. doi: 10.1002/cncr.24546. [DOI] [PubMed] [Google Scholar]
- 30.Felsberg J, Thon N, Eigenbrod S, et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer. 2011;129:659–670. doi: 10.1002/ijc.26083. [DOI] [PubMed] [Google Scholar]
- 31.Hammond A, Norden AD, Lesser GJ, et al. (2011) Phase II study of dose-intense temozolomide in recurrent glioblastoma. J Clin Oncol 29(15S) Abstract no 2038 [DOI] [PMC free article] [PubMed]
- 32.Brandes AA, Tosoni A, Cavallo G, et al. Temozolomide 3 weeks on and 1 week off as first-line therapy for recurrent glioblastoma: phase II study from Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO) Br J Cancer. 2006;95:1155–1160. doi: 10.1038/sj.bjc.6603376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wick A, Felsberg J, Steinbach JP, et al. Efficacy and tolerability of temozolomide in an alternating weekly regimen in patients with recurrent glioma. J Clin Oncol. 2007;25:3357–3361. doi: 10.1200/JCO.2007.10.7722. [DOI] [PubMed] [Google Scholar]
- 34.Sadones J, Michotte A, Veld P, et al. MGMT promoter hypermethylation correlates with a survival benefit from temozolomide in patients with recurrent anaplastic astrocytoma but not glioblastoma. Eur J Cancer. 2009;45:146–153. doi: 10.1016/j.ejca.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 35.Kong DS, Lee JI, Kim JH, et al. Phase II trial of low-dose continuous (metronomic) treatment of temozolomide for recurrent glioblastoma. Neuro Oncol. 2010;12:289–296. doi: 10.1093/neuonc/nop030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stockhammer F, Misch M, Koch A, et al. Continuous low-dose temozolomide and celecoxib in recurrent glioblastoma. J Neurooncol. 2010;100:407–415. doi: 10.1007/s11060-010-0192-y. [DOI] [PubMed] [Google Scholar]
- 37.Perry MS, Perry JR, Belanger K, Mason WP, et al. Phase II trial of continuous dose-intense temozolomide in recurrent malignant glioma: RESCUE study. J Clin Oncol. 2010;28:2051–2057. doi: 10.1200/JCO.2009.26.5520. [DOI] [PubMed] [Google Scholar]
- 38.Pope WB, Xia Q, Paton VE, et al. Patterns of progression in patients with recurrent glioblastoma treated with bevacizumab. Neurology. 2011;76:432–437. doi: 10.1212/WNL.0b013e31820a0a8a. [DOI] [PubMed] [Google Scholar]
- 39.Gállego Pérez-Larraya J, Lahutte M, Petrirena G, Reyes-Botero G, et al. Response assessment in recurrent glioblastoma treated with irinotecan-bevacizumab: comparative analysis of the Macdonald, RECIST, RANO, and RECIST + F criteria. Neuro Oncol. 2012;14:667–673. doi: 10.1093/neuonc/nos070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radbruch A, Lutz K, Wiestler B, et al. Relevance of T2 signal changes in the assessment of progression of glioblastoma according to the Response Assessment in Neurooncology criteria. Neuro Oncol. 2012;14:222–229. doi: 10.1093/neuonc/nor200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wick A, Dörner N, Schäfer N, et al. Bevacizumab does not increase the risk of remote relapse in malignant glioma. Ann Neurol. 2011;69:586–592. doi: 10.1002/ana.22336. [DOI] [PubMed] [Google Scholar]
- 42.Norden AD, Young GS, Setayesh K. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology. 2008;70:779–787. doi: 10.1212/01.wnl.0000304121.57857.38. [DOI] [PubMed] [Google Scholar]
- 43.Iwamoto FM, Abrey LE, Beal K. Patterns of relapse and prognosis after bevacizumab failure in recurrent glioblastoma. Neurology. 2009;73:1200–1206. doi: 10.1212/WNL.0b013e3181bc0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuniga RM, Torcuator R, Jain R, et al. Efficacy, safety and patterns of response and recurrence in patients with recurrent high-grade gliomas treated with bevacizumab plus irinotecan. J Neurooncol. 2009;91:329–336. doi: 10.1007/s11060-008-9718-y. [DOI] [PubMed] [Google Scholar]
- 45.Quant EC, Norden AD, Drappatz J, et al. Role of a second chemotherapy in recurrent malignant glioma patients who progress on bevacizumab. Neuro Oncol. 2009;11:550–555. doi: 10.1215/15228517-2009-006. [DOI] [PMC free article] [PubMed] [Google Scholar]