

Abstract
Cytochrome c (Cyt c) is a small mitochondrial heme protein involved in the intrinsic apoptotic pathway. Once Cyt c is released into the cytosol, the caspase mediated apoptosis cascade is activated resulting in programmed cell death. Herein, we explore the covalent immobilization of Cyt c into mesoporous silica nanoparticles (MSN) to generate a smart delivery system for intracellular drug delivery to cancer cells aiming at affording subsequent cell death. Cyt c was modified with sulfosuccinimidyl-6-[3′-(2-pyridyldithio)-propionamido] hexanoate (SPDP) and incorporated into SH-functionalized MSN by thiol-disulfide interchange. Unfortunately, delivery of Cyt c from the MSN was not efficient in inducing apoptosis in human cervical cancer HeLa cells. We tested whether chemical Cyt c glycosylation could be useful in overcoming the efficacy problems by potentially improving Cyt c thermodynamic stability and reducing proteolytic degradation. Cyt c lysine residues were modified with lactose at a lactose-to-protein molar ratio of 3.7±0.9 using mono-(lactosylamido)-mono-(succinimidyl) suberate linker chemistry. Circular dichroism (CD) spectra demonstrated that part of the activity loss of Cyt c was due to conformational changes upon its modification with the SPDP linker. These conformational changes were prevented in the glycoconjugate. In agreement with the unfolding of Cyt c by the linker, a proteolytic assay demonstrated that the Cyt c-SPDP conjugate was more susceptible to proteolysis than Cyt c. Attachment of the four lactose molecules reversed this increased susceptibility and protected Cyt c from proteolytic degradation. Furthermore, a cell-free caspase-3 assay revealed 47% and 87% of relative caspase activation by Cyt c-SPDP and the Cyt c-lactose bioconjugate, respectively, when compared to Cyt c. This again demonstrates the efficiency of the glycosylation to improve maintaining Cyt c structure and thus function. To test for cytotoxicity, HeLa cells were incubated with Cyt c loaded MSN at different Cyt c concentrations (12.5, 25.0, and 37.5 μg/mL) for 24 to 72 h and cellular metabolic activity determined by a cell proliferation assay. While MSN-SPDP-Cyt c did not induced cell death, the Cyt c-lactose bioconjugate induced significant cell death after 72 h, reducing HeLa cell viability to 67% and 45% at the 25 μg/mL and 37.5 μg/mL concentrations, respectively. Confocal microscopy confirmed that the MSN immobilized Cyt c-lactose bioconjugate was internalized by HeLa cells and that the bioconjugate was capable of endosomal escape. The results clearly demonstrate that chemical glycosylation stabilized Cyt c upon formulation of a smart drug delivery system and upon delivery into cancer cells and highlight the general potential of chemical protein glycosylation to improve the stability of protein drugs.
Keywords: Apoptosis, chemical glycosylation, drug delivery, nanoparticle, protein drug, protein stability, smart release
INTRODUCTION
Cancer is a major public health problem worldwide. Currently, one in four deaths in the United States is due to cancer.1 Treatment options include surgery, radiation treatment, and cytotoxic chemotherapy. One of the problems with the commonly employed conventional cytotoxic chemotherapy is the high systemic toxicity producing undesirable side effects, such as, damage of liver, kidney, and bone morrow.2 Therefore, it is essential to develop novel drug delivery systems that allow for the specific targeting of cancer cells by taking advantage of their particular microenvironment.
One of the hallmarks of cancer is sustained angiogenesis which leads to the tumor vasculature possesses poor architecture with abnormal basement membrane and fissures between the endothelial cells. The so-called leaky vasculature, accompanied by insufficient lymphatic drainage in tumors, leads to the well described enhanced permeability and retention (EPR) effect leading to accumulation of nanoparticles in tumors (Figure 1).3–6 In this work we use mesoporous silica nanoparticles (MSN) as delivery vehicle because they are biocompatible, biodegradable, and generally recognized as safe by the US Food and Drug Administration (FDA).7, 8 MSN have been developed as delivery vehicles for proteins some years ago and it has been established that they are capable of transporting membrane-impermeable proteins (i.e., Cyt c) into the cytoplasm of target cells including the HeLa cell line used herein by us.9,10 Previously, we reported on the improvement of the use of MSN as carriers for protein drugs and by covalently immobilizing the model protein carbonic anhydrase on a thiolated surface.11 We designed a stimulus-responsive controlled release system in which the discharge of the protein only proceeded under intra- but not under extra-cellular redox conditions by linking it to the MSN via a redox-sensitive disulfide bond. Going one step further, herein, we designed a nanoparticulate drug delivery system for the smart delivery of the apoptosis-inducing protein Cyt c to the cytoplasm of cancer cells. We immobilized Cyt c into MSN and conducted studies related to protein stability, caspase activation, cell viability, cellular uptake, endosomal escape, and induction of apoptosis.
Figure 1.
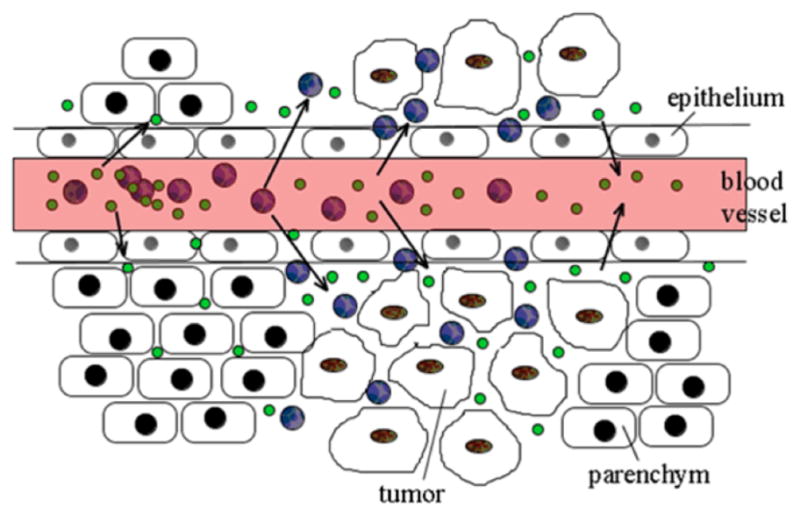
Scheme of the Enhanced Permeability and Retention (EPR) effect. Nanoparticles (blue) can extravasate and accumulate inside the interstitial space. Small molecule drugs or particles of less than 10 nm in diameter (green) will not be retained. The image does not display dimensions proportionally.
Cyt c (EC 232.700.9) is a small mitochondrial electron transport protein (MW = 12 kDa). In addition to its function in the oxidative phosphorylation, the heme protein is a crucial component of the intrinsic apoptosis pathway. To induce apoptosis, Cyt c is translocated to the cytoplasm, where it binds to the apoptotic protease-activating factor 1 (Apaf-1) which promotes assembly of the apoptosome. The apoptosome cleaves procaspase-9 to active caspase-9, which activates the effector caspases 3 and 7 leading to apoptosis.12, 13 Avoidance of apoptosis is a hallmark of cancer.14 Delivering Cyt c into the cytoplasm activates apoptosis downstream from many events which in many cancers have been shown to prevent cancer cells from undergoing apoptosis (e.g., p53 pathway). Experimental evidence for the feasibility has been presented by Santra et al. (2010) who demonstrated that Cyt c induced apoptosis in human lung carcinoma (A549) and breast carcinoma (MCF 7) cells when released from water-soluble hyperbranched polyhydroxyl nanoparticles.15 Additionally, Huang et al. (2012) delivered Cyt c using nanoparticles composed of lipid and apolipoprotein, which provoked a tumor growth retardation effect in H460 xenograft mice.16 In the initial works transport of the membrane impermeable Cyt c into the cytoplasm of target cells via MSN has been reported, but reports on induction of apoptosis are lacking in these works.8,9 There are no reports on delivering Cyt c via stimulus-responsive bonds from MSN as drug delivery system (Figure 2).
Figure 2.
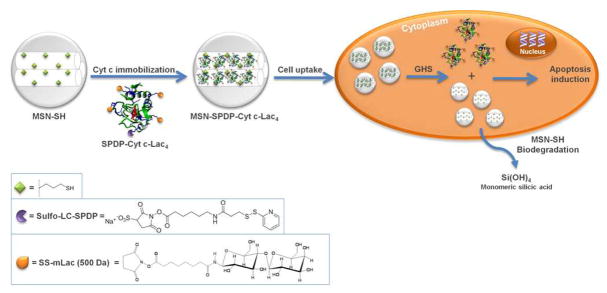
Scheme of the immobilization of Cyt c-Lac4 into MSN-SH via redox-sensitive smart bonds followed by its intracellular delivery into cancer cells.
The main point of this work, however, deals with another pertinent problem frequently encountered in protein drug delivery applications: protein instability during encapsulation, storage, and release. Immobilization of proteins into any material can cause detrimental protein structural and functional changes.17 Indeed, we found this to be the case for our system and consequently, the main point of our work was to find a way to stabilize Cyt c to allow for the smart delivery from MSN. In this context, we investigated the potential of chemical glycosylation to improve the stability of Cyt c during immobilization. Furthermore, proteins are very vulnerable to proteolytic degradation during delivery due to the ubiquitous nature and systemic distribution of proteases.18, 19 Proteolysis can be particularly prominent when delivering proteins to tumors as a result of the tumor microenvironment, which is frequently enriched with a broad spectrum of proteases. Consequently, proteolysis presents a fundamental limitation for protein drugs and is one of the causes of drug resistance in cancer therapy.20 There is some evidence that natural glycosylation increases intracellular protein stability21 but chemical glycosylation has not been tested to improve protein stability in any protein drug delivery system for intracellular delivery. We set out to investigate, for the first time, whether limitations arising from Cyt c instability could be addressed and investigated the chemical modification of Cyt c with lactose in this context. Chemical protein glycosylation has been introduced by us and shown to improve the thermodynamic and kinetic stability in several model enzymes.22–24 We hypothesized that chemical Cyt c glycosylation should be useful in preventing or minimizing Cyt c functional loss upon the chemical immobilization process. Furthermore, glycosylation has been demonstrated to reduce proteolysis,25–28 but this knowledge has previously not been employed to improve any (intracellular) protein drug delivery system.
EXPERIMENTAL PROCEDURES
Propylthiol functionalized mesoporous silica nanoparticles (MSN-SH), FTIC labeled MSN-SH, cytochrome c from equine heart, dimethyl sulfoxide (DMSO), reduced glutathione ethyl ester (GHS-OEt), and a protease inhibitor cocktail were from Sigma-Aldrich (St. Louis, MO). Sulfosuccinimidyl 6-(3′-[2-pyridyldithio]-propionamido)hexanoate (SPDP) was from Proteochem (Denver, CO). Mono-(lactosylamido)-mono-(succinimidyl) suberate (SS-mLac) was from Carbomer (San Diego, CA). Cellulose ester dialysis membranes were from Sprectrum Lab (Rancho Dominguez, CA). 4′, 6-Diamidino-2-phenylindole (DAPI), propidium iodide (PI) and FM-4-64 membrane stain were purchased from Invitrogen (Grand Island, NY). All the reagents were used without further purification. All other chemicals were from various commercial suppliers and were of analytical grade. HeLa cells were purchased from the American Type Culture Collection (Manassas, VA).
Glycosylation of Cyt c
Glycosylation of Cyt c was performed as described by us in detail previously.22, 24 Briefly, 500 mg of Cyt c and 45 mg of SS-mLac were dissolved in 250 mL of 0.1 M borate buffer at pH 9.0. The reaction was performed for 1 h at room temperature under gently stirring. Unreacted lactose was removed by dialyzing thrice against nanopure water at 4°C using 6–8 kDa molecular weight cutoff cellulose ester membranes. The extent of modification was determined by TNBSA assay.28
Cyt c modification with SPDP
Cyt c was chemically modified using the heterobifunctional cross-linker SPDP using the methodology recently described by us.11 Briefly, Cyt c was dissolved in 50 mM PBS and 0.15 M NaCl at pH 7.2 to accomplish a final concentration of 2 mg/mL. SPDP was added directly to the reaction flask and dissolved to reach 0.5 mM concentration. The mixture was reacted for 30 min at room temperature under gently stirring, dialyzed against nanopure water, and lyophilized. The extent of modification with the cross-linker was determined by measuring the release of pyridine-2-thione at 343 nm after addition of 10 μL of 15 mg/mL dithiothreitol (DTT) solution.
Circular dischroism (CD) spectroscopy
CD spectra were recorded using an OLIS DSM-10 UV-Vis CD spectrometer at room temperature. The protein (Cyt c, Cyt c-SPDP or Cyt c-Lac4-SPDP) was dissolved in 10 mM PBS at pH 7.4. CD spectra were acquired from 250 to 320 nm (tertiary structure) and 380 to 450 nm (Soret region) at a concentration of 0.6 mg/mL using 10 mm quartz cuvette. Each spectrum was obtained by averaging six scans. Spectra of buffer blanks were measured prior to the samples and subtracted from the sample spectra.
Cell-free caspase-3 assay
HeLa cells were grown to 80% confluency, harvested, washed, and finally disrupted. For disruption the cells were suspended in a homogenizing buffer containing 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) at pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM sodium EDTA, 1 mM sodium EGTA, 1 mM DTT, 250 mM sucrose, and a cocktail of serine, cysteine, aspartic acid, and metalloprotease inhibitors (1X). As control, the assay was also performed in the absence of the protease inhibitor cocktail. The suspended cells were frozen in liquid N2 for 2 min and thawed in a 37°C water bath and the freeze/thaw cycle repeated thrice. The protein content in the lysate was determined using the Bradford assay.29 The cell-free reactions were performed in homogenizing buffer in a total volume of 100 μL. The reaction was initiated by adding Cyt c or the different Cyt c bioconjugates (i.e., 100 μg/mL of Cyt-SPDP, Cyt-Lac4 or Cyt c-Lac4-SPDP) to freshly purified cytosol (3 mg/mL). The reaction was incubated at 37°C for 150 min.14 The caspase-3 assay was performed following the manufacturer’s protocol (CaspACE™ assay; Promega, Madison, WI). Briefly, 20 μL of the reaction mixtures were withdrawn and added to 78 μL of a mixture containing 100 mM HEPES (pH 7.5), 10% w/v sucrose, 0.1% w/v CHAPS (3-[(3-cholamido-propyl)-dimethylammonio]-1-propane-sulfonate), 10 mM DTT, and 2% v/v DMSO. Afterwards, 2 μL of 10 mM DEVD-pNA substrate was added to each sample. The plate was incubated overnight at room temperature and the absorbance at 405 nm was measured in each well using a microplate reader (Thermo Scientific Multiskan FC). All measurements were performed in triplicate.
Covalent immobilization of Cyt c into MSN-SH
Protein immobilization was performed as described by us.11 Briefly, 3.0 mg of MSN-SH were subjected to ultrasonication at 240 watts for 5 min in the immobilization buffer containing 50 mM PBS, 0.15 M NaCl, and 10 mM EDTA at pH 7.2 in safe-lock tubes to create a homogeneous dispersion. 1 mL of a 3.5 mg/mL stock solution of the protein (Cyt c-SPDP or Cyt c-Lac4-SPDP) was added to 500 μL of the MSN-SH dispersion and the mixture was gently stirred overnight at 4°C. Then, the samples were centrifuged at 14,000 rpm for 15 min. To remove the unreacted enzyme three washing/centrifugation cycles were performed using the immobilization buffer. The amount of immobilized enzyme was determined by depletion, i.e., the amount of the protein immobilized into MSN-SH was calculated from the difference between initial and final protein concentration in the supernatant.
Dynamic light scattering (DLS)
Particle sizes of MSN-SH, MSN-SPDP-Cyt c, and MSN-SPDP-Cyt c-Lac4 were determined by dynamic light scattering (DLS) using a DynaPro Titan. The samples were dispersed in water and subjected to ultrasonication at 240 watts for 30 sec.
Scanning electron microscope (SEM)
SEM of MSN-SH, MSN-SPDP-Cyt c, and MSN-SPDP-Cyt c-Lac4 was performed using a JEOL 5800LV scanning electron microscope at 20 kV. The samples were coated with gold for 10 sec using a Denton Vacuum DV-502A.
Cell culture
HeLa cells were maintained in accordance with the ATCC protocol. Briefly, the cells were cultured in minimum essential medium (MEM) containing 1% L-glutamine, 10% fetal bovine serum (FBS), and 1% penicillin in a humidified incubator with 5% CO2 and 95% air at 37°C. All experiments were conducted before cells reached 25 passages. For the cell viability and confocal miscroscopy experiments, HeLa cells were seeded in 96-well plates or lab-tek chambered coverslides (4 wells) for 24 h in MEM containing 1% L-glutamine, 10% FBS, and 1% penicillin. Subsequently, cell growth was arrested by decreasing the FBS concentration in the medium to 1% for 18 h and the cells were treated thereafter with GHS-OEt for 2 h to ensure an intracellular GHS concentration of 10 mM.30, 31 Cells were washed with PBS, exposed, and incubated with the different bioconjugates.
In vitro release of cytochrome c from MSN
The release of Cyt c from MSN was measured as previously described by us.11 Briefly, 3 mg of the MSN-SPDP-Cyt c-Lac4 bioconjugate was suspended in 1 ml of 50 mM PBS with 1 mM EDTA at pH 7.4 and 0, 1 μM, or 10 mM of glutathione (GHS) added, respectively. After incubation for 18 h at 37 °C, the MSN were pelleted by centrifugation at 14,000 rpm for 20 min. The supernatant was removed and used to determine the concentration of released Cyt c and the pellet was resuspended in fresh release buffer. The released protein was used to construct cumulative release profiles.
Cell viability assay
Mitochondrial function was measured using the CellTiter 96 aqueous non-radioactive cell proliferation assay from Promega (Madison, WI). HeLa cells (5000 cells/well) were seeded in 96-well plates as described above. Cells were incubated with serial dilutions of MSN-SPDP-Cyt c and MSN-SPDP-Cyt c-Lac4 bioconjugates (12.5, 25.0 and 37.5 μg/mL) and MSN-SH (80 μg/mL) for 24 to 72 h. Afterward, 20 μL of 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) and phenazine methosulfate (PMS) was added to each well (333 μg/mL MTS and 25 μM PMS). After 1 h, the absorbance at 492 nm was measured using a microplate reader. HeLa cells treated with 2 μM staurosporin for 6 h were used as positive control and cells without treatment were used as negative control. The relative cell viability (%) was calculated by:
Cellular uptake, endosomal escape and of MSN-SPDP-Cyt c-Lac4 bioconjugate
The internalization of MSN-SPDP-Cyt-c-Lac4 and its ability to avoid endosomal entrapment were determined by confocal laser scanning microscopy (CLSM). To execute these experiments, MSN labeled with FITC were used during the immobilization process. HeLa cells (12 600 cells) were seeded in Lab-tek chambered coverglass (4-wells) as described above. The cells were incubated with FITC-MSN-SPDP-Cyt c-Lac4 bioconjugates at a Cyt c concentration of 37.5 μg/mL and an endosome marker31 (FM-4-64; 10 μg/mL) at 37°C for 72 h. Afterwards, the medium was removed and the cells were washed with PBS thrice followed by fixation of the cells with 3.7% formaldehyde. The coverslips were examined under a Zeiss laser-scanning microscope 510 using a 100x oil immersion objective and excitation at 488 nm. FITC-MSN fluorescence was detected at wavelengths between 513–588 nm and the endosome marker between 598–738 nm.
Investigation of apoptosis induction in HeLa cells by the delivery of Cyt c-Lac4-SPDP
HeLa cells (12,600 cells) were seeded in Lab-tek chambered coverglass (4-wells) as previously described. The cells were incubated with MSN-SPDP-Cyt c-Lac4 bioconjugate at a Cyt c concentration of 37.5 μg/mL at 37°C for 72 h. For detection of apoptosis-dependent nuclear fragmentation, the cells were washed with PBS (1X) and incubated initially with DAPI (300 nM) and thereafter with PI (75 μM) for 5 min each. HeLa cells were then fixed using 3.7% formaldehyde. The coverslips were examined under a Zeiss laser-scanning microscope 510 using a 67x objective. Colocalization of DAPI and PI upon internalization into HeLa cells was determined, which is representative of highly condensed and fragmented chromatin in apoptotic cells.12, 15, 33 DAPI was excited at 405 nm and it emission was detected at 420–480 nm. PI was excited at 488 nm and was detected above 505 nm.
Proteolytic degradation assay
The proteolytic assay was adapted from Reinhardt et al.34 In brief, 0.16 mg/ml of Cyt c and Cyt c bioconjugates were dissolved in 20 mM Tris-HCl at pH 7.4 and incubated for 20 min at 20 °C. Then, 4 mg of trypsin (Sigma) was added to 1 ml of each sample and incubated at 37 °C. The degradation of the Cyt c samples was determined by measuring the absorbance at 408 nm at various time points.
Statistical analysis
Mann Whitney analysis was used for comparison of two independent groups for cell viability and cell-free caspase-3 assay. Difference between control (untreated cells for cell viability and native Cyt c for caspase-3 assay) and experimental group (i.e., MSN-SH, Cyt c-SPDP, Cyt c-Lac4 and Cyt c-Lac4-SPDP bioconjugates) was considered statistically significant at p<0.05. For comparison of multiple groups (Figure 6), ANOVA analysis was used and when differences were found between groups, Scheffe post-hoc analysis was performance (significance was assigned at p<0.05). All the experiments were at least performed in triplicate, the results averaged, and the standard deviation (SD) or standard error of the mean (SEM) calculated.
Figure 6.

Cell-free apoptosis assay with freshly purified HeLa cell cytosol. Asterisk (*) indicates statistical significance (p < 0.05), see Methods section for details.
RESULTS AND DISCUSSION
The goal of this work was to develop a nano-scale drug delivery device to deliver functional Cyt c to cancer cells using smart release to induce apoptosis. Obviously, protein denaturation during incorporation into the drug delivery device has to be avoided. In addition, delivery of proteins to target cells is difficult because proteins may undergo degradation after uptake by the cell. For example, Barnes and Shen35 conjugated Cyt c to oligo-arginine to accomplish intracellular delivery of the protein. Their results demonstrated that the Cyt c-oligo-arginine conjugate was efficiently internalized by HeLa cells but did not show any biological activity. Experiments involving proteasome inhibition demonstrated that this lack of biological activity was due to proteasome Cyt c degradation.35 However, while trivial in cell culture experiments, proteasome inhibition is not feasible in therapeutic approaches. Consequently, herein we explore an alternative strategy to efficiently deliver Cyt c to cancer cells in a bioactive form capable of inducing apoptosis. We argue that decorating Cyt c with a sugar should improve bioactivity by both preventing Cyt c denaturation during incorporation into the MSN and reducing proteolytic Cyt c degradation.
Glycosylation of Cyt c
Chemical glycosylation has been exploited previously by us as a strategy for protein stabilization.22, 24 Therein we demonstrated that chemical protein glycosylation can improve thermodynamic and colloidal protein stability as well as shield the protein surface from potential chemical and biological (i.e., proteolytic) degradation.25
Cyt c was modified with activated lactose (SS-mLac) resulting in lactose bound to the surface lysine residues of Cyt c (Cyt c-Lac). The average lactose molar content attached to the surface lysines of the protein was determined via TNBSA chromogenic assay.28 The results showed that on average 3.7 ± 0.9 molecules of lactose were bound to Cyt c which represents a ca. 20% level of modification of the available surface amino groups. We refer to this bioconjugate as Cyt c-Lac4-SPDP. The chemistry for obtaining the construct is shown in Figure 3.
Figure 3.
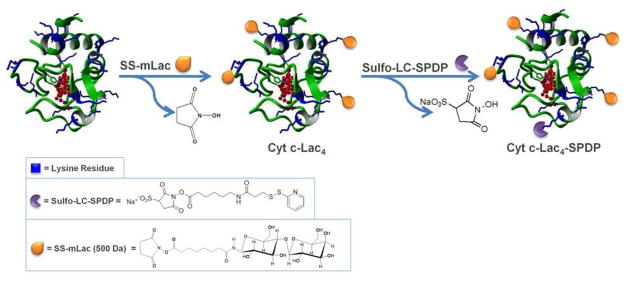
Scheme of the synthesis and structure of the bioconjugate Cyt c-Lac4-SPDP. Available lysine residues for linker attachment are shown in blue. Since all lysines are available for modification, the lactose molecules and SPDP linker can in principle be bound to any of them.
Cyt c and Cyt c-Lac4 immobilization into MSN
The smart release system designed by us involves the generation of redox sensitive disulfide bonds to attach the protein to the MSN. First, a suitable linker must be attached to the protein containing a disulfide bond followed by immobilization of the construct into thiolated MSN by thiol-disulfiode interchange (Figure 2). We employed the cross-linker sulfo-LC-SPDP to activate Cyt c and obtained the constructs Cyt c-SPDP and Cyt c-Lac4-SPDP. Reduction of the bioconjugates with DTT and determination of the released pyridine-2-thione at 343 nm as described by us11 revealed that under the conditions employed Cyt c was modified with the cross-linker at a linker-to-Cyt c molar ratio of 1.2 ± 0.1 and the lactose conjugate at a 1.6 ± 0.2 ratio, respectively.
Electrostatic interactions between the positively charged side of Cyt c containing the exposed heme edge and the WD-40 region of Apaf-1 are critically important to Cyt c-to-Apaf-1 binding and subsequent apoptosis.36 Thus, it is imperative to preserve the Cyt c conformation subsequent to any chemical modification to assure that Cyt c is still able to bind to Apaf-1 and induce apoptosis. Because the linker molecule chosen is quite hydrophobic, it was necessary to investigate whether the chemical modification would cause detrimental tertiary structural changes to Cyt c.
We performed circular dichroism (CD) spectroscopy to investigate Cyt c structural intactness. The CD spectrum in the heme Soret absorption band (380 to 450 nm) is related to the structure of the heme binding pocket. Amino acid side chains in the heme binding pocket are indirectly responsible for the optical activity in the Soret band region because of the coupling of the heme π—π* electronic transition dipole moment with adjacent aromatic amino acid residues.37, 38 The near-UV CD spectrum (250–320 nm) provides insight into protein tertiary structure changes that affect the environment of aromatic amino acids.8, 37 The near-UV CD spectrum of Cyt c is generated by the contribution of four Phe residues, four Tyr residues, one Trp residue, and two thioether bonds.37
The near-UV CD spectrum of native Cyt c presents two minima at 286 and 293 nm which according to the literature correspond to Trp-59.39 These two minima are significantly reduced in the Cyt c-SPDP bioconjugate spectrum clearly demonstrating that changes in Cyt c tertiary structure occurred upon chemical modification (Figure 4).37, 40 Interestingly, in the case of Cyt c-Lac4–SPDP the CD spectrum showed little spectral changes indicating that no significant changes in the tertiary structure occurred in this case. We can conclude that the attachment of lactose to Cyt c prior to attachment of the linker prevented structural changes caused by the hydrophobic linker. This is likely due to thermodynamic stabilization of Cyt c structure upon modification with lactose, which has been recently been demonstrated in our laboratory (Delgado Reyes and Griebenow, unpublished).
Figure 4.

Near-UV CD spectrum of Cyt c, Cyt c-SPDP, and Cyt c-Lac4-SPD
The Soret CD spectrum of native Cyt c shows a negative peak at ca. 422 nm and a positive peak at ca. 410 nm (Figure 5). This change of sign within the band has been interpreted to originate from characteristic heme-polypeptide interactions in the heme biding pocket.41, 42 The bisignate CD spectrum disappeared for the Cyt c-SPDP bioconjugate. This suggests that as a result of tertiary structure changes of Cyt c upon chemical modification the structure of the heme binding pocket changed which provoked a disruption in the coupling between π—π* transitions of the heme group and nearby amino acid residues.37, 41, 43 Changes in the Soret CD spectrum of Cyt c have been observed by others under various denaturing conditions, including, after the addition of urea, at elevated temperatures, in the presence of extrinsic ligands, and pH-induced.37, 40 In agreement with the CD data on tertiary structure (Figure 4), chemical protein glycosylation prevented significant spectral changes in the heme Soret band and thus structural changes in the heme binding pocket. The CD spectra of Cyt c and Cyt c-Lac4-SPDP are quite similar, indicating that heme-polypeptide interactions must be largely the same (Figure 5).
Figure 5.

Soret band CD spectrum of Cyt c, Cyt c-SPDP, and Cyt c-Lac4-SPDP.
We can surmise that both, Soret and near-UV CD spectra, indicate that activation of Cyt c with sulfo-LC-SPDP is detrimental to the tertiary structure. However, those structural changes were completely prevented when Cyt c was chemically modified with lactose prior to the attachment of the cross-linker.
Capability of apoptosis induction in a cell-free system
While Cyt c modification with lactose was efficient in preventing tertiary structural changes, it was unclear whether the Cyt-c-lactose bioconjugate was still capable of interacting with Apaf-1 and to induce apoptosis due to potential steric hindrance. With the aim to experimentally determine if the constructed Cyt c bioconjugates were still able to interaction with Apaf-1 to induce apoptosis, experiments using a cell-free system were performed. Cyt c is a cellmembrane-impermeable protein32 and it was therefore crucial to conduct the experiments in a cell-free system. Cell-free apoptosis assay is a relatively novel approach to mimic and study caspase activation.14 The addition of Cyt c to fresh cytosol should produce caspase activation. Indeed, it has been demonstrated that freshly purified cytosol contains sufficient dATP and ATP (mM) to induce Cyt c initiated caspase activation.12 Figure 6 shows the results of caspase-3 activity after addition of Cyt c, Cyt-c-SPDP, Cyt c-Lac4, and Cyt c-Lac4-SPDP to the freshly prepared cytosol of HeLa cells. Compared to Cyt c the caspase-3 activity induced by Cyt c-SPDP was reduced to only 47%. This result was expected because attachment of the linker produced significant tertiary structural changes. Somewhat counter-intuitive, glycosylation of Cyt c with four molecules of lactose did not significantly impair the Cyt c-Apaf-1 interaction. Attachment of the sulfo-LC-SPDP linker to Cyt c-Lac4 slightly reduced the ability of Cyt c-Lac4-SPDP to interact with Apaf-1 to 87%. The value was, however, much higher than for Cyt-c-SPDP confirming that structural preservation by the modification of Cyt c with lactose improved the apoptosis-inducing capability of Cyt c. Note that a cocktail of protease inhibitors must be added to the reaction mixture during this cell-free apoptosis assay to avoid proteolysis. Thus, the cell-free assay data obtained confirm the results obtained in the studies related to the structural integrity of Cyt c upon chemical modification because they exclude potential protection from proteolysis by the glycosylation. It should be noted that in vitro no difference was found by us for apoptosis induction by Cyt c in the cell-free assay in the presence or absence of the protease inhibitor cocktail.
In vivo, however, many tumor tissues display a high content in an array of proteases limiting the efficiency of protein drugs. We therefore explored whether glycosylation would protect the protein from proteolysis in an in vitro assay. Following a tryptolytic assay described in the literature32 we found this indeed to be the case (Figure 7). Cyt c, Cyt c-Lac4, and Cyt c-Lac4-SPDP show a similar degradation kinetics in the assay but Cyt c-SPDP degrades much faster, likely due to increased susceptibility to proteases by being partially unfolded. Increasing the amount of sugars attached to Cyt c to 9 increased the resistance towards proteolysis. We can conclude that attachment of lactose to Cyt c protects the protein from potential proteolysis.
Figure 7.

Kinetics of the degradation of Cyt c and Cyt c conjugates by 4 mg/ml trypsin at 37 °C.
Covalent immobilization of Cyt c-SPDP and Cyt c-Lac4-SPDP into MSN-SH
In order to evaluate and confirm cellular uptake, endosomal escape, and apoptosis using the designed nanoparticulate MSN drug delivery system, Cyt c-SPDP and Cyt c-Lac4-SPDP bioconjugates were immobilized into thiolated MSN. Under the conditions employed, the immobilization was determined to be 350 ± 79 mg and 356 ± 56 mg of Cyt c per 1.0 g of MSN-SH for Cyt c-SPDP and Cyt c-Lac4-SPDP, respectively (Table 1). We conducted UV-Vis spectroscopy to verify the presence of Cyt c in the MSN. The visible absorption spectrum demonstrated typical absorption bands at 408 nm (Soret) and around 520 and 550 nm (α and β absorption bands) demonstrating the presence of oxidized Cyt c in the MSN (Figure S1). With the aim to determine if the immobilization process had any effect on the morphology and/or size of the MSN-SH we employed SEM and DLS. SEM micrographs revealed no impact of the immobilization on the morphology and/or size of MSN-SH (Figure 8).
Table 1.
Sample characteristics of Cyt c and Cyt c bioconjugates used for encapsulation in MSN and MSN characteristics.
| Sample | Diameter (nm) | Lactose-to-Cyt c molar ratio | SDPD-to-Cyt c molar ratio | Immobilizeda | |
|---|---|---|---|---|---|
| DLS | SEM | ||||
| MSN-SH | 343±20 | 321±68 | - | - | - |
| MSN-SPDP-Cyt | 341±30 | 305±88 | - | 1.2±0.1 | 350±79 |
| MSN-SPDP-Cyt-Lac4 | 325±24 | 312±74 | 3.7±0.9 | 1.6±0.2 | 356±56 |
The amount is given in mg of Cyt c immobilized per gram of MSN-SH.
Figure 8.

SEM micrographs of (A) MSN-SH, (B) MSN-SPDP-Cyt c, and (C) MSN-SPDP-Cyt c-Lac4.
The size of MSN-SH suspended in water was 343 ± 20 nm, 341 ± 30 nm for Cyt c-SPDP, and 325 ± 24 nm for Cyt c-Lac4-SPDP. The nanoparticles sizes determined by DSL were congruent with those obtained using SEM (Table 1).
Controlled Release of Cyt c from Nanoparticles
In vitro release studies were conducted to assess the covalently modification of Cyt c to MSN and their release from nanoparticles by reductive intracellular conditions stimuli. Only intracellular glutathione concentrations (1–10 mM), but not extracellular plasma conditions (1 μM), are sufficient to cleave disulfide bonds.11
We found that about 10–20% of Cyt c was released using no or 1 μM GHS (Figure 9). That confirms that at least 80% of Cyt c was covalently immobilized and not adsorbed at the nanoparticle surface. After 300 h under reducing conditions no more protein was released from MSN. The data are compatible with the bioactivity we found for our constructs (see below).
Figure 9.

Cumulative release of Cyt c from mesoporous silica nanparticles suspended in buffer (●), buffer with 1 μM glutathione (○), and buffer with 10 mM glutathione (▼). Asterisk (*) indicates statistical significance (p < 0.05), see Methods section for details.
Viability of HeLa cells upon exposure to the MSN drug delivery system
Once confirmed by the cell-free assay that the Cyt c bioconjugates had the capability to interact with Apaf-1 and induce apoptosis, the bioconjugates were incorporated into thiolated MSN by thiol-disulfide interchange as described by us.11 These constructs were then employed to study their efficiency in inducing apoptosis in cancer cells using HeLa cells as the model HeLa cells were treated with MSN-SPDP-Cyt c and MSN-SPDP-Cyt c-Lac4 at different concentrations (12.5, 25.0 and 37.5 μg Cyt c/mL) for 24 to 72 h. None of the conjugates induced significant cell death in HeLa cells up to 48 h. However, HeLa cells treated with MSN-SPDP-Cyt c-Lac4 for 72 h showed a statistically significantly reduced viability when compared with untreated cells (control) of 67% and 45% at Cyt c concentrations of 25 μg/mL and 37.5 μg/mL, respectively (Figure 10). The MSN-Cyt c-SPDP bioconjugate induced cell death only at the higher concentration of 37.5 μg/mL (81% cell viability). However, the result was statistically not significant. MSN-SH had no significant effect in HeLa cell viability (Figure 10) in agreement with the literature.43, 44 This confirms that cell death was induced by Cyt c and not by the drug carrier per se. An additional control experiment was performed by adding SPDP-Cyt c and SPDP-Cyt c-Lac4 to the cells (30 μg/ml final concentration) under otherwise identical conditions. As expected, no significant effect on cell viability was observed after 72 h because the cytoplasmic membrane is impermeable for Cyt c.9, 10
Figure 10.

HeLa cell viability in the presence of MSN-SPDP-Cyt c, and MSN-SPDP-Cyt c-Lac4 bioconjugates at different concentrations after incubation for 72 h. The MSN-SH, SPDP-Cyt c and SPDP-Cyt c-Lac4 controls were adjusted to the same MSN or Cyt c concentrations as the corresponding Cyt c immobilized in the MNS. Asterisk (*) indicates statistical significance (p < 0.05), see Methods section for details.
Chemically glycosylated proteins are conformationally more stable18, 22–25 and also resist degradation by proteases46, 47 better than their non-glycosylated forms. They should display enhanced stability under conditions associated with in vivo endosomolytic/lysosomolytic degradation. For this reason, researches have exploited glycosylation as a strategy for improving stability in therapeutic agents48 or drug delivery systems.46 The fact that MSN-SPDP-Cyt c did not induce cell death is in agreement with results by Barnes and Shen (2009) who demonstrated that Cyt c degradation by proteasomes is responsible for the lack of apoptotic activity when delivered to HeLa cells.35 Our results demonstrate that glycosylation is an effective strategy to stabilize Cyt c for delivery into cancer cells, likely by hindering protein degradation.
Cellular uptake and endosomal escape of MSN-SPDP-Cyt c-Lac4
Based on the obtained results for the viability of HeLa cells upon exposure to the MSN drug delivery system and based on the fact that Cyt c is a membrane impermeable protein,9, 10 experiments related to cellular uptake, endosomal escape, and apoptosis induction were only conducted using MSN-SPDP-Cyt c-Lac4 and were only of a confirmatory nature.
With the aim to investigate internalization and endosomal escape capability of MSN-SPDP-Cyt c-Lac4, we used FITC labeled MSN and the endosome marker FM-4-64. The cells were incubated with the bioconjugate and with the endosome marker and thereafter examined by confocal microscopy. Under the chosen conditions, green fluorescence spots observed in the micrographs are due to the internalization of the bioconjugate and red fluorescence spots show endosomes in the cytoplasm. Co-localization of the two dyes because the bioconjugate is entrapped in endosomes results in yellow fluorescence spots.32 As depicted in Figure 11, after 72 h of incubation a significant amount of the bioconjugate is located in the cytoplasm. Some of the bioconjugate was still entrapped in endosomes (yellow spots) in agreement with expected uptake by endocytosis. In the merge micrograph several red spots are still observed indicative of endosomes that have not sequestered any bioconjugate in them or those that have not disintegrated after the bioconjugate was released into the cytoplasm.49 We have not performed extensive time-course experiments, but the confocal micrographs support the idea that the MSN-SPDP-Cyt c-Lac4 bioconjugate is internalized by HeLa cells. This likely involves endocytosis because we find bioconjugates in endosomes followed by their escape into the cytoplasm.
Figure 11.

Internalization of the MSN-SPDP-Cyt c-Lac4 bioconjugate by HeLa cells observed by confocal microscopy. The left image is the autofluorescence image of the cells, the lower left shows the FITC labeled MSN internalized by the cells, the lower right shows the FM-4-64 labeled endosomes, and the upper right micrograph is the merged image.
Apoptosis induction in HeLa cells by the delivery of Cyt c-Lac4-SPDP
Internalization and endosomal escape of the MSN-SPDP-Cyt c-Lac4 bioconjugate is necessary but not sufficient to induce apoptosis in HeLa cells. It is crucial that Cyt c is released from the MSN carrier and binds to Apaf-1 to induce apoptosis. Cells undergoing apoptosis exhibit distinctive characteristics, such as cell shrinkage, membrane blebbing, chromatin condensation, and nuclear fragmentation. To confirm that cell death determined in the cell viability experiments was indeed due to apoptosis, we assessed the occurrence of nuclear segmentation and chromatin condensation in the cell nuclei. HeLa cells were incubated with the MSN-SPDP-Cyt c-Lac4 bioconjugate at a Cyt c concentration of 37.5 μg/mL for 72 h and the cells stained with PI and DAPI. Co-localization of DAPI and PI occurred (Figure 12), which points toward nuclear fragmentation and chromatin condensation in the cells indicative of ongoing apoptosis.15 These results demonstrate that MSN can efficiently immobilize an apoptosis-inducing protein, such as Cyt c, and release the protein under intracellular redox conditions thus inducing apoptosis in HeLa cells. In contrast, no co-localization was observed when using MSN without any protein (Fig. 12C).
Figure 12.
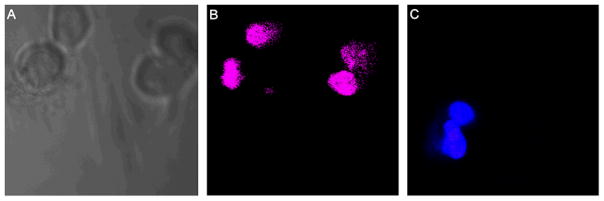
Study of DAPI and propidium iodine (PI) co-localization, for the detection of apoptotic cells after 75 h of incubation. (A) The autofluorescence image of the HeLa cells. (B) Selective induction of apoptosis observed in HeLa cells incubated with the MSN-SPDP-Cyt c-Lac4 bioconjugate. (C) No cellular apoptosis observed in HeLa cells when incubated with MSN alone.
CONCLUSIONS
In this work we delivered a model pharmaceutical protein, Cyt c, to a model cancer cell line (HeLa) from nanosized mesoporous silica particles and induced apoptosis to a varying extent. Cyt c was attached to the particles by a smart linker releasing the payload under intracellular conditions. Importantly we were able to demonstrate that induction of apoptosis was improved by attachment of lactose to the surface of Cyt c. Two mechanisms were identified: stabilization of Cyt c during immobilization into the MSN and reduced susceptibility towards proteolysis. It is conceivable that glycosylated anti-cancer proteins could be generally useful to enable new cancer therapeutic approaches. Experiments are on the way to improve the described system further by employing mutant proteins to enable site-directed glycosylation of the constructs.
Our studies using confocal microscopy confirm that the MSN-SPDP-Cyt c-Lac4 bioconjugate was internalized by HeLa cells and escaped from endosomal entrapment, which allowed the release of its cargo into the cytoplasm to induce apoptosis. Co-localization analysis using DAPI and PI confirmed apoptosis induction in the treated HeLa cells. The system could be further improved by enabling active targeting using cancer cell selective ligands or molecules.
Supplementary Material
Acknowledgments
This publication was made possible by grant number SC1 GM086240 from the National Institute for General Medical Sciences (NIGMS) at the National Institutes of Health (NIH) through the Support of Competitive Research (SCoRE) Program. We also like to acknowledge the contribution to this study by grants ISI0 RR-13705-01 and DBI-0923132 to establish and upgrade the Confocal Microscopy Facility at the University of Puerto Rico (CIF-UPR). The authors like to thank Mr. Bismark Madera, M.T., for his outstanding dedication to support our confocal imaging needs and the superb work provided by him. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS. MM and YD were supported by fellowships from the NIH Research Initiative for Scientific Enhancement (RISE) Program (R25 GM061151) and JM received a fellowship from the Institute for Functional Nanomaterials (IFN) at the University of Puerto Rico.
Footnotes
Supporting Information Available. This information is available free of charge via the Internet at http://pubs.acs.org/. Figure S1: Absorption spectra of MSN and of Cyt c immobilized in MSN.
References
- 1.Siegel R, Ward E, Brawley O, Jemal A. Cancer Statistics, 2011: The Impact of Eliminating Socioeconomic and Racial Disparities on Premature Cancer Deaths. Cancer J Clin. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Jaracz S, Chen J, Kuznetsova LV, Ojima I. Recent Advances in Tumor-Targeting Anticancer Drug Conjugates. Bioorgan Med Chem. 2005;13:5043–5054. doi: 10.1016/j.bmc.2005.04.084. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Ranganathan R, Madanmohan S, Kesavan A, Baskar G, Krishnamoorthy YR, Santosham R, Ponraju D, Rayala SK, Venkatraman G. Nanomedicine: Towards Development of Patient-Friendly Drug-Delivery Systems for Oncological Applications. Int J Nanomed. 2012;7:1043–1060. doi: 10.2147/IJN.S25182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Danhier F, Feron O, Preat V. To Exploit the Tumor Microenvironment: Passive and Active Tumor Targeting of Nanocarriers for Anti-Cancer Drug Delivery. J Contr Rel. 2010;148:135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 6.Maeda H, Fang J, Inutsuka T, Kitamoto Y. Vascular Permeability Enhancement in Solid Tumor: Various Factors, Mechanisms Involved and its Implications. Int Immunopharmacol. 2003;3:319–328. doi: 10.1016/S1567-5769(02)00271-0. [DOI] [PubMed] [Google Scholar]
- 7.He QJ, Shi JL. Mesoporous Silica Nanoparticle Based Nano Drug Delivery Systems: Synthesis, Controlled Drug Release and Delivery, Pharmacokinetics and Biocompatibility. J Mater Chem. 2011;21:5845–5855. [Google Scholar]
- 8.Chen E, Goldbeck RA, Kliger DS. Probing Early Events in Ferrous Cytochrome c Folding with Time-resolved Natural and Magnetic Circular Dichroism Spectroscopies. Curr Protein Pept Sci. 2009;10:464–475. doi: 10.2174/138920309789352001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Caruso F. Mesoporous Silica Spheres as Supports for Enzyme Immobilization and Encapsulation. Chem Mater. 2005;17:953–961. [Google Scholar]
- 10.Slowing II, Trewyn BG, Lin VSY. Mesoporous Silica Nanoparticles for Intracellular Delivery of Membrane-Impermeable Proteins. J Am Chem Soc. 2007;129:8845–8849. doi: 10.1021/ja0719780. [DOI] [PubMed] [Google Scholar]
- 11.Mendez J, Monteagudo A, Griebenow K. Stimulus-Responsive Controlled Release System by Covalent Immobilization of an Enzyme into Mesoporous Silica Nanoparticles. Bioconj Chem. 2012;23:698–704. doi: 10.1021/bc200301a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bratton SB, Salvesen GS. Regulation of the Apaf-1-Caspase-9 Apoptosome. J Cell Sci. 2010;123:3209–3214. doi: 10.1242/jcs.073643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chinnaiyan AM. The Apoptosome: Heart and Soul of the Cell Death Machine. Neoplasia. 1999;1:5–15. doi: 10.1038/sj.neo.7900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandra D, Tang DG. Detection of Apoptosis in Cell-Free Systems. Methods Mol Biol. 2009;559:65–75. doi: 10.1007/978-1-60327-017-5_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santra S, Kaittanis C, Perez JM. Cytochrome c Encapsulating Theranostic Nanoparticles: A Novel Bifunctional System for Targeted Delivery of Therapeutic Membrane-Impermeable Proteins to Tumors and Imaging of Cancer Therapy. Mol Pharm. 2010;7:1209–1222. doi: 10.1021/mp100043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim SK, Foote MB, Huang L. The targeted Intracellular Delivery of Cytochrome c Protein to Tumors Using Lipid-apolipoprotein Nanoparticles. Biomaterials. 2012;33:3959–3966. doi: 10.1016/j.biomaterials.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torchilin V. Intracellular Delivery of Protein and Peptide Therapeutics. Drug Discov Today: Technol. 2008;5:e95–e103. doi: 10.1016/j.ddtec.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 18.Solá RJ, Griebenow K. Glycosylation of Therapeutic Proteins: An Effective Strategy to Optimize Efficacy. BioDrugs. 2010;24:9–21. doi: 10.2165/11530550-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang L, Persky AM, Hochhaus G, Meibohm B. Pharmacokinetic Aspects of Biotechnology Products. J Pharm Sci. 2004;93:2184–2204. doi: 10.1002/jps.20125. [DOI] [PubMed] [Google Scholar]
- 20.Wu W, Luo Y, Sun C, Liu Y, Kuo P, Varga J, Xiang R, Reisfeld R, Janda KD, Edgington TS, Liu C. Targeting Cell-impermeable Prodrug Activation to Tumor Microenvironment Eradicates Multiple Drug-resistant Neoplasms. Cancer Res. 2006;66:970–980. doi: 10.1158/0008-5472.CAN-05-2591. [DOI] [PubMed] [Google Scholar]
- 21.De Virgilio M, Klitzmüller C, Schwaiger E, Klein M, Kreibich DG, Ivessa NE. Degradation of Short-lived Glycoprotein from the Lumen of the Endoplasmic Reticulum: The Role of N-linked Glycans and the Unfolding Protein Response. Mol Cell Biol. 1999;10:4059–4073. doi: 10.1091/mbc.10.12.4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solá RJ, Al-Azzam W, Griebenow K. Engineering of Protein Thermodynamic, Kinetic, and Colloidal Stability: Chemical Glycosylation with Monofunctionally Activated Glycans. Biotechnol Bioeng. 2006;94:1072–1079. doi: 10.1002/bit.20933. [DOI] [PubMed] [Google Scholar]
- 23.Solá RJ, Griebenow K. Chemical Glycosylation: New Insights on the Interrelation Between Protein Structural Mobility, Thermodynamic Stability, and Catalysis. FEBS Lett. 2006;580:1685–1690. doi: 10.1016/j.febslet.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 24.Pagan M, Solá RJ, Griebenow K. On the Role of Protein Structural Dynamics in the Catalytic Activity and Thermostability of Serine Protease Subtilisin Carlsberg. Biotechnol Bioeng. 2009;103:77–84. doi: 10.1002/bit.22221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solá RJ, Griebenow K. Effects of Glycosylation on the Stability of Protein Pharmaceuticals. J Pharm Sci. 2009;98:1223–1245. doi: 10.1002/jps.21504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vegarud G, Christensen TB. Glycosylation of Proteins: A New Method of Enzyme Stabilization. Biotechnol Bioeng. 1975;17:1391–1397. doi: 10.1002/bit.260170918. [DOI] [PubMed] [Google Scholar]
- 27.Lis H, Sharon N. Protein Glycosylation. Structural and Functional Aspects. FEBS J. 1993;218:1–27. doi: 10.1111/j.1432-1033.1993.tb18347.x. [DOI] [PubMed] [Google Scholar]
- 28.Habeeb AF. Determination of Free Amino Groups in Proteins by Trinitrobenzenesulfonic Acid. Anal Biochem. 1966;14:328–336. doi: 10.1016/0003-2697(66)90275-2. [DOI] [PubMed] [Google Scholar]
- 29.Bradford MM. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 30.Liu J, Pang Y, Huang W, Zhu Z, Zhu X, Zhou Y, Yan D. Redox-responsive Polyphosphate Nanosized Assemblies: A Smart Drug Delivery Platform for Cancer Therapy. Biomacromolecules. 2011;12:2407–2415. doi: 10.1021/bm2005164. [DOI] [PubMed] [Google Scholar]
- 31.Mortera R, Vivero-Escoto J, Slowing II, Garrone E, Onida B, Lin VS. Cell-induced Intracellular Controlled Release of Membrane Impermeable Cysteine from a Mesoporous Silica Nanoparticle-based Drug Delivery System. Chem Commun. 2009;22:3219–3221. doi: 10.1039/b900559e. [DOI] [PubMed] [Google Scholar]
- 32.Slowing II, Trewyn BG, Lin VSY. Mesoporous Silica Nanoparticles for Intracellular Delivery of Membrane-Impermeable Proteins. J Am Chem Soc. 2007;129:8845–8849. doi: 10.1021/ja0719780. [DOI] [PubMed] [Google Scholar]
- 33.Shacter E, Williams JA, Hinson RM, Senturker S, Lee YJ. Oxidative Stress Interferes with Cancer Chemotherapy: Inhibition of Lymphoma Cell Apoptosis and Phagocytosis. Blood. 2000;96:307–313. [PubMed] [Google Scholar]
- 34.Reinhardt DP, Ono RN, Sakai LY. Calcium Stabilizes Fibrillin-1 Against Proteolytic Degradation. J Biol Chem. 1997;272:1231–1236. doi: 10.1074/jbc.272.2.1231. [DOI] [PubMed] [Google Scholar]
- 35.Barnes MP, Shen WC. Disulfide and Thioether Linked Cytochrome c-Oligoarginine Conjugates in HeLa Cells. Int J Pharm. 2009;369:79–84. doi: 10.1016/j.ijpharm.2008.10.036. [DOI] [PubMed] [Google Scholar]
- 36.Purring-Koch C, McLendon G. Cytochrome c Binding to Apaf-1: The Effects of dATP and Ionic Strength. Proc Natl Acad Sci US A. 2000;97:11928–11931. doi: 10.1073/pnas.220416197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinheiro TJ, Elove GA, Watts A, Roder H. Structural and Kinetic Description of Cytochrome c Unfolding Induced by the Interaction with Lipid Vesicles. Biochemistry-US. 1997;36:13122–13132. doi: 10.1021/bi971235z. [DOI] [PubMed] [Google Scholar]
- 38.Woody RW, Hsu MC. Origin of the Heme Cotton Effects in Myoglobin and Hemoglobin. J Am Chem Soc. 1971;93:3515–3525. doi: 10.1021/ja00743a036. [DOI] [PubMed] [Google Scholar]
- 39.Davies AM, Guillemette JG, Smith M, Greenwood C, Thurgood AG, Mauk AG, Moore GR. Redesign of the Interior Hydrophilic Region of Mitochondrial Cytochrome c by Site-directed Mutagenesis. Biochemistry-US. 1993;32:5431–5435. doi: 10.1021/bi00071a019. [DOI] [PubMed] [Google Scholar]
- 40.Myer YP. Conformation of cytochromes. III. Effect of Urea, Temperature, Extrinsic Ligands, and pH Variation on the Conformation of Horse Heart Ferricytochrome c. Biochemistry-US. 1968;7:765–776. doi: 10.1021/bi00842a035. [DOI] [PubMed] [Google Scholar]
- 41.Wei W, Danielson ND. Fluorescence and Circular Dichroism Spectroscopy of Cytochrome c in Alkylammonium Formate Ionic Liquids. Biomacromolecules. 2011;12:290–297. doi: 10.1021/bm1008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Myer YP. Conformation of Cytochromes. 3. Effect of Urea, Temperature, Extrinsic Ligands, and pH Variation on the Conformation of Horse Heart Ferricytochrome c. Biochemistry-US. 1968;7:765–776. doi: 10.1021/bi00842a035. [DOI] [PubMed] [Google Scholar]
- 43.Thomas YG, Goldbeck RA, Kliger DS. Characterization of Equilibrium Intermediates in Denaturant-Induced Unfolding of Ferrous and Ferric Cytochromes c Using Magnetic Circular Dichroism, Circular Dichroism, and Optical Absorption Spectroscopies. Biopolymers. 2000;57:29–36. doi: 10.1002/(SICI)1097-0282(2000)57:1<29::AID-BIP5>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 44.Yu K, Grabinski C, Schrand A, Murdock R, Wang W, Gu B, Schlager J, Hussain S. Toxicity of Amorphous Silica Nanoparticles in Mouse Keratinocytes. J Nanopart Res. 2009;11:15–24. [Google Scholar]
- 45.Lin W, Huang YW, Zhou XD, Ma Y. In Vitro Toxicity of Silica Nanoparticles in Human Lung Cancer Cells. Toxicol Appl Pharmacol. 2006;217:252–259. doi: 10.1016/j.taap.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 46.Robinson MA, Charlton ST, Garnier P, Wang XT, Davis SS, Perkins AC, Frier M, Duncan R, Savage TJ, Wyatt DA, Watson SA, Davis BG. LEAPT: Lectin-Directed Enzyme-Activated Prodrug Therapy. Proc Natl Acad Sci US A. 2004;101:14527–14532. doi: 10.1073/pnas.0303574101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Russell D, Oldham NJ, Davis BG. Site-selective Chemical Protein Glycosylation Protects from Autolysis and Proteolytic Degradation. Carbohydr Res. 2009;344:1508–1514. doi: 10.1016/j.carres.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 48.Kim BM, Kim H, Raines RT, Lee Y. Glycosylation of Onconase Increases its Conformational Stability and Toxicity for Cancer Cells. Biochem Biophys Res Com. 2004;315:976–983. doi: 10.1016/j.bbrc.2004.01.153. [DOI] [PubMed] [Google Scholar]
- 49.Guo H, Qian H, Sun S, Sun D, Yin H, Cai X, Liu Z, Wu J, Jiang T, Liu X. Hollow Mesoporous Silica Nanoparticles for Intracellular Delivery of Fluorescent Dye. Chem Cent J. 2011;5:1. doi: 10.1186/1752-153X-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.