

Abstract
Purpose
The MAPK pathway is a crucial regulator of cell proliferation, survival, and resistance to apoptosis. MEK inhibitors are being explored as a treatment option for patients with KRAS mutant colorectal cancer (CRC) who are not candidates for EGFR-directed therapies. Initial clinical results of MEK inhibitors have yielded limited single-agent activity in CRC, indicating that rational combination strategies are needed.
Experimental Design
In this study, we performed unbiased GSEA and synthetic lethality screens with selumetinib, which identified the non-canonical Wnt/Ca++ signaling pathway as a potential mediator of resistance to the MEK1/2 inhibitor selumetinib. To test this, we utilized shRNA constructs against relevant WNT receptors and ligands resulting in increased responsiveness to selumetinib in CRC cell lines. Further, we evaluated the rational combination of selumetinib and WNT pathway modulators and demonstrated synergistic antiproliferative effects in in vitro and in vivo models of CRC.
Results
Importantly, this combination not only demonstrated tumor growth inhibition but also tumor regression in the more clinically relevant patient-derived tumor explant (PDTX) models of CRC. In mechanistic studies, we observed a trend towards increased markers of apoptosis in response to the combination of MEK and WntCa++ inhibitors, which may explain the observed synergistic antitumor effects.
Conclusions
These results strengthen the hypothesis that targeting both the MEK and Wnt pathways may be a clinically effective rational combination strategy for metastatic CRC patients.
INTRODUCTION
Activating mutations in the KRAS proto-oncogene are present in up to 40-50% of colorectal cancer (CRC) patient tumors, while another 10% are reported to have BRAF V600E mutations (1-3). Retrospective clinical studies have confirmed the lack of benefit of epidermal growth factor receptor (EGFR)-directed therapy in patients with KRAS mutations, limiting treatment options to standard chemotherapeutics with or without VEGF pathway-targeted agents (4-8). This creates an urgent need to test new targeted agents such as inhibitors of the MAPK/MEK/ERK pathway. MEK is central to the pathogenesis of CRC as a downstream effector of mutant KRAS as well as in mediating effects of cell surface receptors such as the EGFR and IGF-1R (9-11). Despite the entry of MEK inhibitors into Phase I and II trials, single-agent activity has been limited in CRC, suggesting that targeting resistance pathways through rational combination strategies may lead to greater efficacy (12-15). Previous studies in our laboratory (16) have identified the Wnt signaling pathway as a resistance mechanism to selumetinib, a selective inhibitor of MEK1/2 (AZD6244; ARRAY-142886; AstraZeneca, Inc), that has been investigated as monotherapy and is currently in the clinic evaluating efficacy in combination with chemotherapy for several cancer types (12, 14, 17-20). The wingless/integrated (Wnt) signaling pathway has been described as canonical or noncanonical based on β–catenin dependence (21). The canonical pathway signals via the frizzled (FZD) family of G-protein coupled receptors resulting in the stabilization and cytoplasmic accumulation of β-catenin, which then translocates to the nucleus and interacts with TCF-Lef transcription factors to regulate genes involved in proliferation, migration and survival (21). The noncanonical pathways, also termed β–catenin independent, Wnt/ Ca2+ or planar cell polarity, are also activated by Wnt/FZD ligands and receptors, leading to the release of intracellular calcium. Increased calcium leads to the activation of the serine/threonine phosphatase, calcineurin (CN) (22, 23). Calcineurin induced de-phosphorylation of Nuclear Factor of Activated T-cells (NFAT) results in its translocation to the nucleus and transcriptional regulation of NFAT-dependent genes (21). We initially studied the role of Wnt signaling in mediating resistance to MEK inhibitors based upon the observation of high levels of Wnt pathway gene expression in selumetinib-resistant KRAS mutant CRC cell lines (16).
Recent advances in genome-wide screening technologies have resulted in the identification of new combination targets through the use of synthetic lethality for a number of clinical anticancer agents (24-26). The aim of the current study was to test the hypothesis that pharmacologic modulation of the Wnt signaling pathway, discovered through gene set enrichment analysis and synthetic lethality as mediating resistance to MEK inhibition, would result in synergistic antitumor effects against in vitro and in vivo models of CRC when combined with MEK blockade. Based on these preclinical results, this rational combination could lead to a promising new therapeutic strategy for advanced CRC patients.
MATERIALS AND METHODS
Gene Set Enrichment Analysis
Gene Set Enrichment Analysis (27) was performed on 26 CRC cell lines using the GSEA software as previously reported (16). See Supplementary Methods for details.
Synthetic Lethality Screen Analysis
The Synthetic Lethality Screen Analysis (SLS) was performed using the SW480 (KRAS mutant) and RKO (BRAF V600E mutant) CRC cell lines. A lentiviral-expressed genome-wide human short-hairpin RNA (shRNA) library obtained from Systems Biosciences was utilized. The library contains 3-5 shRNAs per target gene, targeting 47,000 human transcripts. For the screen, the CRC cell lines were infected with the lentiviral shRNA library and subjected to puromycin selection for 2 weeks to obtain a pure population of shRNA expressing cells, and to eliminate shRNAs that targeted essential genes. The cells were then divided into 6 populations: 3 untreated, and 3 treated for 72 hours with selumetinib at a dose equivalent to 1 μm/L. Total RNA was isolated from each group of cells, reverse-transcribed and PCR amplified. The PCR products were then deep-sequenced by the Illumina Genome AnalyzerIIx. Sequences obtained from the sequencer were analyzed by the BiNGS!SL-seq system (28) to identify shRNAs that were depleted in the treated samples, as previously described and validated (26, 29, 30).
Patient-Derived Tumor Explant Models
Animal studies were conducted in accordance with the NIH guidelines for the care and use of laboratory animals in a facility accredited by the American Association for Accreditation of Laboratory Animal Care, and approved by the University of Colorado Institutional Animal Care and Use Committee prior to initiation. PDTX establishment and treatment protocols were conducted under previously described procedures (16, 31). See Supplementary Methods for details.
In vitro Effects of MEK and Wnt inhibitors
Cell culture, proliferation assays, shRNA knockdowns, qRT-PCR, immunoblotting analysis, clonogenic assay, caspase 3/7 assay, Annexin V assay, 3D cell culture, luciferase assay were performed as previously described (16, 29, 31, 32). See Supplementary Methods for details.
In vivo Effects of MEK and Wnt inhibitors
Immunohistochemistry and pharmacokinetic analysis of PDTX tumors were performed as previously described (16, 31, 33-35). See Supplementary Methods for details.
Statistical Analyses
Statistical calculations were performed using Microsoft Excel and GraphPad Prism Software version 5.0 (La Jolla, CA). For comparisons of two groups un-paired t-test was performed. When comparisons of multiple groups were needed an ANOVA was performed. The specific tests applied are included in the figure legends.
RESULTS
Integrative Analysis of GSEA and SLS
To identify pathways enriched in selumetinib-resistant CRC cell lines, we previously performed an unbiased GSEA analysis of a panel of 26 CRC cell lines (16). Within the 20 pathways identified as enriched in selumetinib-resistant lines (Table S1), the Wnt signaling pathway was one of the top scoring pathways (p = 0.03). As depicted in Figure 1, core genes involved in the canonical and noncanonical Wnt signaling pathways were enriched, consistent with our previous published gene expression analysis on 11 KRAS mutant CRC cell lines (16). Next, we employed a complementary approach and performed an unbiased genome-wide SLS on two CRC cell lines (SW480 and RKO) to identify genes and pathways that when inhibited may increase response to selumetinib. Interestingly, we found that this functional genomic approach independently identified shRNAs in the canonical and non-canonical Wnt pathway as being synthetically lethal for selumetinib (Figure 1). Of note, concordant with previously published data, FZD2 was identified as a top-scoring gene in both GSEA and SLS analyses (16, 36).
Figure 1.
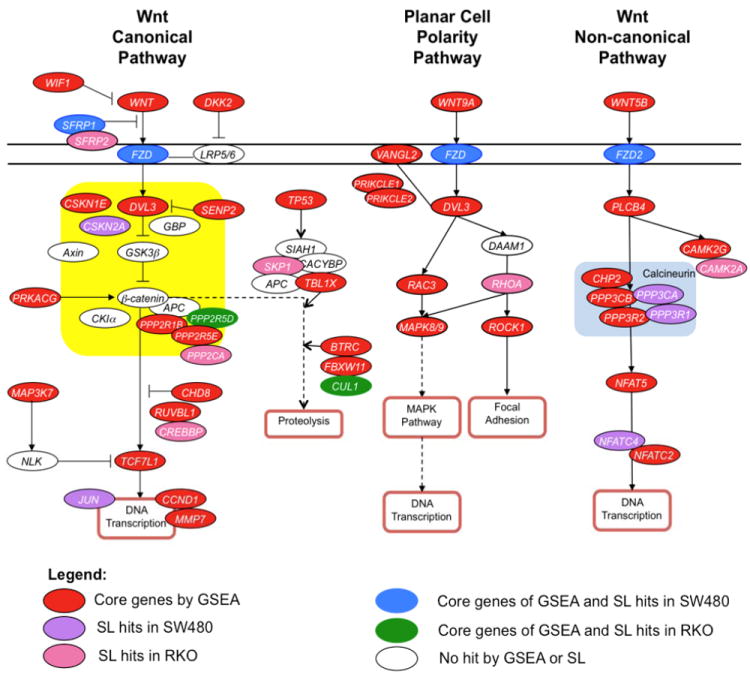
Canonical (A) and noncanonical (B) Wnt signaling pathway represented by KEGG analysis. Red ovals represent core genes in the Wnt pathway with increased expression in CRC cells resistant to selumetinib. Purple and pink ovals represent synthetically lethal genes for SW480 and RKO CRC cell lines respectively. Blue and green ovals represent top-scoring genes in both GSEA and SLS analyses for SW480 and RKO respectively.
Wnt pathway inhibition modulates responsiveness to selumetinib in CRC cell lines
To test the hypothesis that components of the Wnt pathway mediate resistance to selumetinib, shRNA knockdown (KD) experiments were performed to modulate Wnt pathway gene expression. Four Wnt pathway genes, FZD2, FZD3, Wnt2B and Wnt5B, were knocked down in the selumetinib-resistant CRC cell lines SW480 and RKO. These genes, receptors and ligands respectively, were identified by GSEA to be upregulated in selumetinib-resistant CRC cell lines. Moreover, FZD2 was one of the top scoring SL hits in the screen. The relative expression levels of FZD2 and Wnt5B mRNA were significantly reduced relative to a control in the RKO CRC cell line, as measured by qRT-PCR (Suppl. Figure 1A). Moreover, shRNAs targeting the Wnt pathway receptor FZD3 significantly reduced relative levels of this mRNA in both RKO and SW480 CRC cell lines. However, when we attempted to knock down the Wnt pathway ligand, Wnt2B, we observed a significant reduction in this mRNA only in SW480 cells (Suppl. Figure 1 B, C). Possibly due to the low basal expression levels of Wnt ligands and FZD receptors, immunoblot analysis demonstrated only modest or no reduction of Wnt receptors and ligands at the protein level (Suppl. Figure 1D).
To evaluate the effect of these KD experiments on drug responsiveness, proliferation assays were performed in parental, and shRNA transfected RKO and SW480 CRC cell lines treated with selumetinib. Knockdown of Wnt pathway receptors, FZD2 and FZD3 consistently resulted in statistically significant increases in percent inhibition in response to selumetinib in RKO and SW480 cell lines. In addition, KD of Wnt2B showed a trend toward increased percent inhibition that was not statistically significant, while no increase in responsiveness to selumetinib in the Wnt5B KD cells was observed (Suppl. Figure 2). Interestingly, when the same genes were knocked down in the selumetinib-sensitive SW620 CRC cell line, no increase in responsiveness was observed, reinforcing our hypothesis that modulation of sensitivity through Wnt signaling may be restricted to the resistant CRC cell lines (data not shown). To further evaluate the role of Wnt signaling in resistance to selumetinib, we tested the effects of stable expression of Wnt5B, Wnt2B, FZD2, FZD3, or a dominant negative TCF on sensitivity in the selumetinib sensitive CRC cell lines, LS513 and SW620. As depicted in Supplementary Figure 3, we did not observe a significant change in sensitivity when these lines were exposed to increasing concentrations of selumetinib. These results are perhaps not surprising since our hypothesis is that CRC cell lines that are sensitive to selumetinib exhibit low levels of Wnt pathway activation and thus overexpression of a single component of the pathway is not sufficient to elicit a phenotypic change. Likewise, expression of a dominant negative TC4 construct resulted in no significant increase in resistance to selumetinib. Again, this could be the result of low Wnt signaling in these selumetinib sensitive cell lines, whose main pathway addiction is likely the Ras/MAPK pathway and not Wnt.
Synergistic antiproliferative effects of selumetinib and the Wnt inhibitors CsA and TNP-470 in CRC cell lines
Recent reports have demonstrated that the noncanonical Wnt pathway confers resistance to Bcr-Abl inhibition in leukemia models (32). Moreover, our genomic screens and mechanistic studies have shown that this β-catenin independent pathway may mediate responsiveness to MEK inhibitors in CRC. As there are currently no selective noncanonical Wnt inhibitors available, we utilized TNP-470 and CsA, as noncanonical Wnt pathway probes. These compounds have been previously investigated as Wnt inhibitors, although they also exhibit other functions as well (32, 37). Appropriate doses of CsA (0-5 μmol/L) and TNP-470 (0-10 μmol/L) were obtained by exposing a panel of 15 CRC cell lines to the each agent and assessing antiproliferative effects by the SRB method (data not shown). Based on previously published data, three selumetinib-resistant (SW480, RKO, NCI-H508) and one selumetinib-sensitive (SW620) CRC cell lines were chosen for further combination studies (16). After exposure to selumetinib and CsA, synergistic antiproliferative effects were observed at most of the combination doses in RKO and SW480 cells (Figure 2A; Table S2). In contrast, the combination of selumetinib and TNP-470 resulted in synergistic antiproliferative effect in RKO, but not in the SW480 CRC cell line perhaps due to the strong single agent effect of TNP-470. Of note, the dose range of selumetinib used in these combination studies was reduced at least 50% compared to the IC50 of single agent selumetinib for SW480 and RKO cell lines, as determined by SRB.
Figure 2.
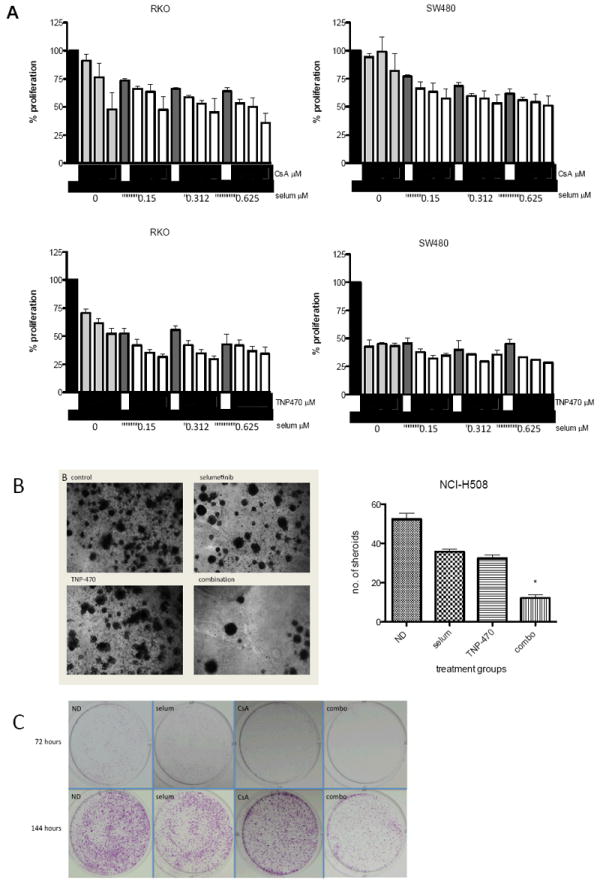
Antiproliferative effects of selumetinib in combination with CsA or TNP-470 in SW480, RKO and NCI-H508 cells. (A) Results of SRB proliferation assay after 72 hours of exposure to indicated concentrations of selumetinib alone or in combination with CsA (1.25, 2.5, 5 μm/mL), or TNP-470 (2.5, 5, 10 μm/mL) as indicated by triangles. Data are expressed as percent proliferation compared to no drug controls ± SD.
(B) Representative images of 3D cultures of NCI-H508 CRC cell line spheroids treated with selumetinib and TNP-470 for 3 weeks, as described in Materials and Methods. Antiproliferative effects for each treatment group were quantified by averaging the number of spheroids in four separate fields per chamber at 4X magnification. Bar graph depicts mean of three separate experiments ± SD. (C) Representative images of clonogenic assays of SW480 cell line treated with either single agents selumetinib (0.3 μm/mL) or CsA (5 μm/mL), or in combination for 72 hours. Cells were then stained with crystal violet at 72 h (top panel) or allowed to re-grow for an additional 72 hours before staining (144 h; bottom panel).
To further evaluate the synergistic antiproliferative effect of selumetinib in combination with inhibitors of the Wnt signaling pathway, and to test the activity of TNP-470 as a noncanonical Wnt inhibitor in a more tumor-like, non-angiogenic environment, three-dimensional (3D) spheroid assays were performed. As depicted in Figure 2B, synergy was detected with the combination of selumetinib and TNP-470. These data suggest that in vitro, the synergistic antiproliferative effects were the result of inhibition of the MEK and Wnt signaling pathways rather than effects of TNP-470 on angiogenic targets.
Futher, these synergistic antiproliferative were confirmed with a clonogenic assay using the SW480 CRC cell line. Cells were exposed to selumetinib, CsA, or the combination, stained with crystal violet or allowed to re-grow in fresh media for an additional 72 hours before staining and analysis. At 72 hours, there was a clear combination effect which was even more pronounced after the recovery growth period of an additional 72 hours (Figure 2C). These results suggest a cytotoxic effect of the combination rather than cytostatic.
Modulation of NFAT promoter activity by CsA and TNP-470
To assess the activity of CsA on the noncanonical Wnt signaling pathway, SW480, RKO, SW620 and NCI-H508 CRC cell lines were transduced with an NFAT-Luc reporter construct and treated with selumetinib, CsA, or TNP-470 as single agents or in combination. Experiments were performed in the presence or absence of the calcium ionophore, ionomycin, as a positive control for activation of the Wnt calcium pathway and to increase the dynamic range of the luciferase assay. As depicted in Figure 3A, a statistically significant decrease in luciferase light units was measured between vehicle treated RKO cells and those treated with CsA alone or in the combination group (p value ≤0.02). Similar results were observed in NCI-H508 cells treated with TNP-470 (p<0.01) (Figure 3B). To varying degrees, luciferase activity was reduced by CsA alone in the other two other cell lines (SW480, SW620), which was maintained in the combination (Figure 3A). To verify transduction efficiency, immunoblotting of the E2A protein, an immediate early gene product of adenoviral infection, was assessed. As depicted in Figure 3C, E2A was present only in transduced cells, as compared to the non-transduced parental cells, and its expression did not change in response to treatment, suggesting relatively equal transfection efficiency between groups. Of note, exposure to single agent selumetinib was associated with luciferase activity that was either comparable, or induced, relative to the control in the RKO, SW480, or SW680 cell lines (Figure 3A). Moreover, as an additional approach to verify that the Wnt pathway probes were acting via a β-catenin independent mechanism, subcellular localization of NFAT1 and β-catenin was determined by immunoblot analysis. As depicted in Figure 3D, in the presence of TNP-470, NFAT1 was retained in the cytoplasm, while β-catenin was equally distributed between the cytoplasmic and nuclear compartments. The distribution of NFAT1 to the cytoplasm was also pronounced in the combination group. These results suggest that TNP-470 preferentially inhibits the noncanonical Wnt signaling pathway preventing the nuclear translocation of NFAT1 from the cytoplasm while having no effect on β-catenin localization. Finally, to exclude the possibility that CsA and TNP-470 were acting on the canonical Wnt signaling pathway, we performed a promoter reporter assay using the β-catenin dependent TOP-FLASH reporter plasmid. As depicted in Figure 3E, no change in luciferase activity was observed in the treatment groups as compared to control, in the SW480 CRC cell line.
Figure 3.
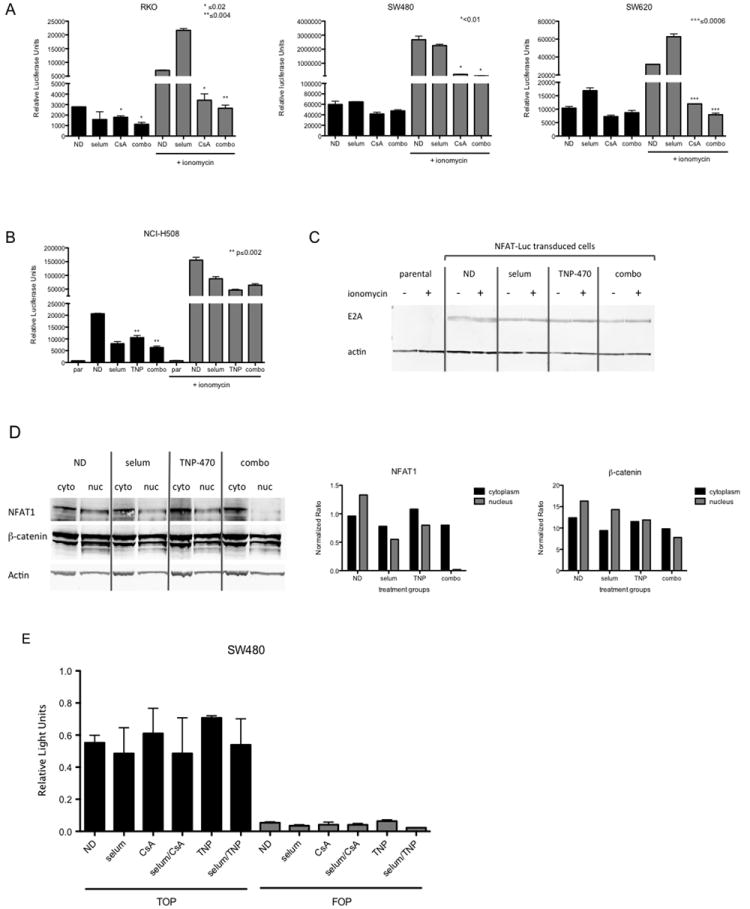
Treatment of CRC cells with CsA and TNP-470 results in inhibition of the non-canonical Wnt pathway. (A) CsA inhibits activity of the non-canonical Wnt-associated NFAT promoter in SW480, RKO, and SW620 CRC cell lines. Bar graphs represent average relative luciferase light units ± SD in each respective cell line (n=3). (B, Left Panel) Effects of TNP-470 on NFAT promoter activity in NCI-H508 cells treated with single agents selumetinib (0.3 μm/mL), TNP-470 (5 μm/mL) and in combination, alone or in the presence of ionomycin (1 μg/mL) for 24 hours. Bar graphs represent average relative luciferase light units ± SD (n=3). par = parental non-transfected control. (B, Right Panel) Immunoblotting analysis of early adenoviral E2A protein as a readout of transduction efficiency. (C) Immunoblot analysis of NFAT1 protein levels in the cytoplasm and nucleus in response to TNP-470 in NCI-H508 cells. C= cytoplasmic fraction; N= nuclear fraction. (D) Treatment with selumetinib, CsA or TNP-470 has no effect on the Wnt β-catenin dependent TOPflash (TOP) promoter construct. The FOPflash (FOP) promoter contains a mutation in the β-catenin binding element as a control for specificity (*=p< 0.01; **=p< 0.001).
In vivo antitumor activity of selumetinib and CsA
As a means of translating our observations into a clinically relevant model, in vivo studies were performed using patient-derived CRC tumor explant (PDTX) models in nude mice. Interestingly, we observed that FZD2 expression, as measured by qRT-PCR, increased significantly in the selumetinib treated group compared to baseline levels in three out of five previously defined selumetinib-resistant patient-derived CRC tumors (16) (Suppl. Figure 4A). Of note, PDTX CRC021 and CRC027 demonstrated no significant change in FZD2 expression upon treatment with selumetinib, indicating the presence of other resistance mechanisms. Therefore, we selected CRC006 and CRC007 for the in vivo combination drug studies. CRC006 is derived from peritoneal metastases of a colon cancer patient carrying a G12D KRAS activating mutation and WT PI3K. CRC007 is derived from the primary site of a CRC tumor, and harbors the G13D KRAS and the 3’UTR PI3K mutations. After 62 days of treatment, CRC006 demonstrated no tumor growth inhibitory effect to single agent selumetinib or CsA, as expected (TGI 30% and 16% respectively). However, the combination of selumetinib and CsA led to a statistically significant tumor growth inhibition (82% TGI, p<0.0001), demonstrating a dramatic synergistic effect (Figure 4A). These results were confirmed in PDTX model CRC007, which also demonstrated significant antitumor activity in the combination group as compared to the vehicle and single agents (Figure 4A). To further confirm and characterize the activity of MEK blockade and Wnt modulation, at the end of the initial study, selumetinib was added to the CsA single agent group, and CsA was added to the selumetinib single agent group in both PDTX models (Figure 4B). The introduction of the respective drugs to either single agents resulted in a striking tumor regression, demonstrating that this combination can also cause tumor volume reduction in addition to tumor growth inhibition. Additionally, in the CRC006 study, after 200 days of treatment, drugs were withdrawn in order to monitor tumor regrowth kinetics. Notably, no tumor regrowth was observed up to 65 days after drug withdrawal indicating a sustained antitumor response (Suppl. Figure 4B). No animals in these studies exhibited obvious signs of toxicity as indicated by outward morbidity or weight loss compared with vehicle controls or single agents, indicating that the combination of selumetinib and CsA was well tolerated in mice.
Figure 4.
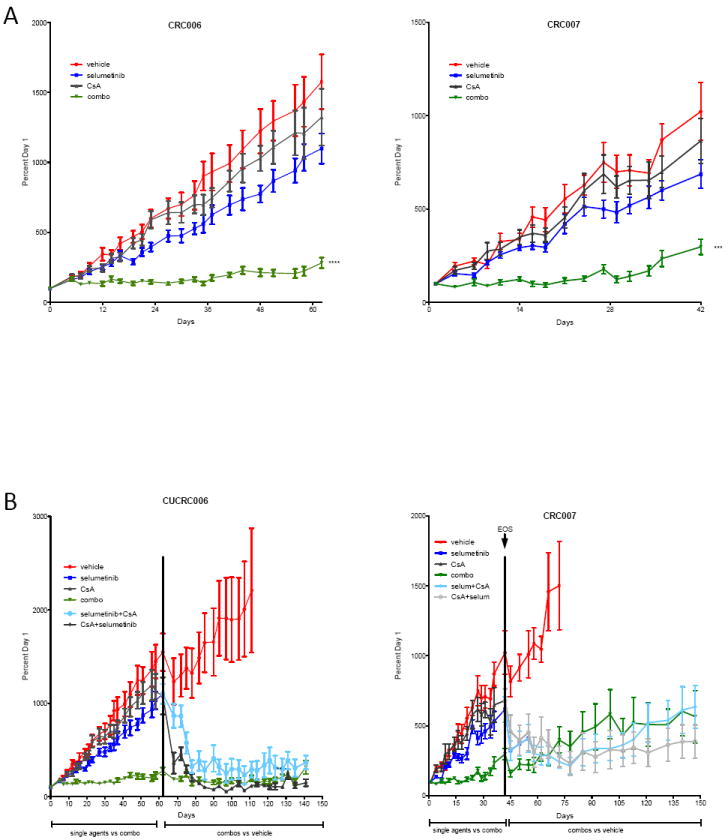
Antitumor activity of selumetinib and CsA on patient-derived tumor xenograft (PDTX) models of CRC. (A) Mice bearing tumors from CRC006 and CRC007 were treated with single agents selumetinib (25 mg/kg BID), or CsA (25 mg/kg QD) and the combination. Tumor growth index (TGI) was calculated by relative tumor growth of treated mice divided by relative tumor growth of control mice × 100. (B) Tumor regressive effects of the addition of respective combination agents to single agent arms in CRC006 and CRC007 PDTX models at end of study (EOS: days 62 and 42 respectively). Potent antitumor effects were maintained up to 200 days in CRC006 and 150 days in CRC007 after the switch to combination treatments in the single agent arms. EOS=end of study.
Tumor and Plasma Measurement of Selumetinib
CsA has been shown to inhibit P-glycoprotein (Pgp) drug efflux pumps, which could lead to prolonged intracellular exposure to pharmaceutical compounds (38). To verify that the potent antitumor activity of selumetinib and CsA was due to the dual targeting of the MAPK and Wnt pathways and not enhanced selumetinib exposure, liquid chromatography tandem mass spectrometry analysis was performed on plasma, tumor and liver at the end of the study (day 62). Because selumetinib is metabolized via N-demethylation to an active compound that inhibits cellular growth in vitro at approximately three times the potency of the parent (data not shown), N-desmethyl-selumetinib was measured as well. As shown in Figure 5, the plasma (A), liver (B), and tumor (C) concentrations of selumetinib were not significantly different when compared to single agent samples. Specifically, no significant differences were measured in any of the compartments at 1-hour post treatment and at the 8-hour trough time point for both selumetinib and its metabolite N-desmethylselumetinib versus combinations.
Figure 5.
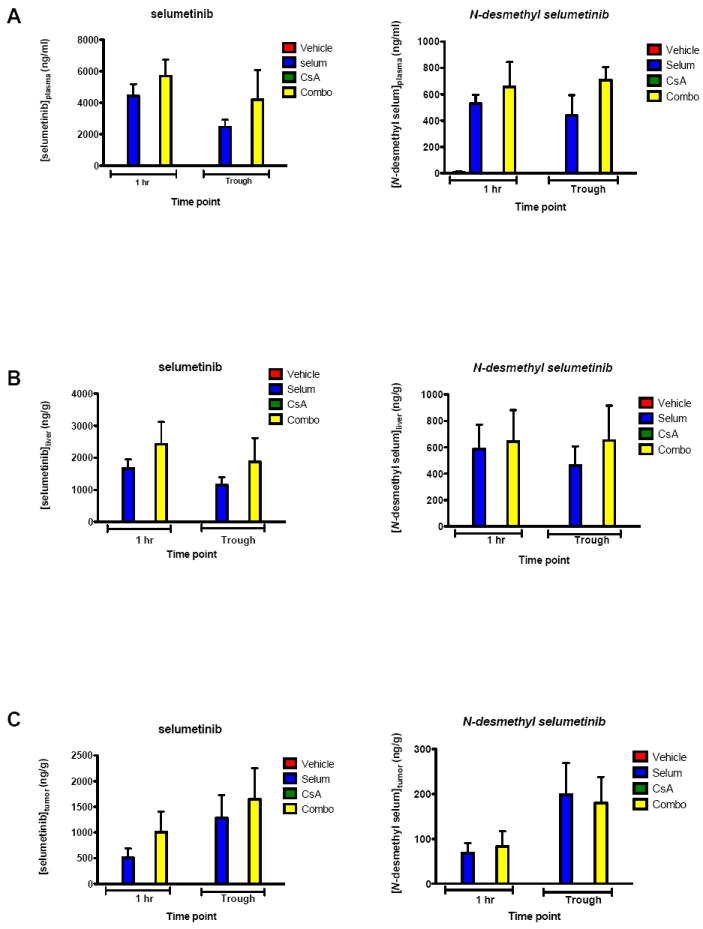
Plasma, tumor and tissue distribution of selumetinib and selumetinib N-desmethyl in mice treated with selumetinib, CsA or the combination. Mice bearing tumor CRC006 were administered a daily (BID) oral dose of 25 mg/kg selumetinib, as a single agent or in combination with daily oral dose of CsA (25 mg/kg) for 62 days. Samples of plasma (A), liver (B) and tumor (C) were taken at the indicated time after the last dose (mean, SD, n = 4). No significant differences were measured in any of the compartments at 1-hour post treatment and at the 8 hour trough time point for both selumetinib and its metabolite N-desmethyl selumetinib versus combinations (p > 0.05).
Pharmacodynamics and Antitumor Effects of Selumetinib and CsA on PDTX Models
To evaluate downstream effectors in response to treatments, tumor samples from PDTX models CRC006 and CRC007 were collected at end of study for immunoblotting and immunohistochemical (IHC) analyses. As shown in Figure 6A, immunoblotting of CRC006 demonstrated a modest decrease in pERK in the combination group only. The pERK inhibition was more pronounced in CRC007 in both the selumetinib single agent and combination groups (Figure 6B). These results are concordant with our previous findings (16), and demonstrate that resistance is not simply a result of the inability of selumetinib to inhibit its target (MEK 1/2).
Figure 6.
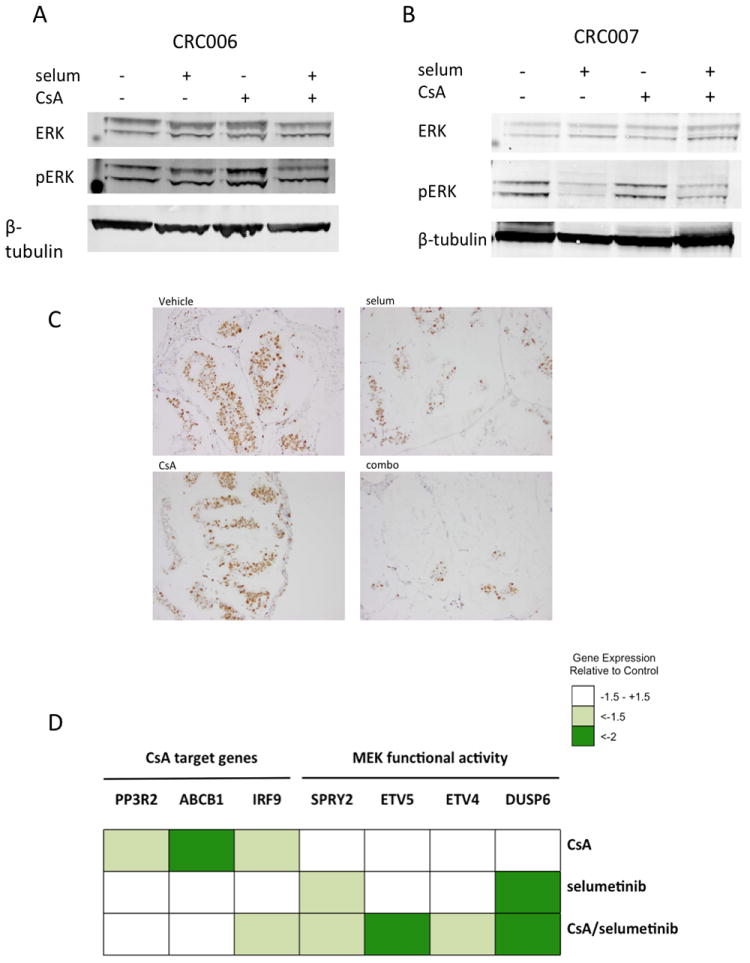
Pharmacodynamics of selumetinib and CsA on PDTX models at EOS. Inhibition of pERK levels in tumors from CRC006 (A) and CRC007 (B) as measured by immunoblot. (C) Inhibition of cell proliferation as measured by immunohistochemical staining of Ki-67 in CRC006 PDTX models treated with the indicated agents. (D) Heatmap depicting relative expression levels of CsA and MEK target genes.
To assess the in vivo mechanism of the synergy, we evaluated proliferation in response to drug treatments in the PDTX model, using Ki67 as a marker of mitotic cells. As depicted in Figure 6C, a dramatic reduction in tumor cell nuclear staining of Ki67 was observed in the combination group (20%) as compared to either single agents selumetinib (75%) and CsA (85%), and vehicle group where 90% of the cells exhibited proliferation (staining intensity: 2-3/3).
To further explore the mechanistic effects of modulation of the Wnt and MAPK pathways, we performed gene array analysis on tumors from the CRC007 PDTX model, treated with selumetinib, CsA or the combination. As depicted in Figure 6D, a heatmap of the relative expression of MEK activity and CsA target genes demonstrated inhibition of the MAPK pathway by single agent selumetinib, which was enhanced in the combination group. CsA single agent also inhibited its target genes, IRF9 (interferon regulatory factor 9), ABCB1 and PPP3R2. Moreover IRF9 inhibition was also maintained in the combination group. CsA alone inhibited the ABCB1 multi-drug resistance pump expression as has been well-documented (38), although in our study we did not observe increased intratumoral selumetinib concentrations. Based on our in vitro findings, we hypothesized that differentially expressed genes in the CsA and combination arms would be enriched with NFAT-signaling target genes. Thus, we utilized the NFAT binding motif of the genes (+2Kb,-2Kb), to perform an enrichment analysis on the differentially expressed genes (defined as ≥ or ≤ 1.5 fold change compared to control). Interestingly, we found that approximately 7% (55/808) of the differentially expressed genes in the combination arm contained the NFAT binding motif. Some of the differentially expressed genes in the selumetinib group also contained this motif, and their expression levels were either maintained or enriched in the combination arm, reinforcing the hypothesis of cross-talk between the two pathways, as has been previously described (39-41) (Suppl. Figure 5). To further confirm modulation of MAPK and Wnt pathways in our treatment groups, we compared gene signature patterns observed after dual inhibition of RAS and Wnt and demonstrated similar transcriptional modifications of Wnt, RAS, and apoptotic pathways (42) (Suppl. Fig.5). Additionally, we found that expression of the MYC oncogene was downregulated in the combination treatment arm as compared to single agents (Suppl. Fig. 5).
Induction of Apoptosis
To further evaluate the mechanism behind the synergistic antiproliferative effects of the dual inhibition of MEK and Wnt pathways, in vitro and in vivo induction of apoptosis was evaluated through caspase 3/7 activation, immunoblotting and immunohistochemical analyses for apoptotic markers. Although caspase 3/7 activity in SW480 was not significantly elevated after treatment with single agent selumetinib or CsA or the combination, a maximum 1.5-fold increase in caspase 3/7 activity was observed in RKO cells treated with the combination (Suppl. Figure 6A). Immunoblotting of these cell lines revealed an increase of cleaved PARP in SW480 cells (Suppl. Figure 6B). We also assessed apoptotic activity by Annexin V staining and flow cytometry in RKO and SW480 cell lines at 24 and 72 hours post treatment, but did not observe either single-agent or combination effects (data not shown). To assess apoptosis in vivo, protein extracts were prepared from tumors in both PDTX models at end of study for immunoblot analysis. As shown in Suppl. Figure 6C, cleaved caspase 3 was apparent but not increased by combination treatment as compared to single agents, perhaps reflecting the long duration of exposure of the drugs in these in vivo models.
DISCUSSION
Small molecule MEK 1/2 inhibitors have been evaluated in both preclinical and clinical studies as a potential therapeutic option for advanced CRC patients carrying KRAS activating mutations (12, 14, 16, 36, 43). However, single agent efficacy has been limited, potentially due to inherent resistance mechanisms. The multiple gene abnormalities and molecular alterations that are a hallmark of cancer affect proliferation and survival may be specifically targeted by novel anticancer agents (44-46). Previous studies have shown that identification of signal transduction pathways mediating resistance can be used to identify effective drug combination treatments (47-49). However, recent data have shown that the cancer phenotype may be defined by changes in expression of groups of genes, which result in pleiotropic responses to specific drugs (16, 31, 50). Gene array analysis in conjunction with GSEA is a powerful strategy to identify interconnected signaling pathways that maintain cancer cell growth and may lead to new rational combination treatments (16, 31, 50). In our study, we performed baseline GSEA and found that Wnt pathway-related genes were upregulated in selumetinib-resistant CRC cell lines. However, as these pathways may not necessarily be overexpressed in resistant tumors, a synthetic lethality approach was utilized. SLS has emerged as a powerful platform to systematically search for cancer addiction pathways and clinically relevant interactions and mechanisms of drug resistance in cancer cells (26, 29, 51, 52). Moreover, this approach identifies genes, which, when suppressed, mediate response to a specific treatment, leading to actionable targets. In our study, combining these novel, complementary approaches we found that several genes involved in the Wnt signaling pathway may be mediators of responsiveness to the MEK inhibitor selumetinib.
The significance of these results is highlighted by the crosstalk between the MAPK and Wnt signaling pathways identified by others and suggests a role for Wnt in maintaining CRC cell survival and escaping MEK inhibition. A report by Horst et al. demonstrated that both CRC cell line xenografts and patient-derived CRC tumor xenografts were heterogeneous with respect to β-catenin expression, and that these areas co-localized with pERK expression (39). Moreover, a recent report described a KRAS-dependent gene signature in CRC models that was characterized by enhanced Wnt activity through activation of BMP-7 and TAK-1 kinase (49). Indeed, we observed that inhibition of MAPK signaling with selumetinib led to decreased expression of NFAT-dependent genes in a PDTX model of CRC, providing further evidence that these two pathways are interconnected.
While the β-catenin dependent Wnt pathway and its dysregulation in CRC is well studied, the noncanonical Wnt pathway is largely uncharacterized in mammalian cells, and its role in oncogenesis still remain controversial (53). We found that the shRNA-mediated inhibition of numerous components of the Wnt signaling pathway, which were either upregulated or synthetically lethal in CRC cell lines, enhanced responsiveness to selumetinib. Because the noncanonical Wnt pathway is less characterized as a therapeutic target, there are presently no clinically selective Wnt/Ca++ pathway inhibitors available for clinical use. Although CsA and TNP-470 have been described as immunosuppressant and anti-angiogenic agents, respectively, we utilized them as pharmacologic probes of the Wnt/Ca++ pathway. A recent report demonstrated that CsA targets the noncanonical Wnt pathway in mouse Bcr-Abl+ leukemia models, which, when combined with dasatinib, resulted in a complete eradication of the leukemia cells, compared to the relapse and mortality observed in mice treated with single agent dasatinib or CsA (32). Likewise, TNP-470 acts on the β–catenin independent Wnt pathway, targeting a site downstream of FZD2 receptor (37). In our study, we documented that the combination of selumetinib and CsA, or TNP-470 resulted in a synergistic antiproliferative effect in CRC cell lines in both 2D and 3D culture conditions, as well as in vivo PDTX models where post-treatment gene array analysis showed modulation of CsA and selumetinib target genes.
Several studies have extensively analyzed selumetinib as a single agent against numerous cancer types, and concluded that its anticancer activity is mainly cytostatic (54, 55). These data are concordant with our results, which showed reversible cell growth suppression with single agent selumetinib exposure, but not in the combination groups with CsA or TNP-470. Therefore, the synergy observed with selumetinib and CsA or TNP-470 may be associated with cytotoxicity. In support of these data, selumetinib has been shown to cause tumor regression and induction of apoptosis in vivo when combined with docetaxel in preclinical models of melanoma (55, 56). As it has been reported that CsA can inhibit Pgp it could be hypothesized that the synergistic effects of CsA and selumetinib are due to inhibition of this efflux pump resulting in higher intratumoral concentrations of selumetinib and not on direct inhibition of CN/NFAT activity (38). However, we demonstrated that the synergistic antiproliferative effects are likely due to CsA-mediated CN inhibition, as we did not see accumulation of selumetinib in the presence of CsA.
The development of more reliable preclinical models is a crucial step towards improving the success rate of novel cancer therapeutics in the clinic. PDTX models in nude mice may represent a more clinically relevant model as they more accurately reflect the complex molecular, genetic and histologic heterogeneity of patient tumors as compared to standard cell line xenografts (57). Our in vivo studies utilizing PDTX models of KRAS mutant CRC demonstrated that selumetinib combined with CsA was a potent inhibitor of tumor growth and could induce tumor regression in models that are resistant to single-agent selumetinib. Previous studies have shown that dual inhibition of Wnt and KRAS signaling pathways resulted in a distinct gene expression signature with respect to to alterations involved in Wnt, RAS and apoptosis pathways (42). Interestingly, we observed similar gene expression modulation in our PDTX models after combination treatment with selumetinib and CsA. The MYC oncogene has been recently shown to play a key role in activating Wnt signaling (58). Consistent with this finding, we also observed MYC downregulation in the combination arm as compared to both single agents suggesting a MYC-mediated suppression of Wnt targeted genes.
To our knowledge, this is the first study in which the combination of MEK blockade and Wnt pathway modulation has demonstrated synergistic antiproliferative effects in both in vitro and in vivo preclinical CRC models. While we focused our studies largely on the KRAS/BRAF mutant CRC population, this does not exclude activity in KRAS wide type tumors, which deserves further study. Interestingly, although the data is limited, several studies have suggested that the results of treatment of explant models correlates with actual clinical outcome, providing early clinical validation of this approach (35, 57, 59).
In summary, we utilized an unbiased molecular genetic approach to identify rational combination partners for MEK inhibitors in CRC. These genetic screens yielded a dual targeting strategy of the Wnt pathway and MEK, which resulted in robust antitumor activity and tumor regression in preclinical models of CRC. The results of these studies further validate our previous findings (16) and support our hypothesis that co-targeting of resistance pathways may result in more effective personalized medicine strategies. While we acknowledge the challenges in utilizing a promiscuous agent such as CsA to definitively elucidate the cross-talk between the MEK and Wnt pathways, nonetheless the data is supportive of further preclinical studies in this area, particularly utilizing more selective Wnt inhibitors, which is ongoing in our lab. These findings have led to the design of a Phase I/IB clinical trial exploring the combination of selumetinib and CsA with an expanded cohort of patients with advanced treatment-refractory CRC.
Supplementary Material
Translational Relevance.
There is an urgent need for the development of novel, individualized therapies for patients with advanced treatment-refractory colorectal cancer in order to advance progress in the treatment this disease. The recent development of genome-wide screening approaches and integrative bioinformatics methods has accelerated the discovery of clinically relevant pathway interactions and mechanisms of drug resistance in numerous cancer types. In this study, we utilized two powerful and complementary molecular genetic approaches, GSEA and synthetic lethality screens, to identify potential resistance mechanisms to the MEK inhibitor, selumetinib which yielded a dual targeting strategy of the Wnt/Ca++ pathway and MEK, resulting in robust antitumor activity and tumor regression in preclinical models of CRC. These findings strongly support further clinical studies to test the efficacy of the combination of MEK and Wnt inhibitors in CRC patients.
Acknowledgments
We would like to thank Drs. Paul D. Smith, Christopher Porter, Erica L. Bradshaw-Pierce, and Ms. Michelle VanScoyk for their helpful discussions and scientific review of this manuscript. We would also like to thank AstraZeneca, Inc. for supplying selumetinib for these studies.
Grant Support: A. Spreafico, Astra Zeneca post-doctoral fellowship. S.G. Eckhardt, grants CA106349 and CA046934.
Footnotes
Conflict of Interest: AstraZeneca research funding to S. G. Eckhardt; AstraZeneca post-doctoral research grant to A. Spreafico.
References
- 1.Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer research. 2010;70:288–98. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 2.Fransen K, Klintenas M, Osterstrom A, Dimberg J, Monstein HJ, Soderkvist P. Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas. Carcinogenesis. 2004;25:527–33. doi: 10.1093/carcin/bgh049. [DOI] [PubMed] [Google Scholar]
- 3.Vaughn CP, Zobell SD, Furtado LV, Baker CL, Samowitz WS. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer. 2011;50:307–12. doi: 10.1002/gcc.20854. [DOI] [PubMed] [Google Scholar]
- 4.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26:1626–34. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 5.Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48:1466–75. doi: 10.1016/j.ejca.2012.02.057. [DOI] [PubMed] [Google Scholar]
- 6.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 7.Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer research. 2006;66:3992–5. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 8.Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer. 2009;101:715–21. doi: 10.1038/sj.bjc.6605177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertrand FE, Steelman LS, Chappell WH, Abrams SL, Shelton JG, White ER, et al. Synergy between an IGF-1R antibody and Raf/MEK/ERK and PI3K/Akt/mTOR pathway inhibitors in suppressing IGF-1R-mediated growth in hematopoietic cells. Leukemia. 2006;20:1254–60. doi: 10.1038/sj.leu.2404217. [DOI] [PubMed] [Google Scholar]
- 10.Corcoran R, Settleman J, Engelman J. Potential therapeutic strategies to overcome acquired resistance to BRAF or MEK inhibitors in BRAF mutant cancers. Oncotarget. 2011;2:336–46. doi: 10.18632/oncotarget.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turke AB, Song Y, Costa C, Cook R, Arteaga CL, Asara JM, et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer research. 2012;72:3228–37. doi: 10.1158/0008-5472.CAN-11-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26:2139–46. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balmanno K, Chell SD, Gillings AS, Hayat S, Cook SJ. Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int J Cancer. 2009;125:2332–41. doi: 10.1002/ijc.24604. [DOI] [PubMed] [Google Scholar]
- 14.Bennouna J, Lang I, Valladares-Ayerbes M, Boer K, Adenis A, Escudero P, et al. A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Invest New Drugs. 2011;29:1021–8. doi: 10.1007/s10637-010-9392-8. [DOI] [PubMed] [Google Scholar]
- 15.Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal. 2010;3:ra84. doi: 10.1126/scisignal.2001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tentler JJ, Nallapareddy S, Tan AC, Spreafico A, Pitts TM, Morelli MP, et al. Identification of predictive markers of response to the MEK1/2 inhibitor selumetinib (AZD6244) in K-ras-mutated colorectal cancer. Molecular cancer therapeutics. 2010;9:3351–62. doi: 10.1158/1535-7163.MCT-10-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bekaii-Saab T, Phelps MA, Li X, Saji M, Goff L, Kauh JS, et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:2357–63. doi: 10.1200/JCO.2010.33.9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayes DN, Lucas AS, Tanvetyanon T, Krzyzanowska MK, Chung CH, Murphy BA, et al. Phase II efficacy and pharmacogenomic study of Selumetinib (AZD6244; ARRY-142886) in iodine-131 refractory papillary thyroid carcinoma with or without follicular elements. Clin Cancer Res. 2012;18:2056–65. doi: 10.1158/1078-0432.CCR-11-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janne P, Shaw A, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Phase II doubleblind, randomized study of selumetinib (SEL) plus docetaxel (DOC) versus DOC plus placebo as second-line treatment for advanced KRAS mutant non-small cell lung cancer (NSCLC) Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30 [Google Scholar]
- 20.O’Neil BH, Goff LW, Kauh JS, Strosberg JR, Bekaii-Saab TS, Lee RM, et al. Phase II study of the mitogen-activated protein kinase 1/2 inhibitor selumetinib in patients with advanced hepatocellular carcinoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:2350–6. doi: 10.1200/JCO.2010.33.9432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Staal FJ, Luis TC, Tiemessen MM. WNT signalling in the immune system: WNT is spreading its wings. Nat Rev Immunol. 2008;8:581–93. doi: 10.1038/nri2360. [DOI] [PubMed] [Google Scholar]
- 22.Jessen JR. Noncanonical Wnt signaling in tumor progression and metastasis. Zebrafish. 2009;6:21–8. doi: 10.1089/zeb.2008.0571. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y. Wnt/Planar cell polarity signaling: a new paradigm for cancer therapy. Molecular cancer therapeutics. 2009;8:2103–9. doi: 10.1158/1535-7163.MCT-09-0282. [DOI] [PubMed] [Google Scholar]
- 24.Martin SA, Hewish M, Sims D, Lord CJ, Ashworth A. Parallel high-throughput RNA interference screens identify PINK1 as a potential therapeutic target for the treatment of DNA mismatch repair-deficient cancers. Cancer research. 2011;71:1836–48. doi: 10.1158/0008-5472.CAN-10-2836. [DOI] [PubMed] [Google Scholar]
- 25.Mendes-Pereira AM, Sims D, Dexter T, Fenwick K, Assiotis I, Kozarewa I, et al. Genome-wide functional screen identifies a compendium of genes affecting sensitivity to tamoxifen. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2730–5. doi: 10.1073/pnas.1018872108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porter CC, Kim J, Fosmire S, Gearheart CM, van Linden A, Baturin D, et al. Integrated genomic analyses identify WEE1 as a critical mediator of cell fate and a novel therapeutic target in acute myeloid leukemia. Leukemia. 2012;26:1266–76. doi: 10.1038/leu.2011.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J, Tan AC. BiNGS!SL-seq: a bioinformatics pipeline for the analysis and interpretation of deep sequencing genome-wide synthetic lethal screen. Methods Mol Biol. 2012;802:389–98. doi: 10.1007/978-1-61779-400-1_26. [DOI] [PubMed] [Google Scholar]
- 29.Sullivan KD, Padilla-Just N, Henry RE, Porter CC, Kim J, Tentler JJ, et al. ATM and MET kinases are synthetic lethal with nongenotoxic activation of p53. Nat Chem Biol. 2012;8:646–54. doi: 10.1038/nchembio.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casas-Selves M, Kim J, Zhang Z, Helfrich BA, Gao D, Porter CC, et al. Tankyrase and the canonical Wnt pathway protect lung cancer cells from EGFR inhibition. Cancer research. 2012;72:4154–64. doi: 10.1158/0008-5472.CAN-11-2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pitts TM, Tan AC, Kulikowski GN, Tentler JJ, Brown AM, Flanigan SA, et al. Development of an integrated genomic classifier for a novel agent in colorectal cancer: approach to individualized therapy in early development. Clin Cancer Res. 2010;16:3193–204. doi: 10.1158/1078-0432.CCR-09-3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gregory MA, Phang TL, Neviani P, Alvarez-Calderon F, Eide CA, O’Hare T, et al. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell. 2010;18:74–87. doi: 10.1016/j.ccr.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Molecular cancer therapeutics. 2007;6:2209–19. doi: 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- 34.Denton CL, Gustafson DL. Pharmacokinetics and pharmacodynamics of AZD6244 (ARRY-142886) in tumor-bearing nude mice. Cancer Chemother Pharmacol. 2011;67:349–60. doi: 10.1007/s00280-010-1323-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morelli MP, Tentler JJ, Kulikowski GN, Tan AC, Bradshaw-Pierce EL, Pitts TM, et al. Preclinical activity of the rational combination of selumetinib (AZD6244) in combination with vorinostat in KRAS-mutant colorectal cancer models. Clin Cancer Res. 2012;18:1051–62. doi: 10.1158/1078-0432.CCR-11-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dry JR, Pavey S, Pratilas CA, Harbron C, Runswick S, Hodgson D, et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244) Cancer research. 2010;70:2264–73. doi: 10.1158/0008-5472.CAN-09-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Yeh JR, Mara A, Ju R, Hines JF, Cirone P, et al. A chemical and genetic approach to the mode of action of fumagillin. Chem Biol. 2006;13:1001–9. doi: 10.1016/j.chembiol.2006.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Illmer T, Schaich M, Platzbecker U, Freiberg-Richter J, Oelschlagel U, von Bonin M, et al. P-glycoprotein-mediated drug efflux is a resistance mechanism of chronic myelogenous leukemia cells to treatment with imatinib mesylate. Leukemia. 2004;18:401–8. doi: 10.1038/sj.leu.2403257. [DOI] [PubMed] [Google Scholar]
- 39.Horst D, Chen J, Morikawa T, Ogino S, Kirchner T, Shivdasani RA. Differential WNT activity in colorectal cancer confers limited tumorigenic potential and is regulated by MAPK signaling. Cancer research. 2012;72:1547–56. doi: 10.1158/0008-5472.CAN-11-3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim D, Rath O, Kolch W, Cho KH. A hidden oncogenic positive feedback loop caused by crosstalk between Wnt and ERK pathways. Oncogene. 2007;26:4571–9. doi: 10.1038/sj.onc.1210230. [DOI] [PubMed] [Google Scholar]
- 41.Wang IC, Snyder J, Zhang Y, Lander J, Nakafuku Y, Lin J, et al. Foxm1 mediates cross talk between Kras/mitogen-activated protein kinase and canonical Wnt pathways during development of respiratory epithelium. Mol Cell Biol. 2012;32:3838–50. doi: 10.1128/MCB.00355-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mologni L, Brussolo S, Ceccon M, Gambacorti-Passerini C. Synergistic effects of combined Wnt/KRAS inhibition in colorectal cancer cells. PLoS One. 2012;7:e51449. doi: 10.1371/journal.pone.0051449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoon J, Koo KH, Choi KY. MEK1/2 inhibitors AS703026 and AZD6244 may be potential therapies for KRAS mutated colorectal cancer that is resistant to EGFR monoclonal antibody therapy. Cancer research. 2011;71:445–53. doi: 10.1158/0008-5472.CAN-10-3058. [DOI] [PubMed] [Google Scholar]
- 44.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26:5705–12. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 45.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lievre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26:374–9. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 47.Gioeli D, Wunderlich W, Sebolt-Leopold J, Bekiranov S, Wulfkuhle JD, Petricoin EF, 3rd, et al. Compensatory pathways induced by MEK inhibition are effective drug targets for combination therapy against castration-resistant prostate cancer. Molecular cancer therapeutics. 2011;10:1581–90. doi: 10.1158/1535-7163.MCT-10-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi H, Kong X, Ribas A, Lo RS. Combinatorial treatments that overcome PDGFRbeta-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer research. 2011;71:5067–74. doi: 10.1158/0008-5472.CAN-11-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh A, Sweeney MF, Yu M, Burger A, Greninger P, Benes C, et al. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell. 2012;148:639–50. doi: 10.1016/j.cell.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jimeno A, Tan AC, Coffa J, Rajeshkumar NV, Kulesza P, Rubio-Viqueira B, et al. Coordinated epidermal growth factor receptor pathway gene overexpression predicts epidermal growth factor receptor inhibitor sensitivity in pancreatic cancer. Cancer research. 2008;68:2841–9. doi: 10.1158/0008-5472.CAN-07-5200. [DOI] [PubMed] [Google Scholar]
- 51.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007;446:815–9. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- 53.Pukrop T, Binder C. The complex pathways of Wnt 5a in cancer progression. J Mol Med (Berl) 2008;86:259–66. doi: 10.1007/s00109-007-0266-2. [DOI] [PubMed] [Google Scholar]
- 54.Ball DW, Jin N, Rosen DM, Dackiw A, Sidransky D, Xing M, et al. Selective growth inhibition in BRAF mutant thyroid cancer by the mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244. J Clin Endocrinol Metab. 2007;92:4712–8. doi: 10.1210/jc.2007-1184. [DOI] [PubMed] [Google Scholar]
- 55.Haass NK, Sproesser K, Nguyen TK, Contractor R, Medina CA, Nathanson KL, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008;14:230–9. doi: 10.1158/1078-0432.CCR-07-1440. [DOI] [PubMed] [Google Scholar]
- 56.Holt SV, Logie A, Odedra R, Heier A, Heaton SP, Alferez D, et al. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br J Cancer. 2012;106:858–66. doi: 10.1038/bjc.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. Patient-derived tumour xenografts as models for oncology drug development. Nature reviews Clinical oncology. 2012;9:338–50. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.TCGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hidalgo M, Bruckheimer E, Rajeshkumar NV, Garrido-Laguna I, De Oliveira E, Rubio-Viqueira B, et al. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Molecular cancer therapeutics. 2011;10:1311–6. doi: 10.1158/1535-7163.MCT-11-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.