

Abstract
The formation of acellular capillaries in the retina, a hallmark feature of diabetic retinopathy, is caused by apoptosis of endothelial cells and pericytes. The biochemical mechanism of such apoptosis remains unclear. Small heat shock proteins play an important role in the regulation of apoptosis. In the diabetic retina, pro-inflammatory cytokines are upregulated. In this study, we investigated the effects of pro-inflammatory cytokines on small heat shock protein 27 (Hsp27) in human retinal endothelial cells (HREC). In HREC cultured in the presence of cytokine mixtures (CM), a significant downregulation of Hsp27 at the protein and mRNA level occurred, with no effect on HSF-1, the transcription factor for Hsp27. The presence of high glucose (25 mM) amplified the effects of cytokines on Hsp27. CM activated indoleamine 2,3-dioxygenase (IDO) and enhanced the production of kynurenine and ROS. An inhibitor of IDO, 1-methyl tryptophan (MT), inhibited the effects of CM on Hsp27. CM also upregulated NOS2 and, consequently, nitric oxide (NO). A NOS inhibitor, L-NAME, and a ROS scavenger blocked the CM-mediated Hsp27 downregulation. While a NO donor in the culture medium did not decrease the Hsp27 content, a peroxynitrite donor and exogenous peroxynitrite did. The cytokines and high glucose-induced apoptosis of HREC were inhibited by MT and L-NAME. Downregulation of Hsp27 by a siRNA treatment promoted apoptosis in HREC. Together, these data suggest that pro-inflammatory cytokines induce the formation of ROS and NO, which, through the formation of peroxynitrite, reduce the Hsp27 content and bring about apoptosis of retinal capillary endothelial cells.
Keywords: Hsp27, retinal endothelial cells, cytokines, ROS, apoptosis
1. Introduction
According to the International Diabetes Federation, there were 311 million people with diabetes worldwide in 2011 and this number is projected to increase to 552 million by 2030 [1]. One of the devastating long-term complications of this disease is diabetic retinopathy, which is the leading cause of visual impairment in working-class adults in the US and other developed countries. In both type 1 and type 2 diabetes, diabetic retinopathy becomes progressively worse with duration of the disease; it is estimated that after 20 years of diabetes, nearly 75% of patients show clinical signs of the disease [2].
Vascular, glial and neuronal abnormalities are the earliest changes in the diabetic retina [3, 4]. Retinal capillary cells become increasingly permeable to macromolecules, leading to macular edema. The extracellular matrix of capillary cells becomes thicker and localized sacular microaneurysms develop on capillaries. Along with these changes, pericytes and endothelial cells, the two cell types in retinal capillaries, die early in the disease, causing formation of acellular capillaries that lead to local ischemia in the retina [reviewed in [5]]. A number of mechanisms have been put forward to explain these histopathological changes, including pro-inflammatory signals [reviewed in [6]]. Chronic low-grade inflammation appears to play a role in the pathogenesis of the disease [7, 8].
Several pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and interferon-γ (IFN-γ), are elevated in the diabetic retina [9–11]. These cytokines upregulate nuclear factor-kappaB (NF-κB), which, in turn, can promote the synthesis of cytokines [12]. Along with these changes, inflammatory markers, including vascular cell adhesion molecule-1 (VCAM-1), intracellular adhesion molecule-1 (ICAM-1), nitric oxide synthase-2 (NOS2), cyclooxygenase-2 (COX2), and monocyte chemotactic protein-1 (MCP-1), are upregulated in the diabetic retina [reviewed in [6]].
Several studies have provided direct evidence for a role of inflammation in diabetic retinopathy. These include a demonstration that pharmacological suppression of inflammation leads to inhibition of ICAM-1 expression and leukostasis (attachment of leukocytes to endothelial cells, a characteristic feature of inflammation) and that the absence of TNF-α leads to suppression of blood retinal barrier breakdown in diabetic retina [13, 14]. In addition, the inhibition of caspase-1, which activates IL-1β, inhibits capillary degeneration [15], and the inhibition of NF-κB leads to inhibition of ICAM-1 and vascular endothelial growth factor-A production in the diabetic retina [16]. Finally, the inhibition of COX2 and NOS2 blocks capillary cell death and reduces leukostasis and blood retinal barrier breakdown, respectively, in the retinas of diabetic rodents [17, 18].
Despite clear evidence of an increased inflammatory response in diabetic retinopathy, the source(s) of inflammatory cytokines in the retina are unclear. While capillary cells can produce cytokines in small amounts, it is believed that retinal glia, Muller cells and microglia, as well as retinal pigmented epithelial cells, are capable of producing these cytokines. Cellular stresses, such as endoplasmic reticulum stress and stress imposed by ROS, promote the synthesis of cytokines [8, 19]; however, it remains unclear whether these cells synthesize inflammatory cytokines in response to hyperglycemia in diabetes or through hyperglycemia-driven processes. Nevertheless, there is a clear link between inflammation and capillary cell death in experimental diabetic retinopathy and in cultured retinal capillary cells. Despite these advances, the exact mechanism by which inflammation brings about apoptosis of retinal capillary cells remains unknown. It has been suggested that ROS, produced from activated NADPH oxidase and leakage of electrons from the mitochondrial electron transport chain, and nitric oxide, produced from upregulated NOS, are involved, but the actual mechanism is largely unknown. We also demonstrated that IFN-γ upregulates indoleamine 2,3-dioxygenase (IDO) through the JAK/STAT pathway in human retinal endothelial cells, leading to the formation of kynurenines, which spontaneously produce ROS [20].
Small heat shock proteins are ubiquitously present in cells and they protect cells from stresses through their chaperone, anti-apoptotic and anti-inflammatory activities. This family is composed of more than 10 members [21], among which αA- and αB-crystallin and Hsp27 are present in retinal cells [22–25]. Both αB-crystallin and Hsp27 have been implicated in angiogenesis [26, 27]. Despite these findings, whether there are alterations in their levels in capillary cells in diabetes is unknown. In this study, we investigated the effects of pro-inflammatory cytokines and high glucose on Hsp27 expression and present data to show that downregulation of this protein leads to apoptosis of human retinal endothelial cells.
2. Materials and Methods
1-methyl-DL-tryptophan, DETA NONOate [2,2′-(hydroxynitrosohydrazino)bis-ethanamine], L-NAME (Nω-nitro-L-arginine methyl ester hydrochloride), 1-oxyl-2,2,6,6-tetramethyl-4-hydroxypiperidine (TEMPOL), DL-kynurenine, and D-glucose were obtained from Sigma-Aldrich Chemical Co. LLC (St. Louis, MO, USA). TNF-α and IL-1β were obtained from Invitrogen (Grand Island, NY). IFN-γ was obtained from R and D systems (Minneapolis, MN). N-Acetyl-Asp-Glu-Val-Asp-7-amido-4-trifluoromethylcoumarin (Ac-DEVD-AFC) was from BD Biosciences, San Jose, CA. The peroxynitrite donor, 3-morpholinosydnonimine (SIN-1) was obtained from Acros Organics, NJ. Peroxynitrite was from Calbiochem, EMD Biosciences, San Diego, CA and 5-(and-6)-chloromethyl-2′,7′-dichlorofluorescein diacetate, acetyl ester (CM-D2DCFDA) was from Molecular Probes, Eugene, OR. All other chemicals were of analytical grade.
2.1. Cell Culture and treatment
Human eyes from 46–55 year-old donors without systemic diseases were obtained from the National Disease Research Interchange (Philadelphia, PA). Human tissue research adhered to the tenets of the Declaration of Helsinki. HREC were isolated and cultured using a previously published method [28]. The cells were characterized by their characteristic uptake of DiI-Ac-LDL (acetylated low density lipoprotein, labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl indocarbocyanine perchlorate) and CD31 staining and were found to be 95–99% pure. Cells in passage numbers between 3 and 8 were used in all experiments. They were grown in six-well plates, coated with 0.1% gelatin, in Dulbecco’s modified Eagle’s medium/F12, supplemented with 10% fetal bovine serum, 5% endothelial cell growth supplements, 1% penicillin/streptomycin and 1% insulin-transferrin-selenium at 37°C in humidified 95% air/5% CO2. All experiments were conducted with cells that had attained 60–70% confluency.
For individual cytokine treatment, HREC were cultured with TNF-α (10 or 20 ng/mL), IL-1β (10 or 20 ng/mL) or IFN-γ (50 or 100 U/mL) for 48 h without replacing the medium. For cytokine mixture (CM) treatment, HREC were incubated with two combinations of cytokines (CM1= 10 ng/mL of TNF-α + 10 ng/mL of IL-1β + 50 U/mL of IFN-γ or CM2= 20 ng/mL of TNF-α + 20 ng/mL of IL-1β + 100 U/mL of IFN-γ) with or without 25 mM D-glucose for 48 h without replacing the medium. To study the effect of kynurenine, cells were cultured with 100 μM DL-kynurenine for 48 h without replacing the medium.
To determine the role of indoleamine 2, 3-dioxygenase (IDO), we added an inhibitor of IDO, 1-methyl-DL-tryptophan (MT), at 20 μM, with or without cytokines to the culture medium and cells were incubated for 48 h. In other experiments, to determine the role of nitric oxide (NO), cells were incubated for 48 h with a nitric oxide synthase inhibitor, L-NAME at 500 μM or a nitric oxide donor DETA NONOate at 500 μM. For peroxynitrite treatment, culture media was replaced with PBS, and peroxynitrite was added at 200 μM concentration. Cells were then incubated for 20–30 min, after which PBS was replaced with culture media; the cells were subsequently incubated for 0, 24 or 48 h. To determine the effect of ROS and NO on Hsp27, we added TEMPOL (100 μM) and L-NAME (500 μM), respectively, along with inflammatory cytokines. We also tested the role of peroxynitrite by adding a peroxynitrite donor, SIN-1, to serum-free medium at 1 or 2 mM and cells were incubated for 3 h, followed by culturing for an additional 48 h in the complete media.
2.2. SDS-PAGE and Western blot analysis
Trypsinized cells were lysed with Mammalian Protein Extraction Reagent (M-PER, from Thermo Scientific, Rockford, IL) containing a protease inhibitor cocktail (1:100; Sigma, St. Louis, MO). Protein concentration was determined using the BCA Protein Assay Kit with BSA as the standard (Thermo Scientific). HREC proteins were resolved by SDS-PAGE, and Western blotting was carried out with 10 μg cell lysate protein, using one of the following antibodies: Hsp27 mAb (1: 1,000 dilution, Cell Signaling Technologies, Danvers, MA), p(S82)Hsp27 mAb (1:1,000 dilution, Cell Signaling), NOS2 mAb (1:1,000 dilution, BD Biosciences, San Jose, CA), HSF-1 pAb (1:1,000 dilution, Cell Signaling) or αB-crystallin (1:1,000 dilution, Enzo Life Sciences, Farmingdale, NY). Secondary antibodies were HRP-conjugated goat anti-mouse (1:5,000 dilution) or goat anti-rabbit antibody (1:5,000 dilution). Western blots were developed using an Enhanced Chemiluminescence Detection Kit (Thermo Scientific). The membrane was re-probed for the loading control with an antibody for GAPDH (1:1,000 dilution, Millipore, Billerica, MA), histone H3 (1:1,000 dilution, Cell Signaling Technologies) or β-actin (1:1,000 dilution, Cell Signaling Technologies).
2.3. RT-PCR analysis of Hsp27 and HSF-1
RNA was extracted from cells using the RNeasy mini kit and RT-PCR was carried out using the One step RT-PCR Kit (Qiagen). The following forward and reverse primers were used: Hsp27-FP-5′-ATG ACC GAG CGC CGC GTC CCC TTC TC-3′, RP-5′-TTA CTT GGC GGC AGT CTC ATC GGA TT-3′, HSF-1- FP-5′-ACA GCA TCA GGG GCA TA-3′, RP-5′-ATG GCC AGC TTC GTG CG -3′ and β-actin - FP-5′-CAG CTC ACC ATG GAT GAT GAT-3′ RP-5′-CTC GCC CGT GGT GGT GAA GCT-3′.
PCR products were visualized by electrophoresis in ethidium bromide-stained 1% Agarose gels with the β-actin amplification as the loading control.
2.4. Measurement of ROS and nitrite levels
ROS were measured as described previously using 5-(and-6)-chloromethyl-2′,7′-dichlorodihydro-fluorescein diacetate (CM-H2DCFDA) [29]. Nitrite levels were measured in media using Measure-IT High-Sensitivity Nitrite Assay Kit (Invitrogen, Grand Island, NY) according to the manufacturer’s instructions. Samples were read in a Spectramax Gemini XPS spectrofluorometer (Molecular Devices, Sunnyvale, CA) at excitation/emission wavelengths of 365/450 nm.
2.5. Measurement of IDO activity
IDO activity was measured by HPLC, as described previously [29]. Briefly, 50 μg of lysate was added to the reaction mixture (50 mM sodium phosphate buffer pH 6.5, 20 mM ascorbic acid sodium salt, 200 μg/mL bovine pancreatic catalase, 10 μM methylene blue and 400 μM L-tryptophan) and incubated at 37°C for 1 h. After stopping the reaction with 30%TCA, the samples were incubated at 65°C for 15 min to convert N-formyl kynurenine to kynurenine. The reaction mixture was analyzed by HPLC for the product, kynurenine.
2.6. Measurement of Kynurenines
Kynurenines were measured by HPLC, as described previously [29]. For these measurements 50 μg protein from HREC lysate was used.
2.7. Electrophoretic mobility shift assay
Electrophoretic mobility shift assay for HSF-1 was carried out using the Light Shift Chemiluminescent EMSA Kit (Thermo Scientific) according to the manufacturer’s instructions. HREC were treated with CM ± HG or peroxynitrite, as above. Cytosolic and nuclear extracts were separated using NE-PER nuclear and cytosolic extraction reagents (Thermo Scientific). Nuclear extract corresponding to 2.5 μg protein was used for the assay. The following biotinylated probe and its complementary sequence were used for the assay:
5′-AGC CGA CCT TAT AAG GGC TGG ACC GTC GGC T-3′
2.8. Cell treatment with Hsp27 siRNA
HRECs were treated with control or Hsp27 siRNA (Cell Signaling Technologies) according to the manufacturer’s instructions. Transfection was carried out with the Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, Grand Island, NY). Transfection was confirmed after 24 h with fluorescein-conjugated control siRNA by florescence microscopy (data not shown). Apoptosis was measured as below. Western blotting was carried out to confirm the downregulation of Hsp27 in HREC.
2.9. Overexpression of Hsp27 in HREC
Human Hsp27 was cloned into pcDNA 3.1 vector as previously described [30] Plasmid DNA (2.5 μg) was transfected into HREC (1 × 105 cells) using Superfect Transfection Reagent following the manufacturer’s protocol (Qiagen, Valencia, CA). Hsp27 overexpression was confirmed by Western blotting. Transfected cells were treated with CM1 and CM2 ± HG for 48 h as above. Apoptotic cells were quantified after staining with Hoechst reagent (see below).
2.10. Measurement of apoptosis
After the treatments described above, cells were washed with PBS, fixed in 4% paraformaldehyde and permeabilized with 80% methanol, and apoptotic cells were detected by their fragmented chromatin after staining with Hoechst 33258 reagent and counted using a fluorescent microscope. Caspase-3 activity in cell lysates (= 10 μg protein) was determined as previously described [29].
2.11. Statistical analysis
The results are given as means ± SD. Statistical significance was determined by two-tailed t-test. Values of p < 0.05 were considered to indicate statistical significance.
3. Results
3.1. Effects of individual cytokines on Hsp27 in HREC
To determine whether individual pro-inflammatory cytokines affected Hsp27 levels in HREC, cells were treated with TNF-α, IL-1β or IFN-γ for 48 h. None of the cytokines, at the two concentrations tested, had any effect on the level of Hsp27 (Fig. 1).
Figure 1. Individual cytokines do not affect Hsp27 levels in HREC.

HREC cultures were incubated with 10 and 20 ng/mL TNF-α (A), 10 and 20 ng/mL IL-1β (B), and 50 and 100 units/mL of IFN-γ (C) for 48 h. Hsp27 was measured by Western blotting and densitometry. The bars represent means ± SD of three independent experiments. GAPDH was used as the loading control. Cont =Control.
3.2. CM and high glucose (HG) downregulate Hsp27 and its phosphorylation (ser82) in HREC
Because all three cytokines are simultaneously elevated in the diabetic retina, we tested whether a combination of cytokines would have an effect on the expression and phosphorylation of Hsp27. The cells were treated with two concentrations of cytokine mixtures (CM1 and CM2). CM1 reduced the Hsp27 levels by 16% (relative to untreated controls), which was further reduced significantly by CM2 (p < 0.005, Fig. 2A). We also tested a mixture of cytokines in which IFN-γ was 50 U/ml and the other two cytokines were each 20 ng/ml (CM3). This combination of the cytokines also resulted in a significant reduction (p < 0.0005) in Hsp27 levels (Supplemental Figure 1). To determine whether high concentrations of glucose (to mimic diabetes conditions) would influence the cytokine-mediated downregulation of Hsp27, cells were treated with 25 mM of D-glucose (referred to as high glucose or HG) along with cytokines. HG alone showed a slight but statistically insignificant reduction in Hsp27. However, in the presence of CM, there was a significant steep drop in Hsp27 level (p < 0.005; Fig. 2B). We then determined whether the downregulation of Hsp27 occurred at the transcription level. RT-PCR analysis showed that CM reduced the Hsp27 mRNA levels (Fig. 2C). The effect was exacerbated when cytokines were co-administered with HG (p < 0.0005; Fig. 2D). The downregulation of Hsp27 was accompanied by reduced phosphorylation at S82 (pS82) of Hsp27 (Fig. 2E). HG alone also reduced Hsp27 phosphorylation. Together, these data suggest that under diabetic conditions, the combined actions of HG and cytokines could markedly deplete Hsp27 and its phosphorylation in HREC, consistent with our previous data of reduction of the rodent homolog, Hsp25 in the STZ-mouse model [31]. We also tested whether cytokines ± HG treatments as above altered the αB-crystallin levels in HREC. Unlike Hsp27, αB-crystallin levels were unaltered by CM ± HG treatments (Supplemental Figure 2).
Figure 2. CM and HG downregulate Hsp27 in HREC.

HREC cultures were incubated with cytokine mixtures, CM1=10 ng/mL TNF-α, 10 ng/mL IL-1β and 50 units/mL IFN-γ or CM2 = 20 ng/mL TNF-α, 20 ng/mL IL-1β and 100 units/mL of IFN-γ for 48 h. Hsp27 was measured by Western blotting and densitometry. CM downregulated Hsp27 protein (A) and mRNA (C) and in the presence of high glucose (HG, 25 mM glucose) further downregulated both Hsp27 protein (B) and mRNA levels (D). HG alone showed a slightly lower but statistically insignificant reduction. Hsp27 phosphorylation at S82, assessed by Western blotting was also reduced by CM ± HG (E). In all figures, representative Western blots/gels are shown at the top left. β-actin was used as the loading/internal control. The bars represent means ± SD of three independent experiments, but in the case of pHsp27 they represent mean values of two independent experiments. * p < 0.05, ** p < 0.005 and *** p < 0.0005, versus control. Cont =Control.
3.3. CM does not affect HSF-1 in HREC
HSF-1 is a transcription factor that regulates the expression of Hsp27. The cytokines with or without HG did not significantly affect HSF-1 mRNA (Fig. 3A and B) or protein levels in HREC (Fig. 3C). We then investigated whether CM and HG inhibited nuclear translocation of HSF-1, which is necessary for the transcription of Hsp27. We measured the HSF-1 content in the cytosol and nuclear fractions of HREC by Western blotting. This analysis showed no difference in HSF-1 content in the cytosolic or nuclear fractions (Fig. 3D, E). We then tested whether HSF-1 binding to DNA was negatively affected by CM using an electrophoretic mobility shift assay, which showed that DNA binding of HSF-1 was unaffected by the CM in the presence or absence of HG (Fig. 3F). Together, these results indicate that the CM-mediated Hsp27 downregulation was not due to reduced expression, reduced nuclear translocation, or decreased DNA binding of HSF-1.
Figure 3. CM does not downregulate or functionally impair HSF-1.

Cells were cultured and treated with cytokines as in Figure 2. HSF-1 mRNA levels were unaffected by CM (A) or CM and HG (B). Similarly, HSF-1 protein levels were unaltered (C). Cells were fractionated into cytosolic and nuclear fractions and Western blotted for HSF-1. HSF-1 levels in the cytosolic (D) and nuclear (E) fractions were unchanged by CM and HG treatments. In all figures, representative Western blots/gels are shown at the top left. β-actin for cytosolic and histone for nuclear proteins were used as the loading/internal control. The DNA binding of HSF-1 was assessed by an EMSA (F). Lane 1, control, lane 2, treated with CM1, lane 3, treated with CM2, lane 4, treated with HG, lane 5, treated with CM1 + HG and lane 6, treated with CM2 + HG. The DNA binding of HSF-1 was similar in all treatments. Arrow indicates DNA-bound HSF-1. FP=free probe.
3.4. CM upregulates indoleamine 2,3-dioxygenase (IDO) and increases the ROS content in HREC
IDO is a cytosolic protein that catalyzes the degradation of tryptophan, and has been shown to be upregulated by inflammatory cytokines. The CM upregulated IDO activity in HREC (Fig. 4A). HG enhanced the effect of CM. Such activation led to the formation of kynurenine (Fig. 4B), which is one of the cytotoxic oxidation products of tryptophan. Whether the IDO-mediated kynurenine pathway was responsible for the downregulation of Hsp27 by the CM was tested using MT, an inhibitor of IDO. MT treatment inhibited kynurenine production (Fig. 4B), and inhibited the CM-mediated Hsp27 downregulation (Fig. 4C); this occurred even in the presence of HG (Fig. 4D). Since kynurenines spontaneously generate ROS, we tested whether the IDO-mediated kynurenine pathway was responsible for ROS production in HREC. HG alone increased ROS levels, an effect that was exacerbated by the addition of cytokines (Fig. 4E). This increase in ROS was inhibited by the addition of the IDO inhibitor, MT. We then tested whether kynurenine alone (in the absence of cytokines) was responsible for the downregulation of Hsp27. Our results showed that kynurenine alone had no effect on the Hsp27 level (Fig. 4F). Together, these data demonstrate that cytokine-mediated induction of IDO in HREC leads to the formation of kynurenines, followed by ROS production during downregulation of Hsp27, but ROS alone is not responsible for such downregulation.
Figure 4. Downregulation of Hsp27 by CM is suppressed by an inhibitor of indoleamine 2,3-dioxygenase.

Cultures of HREC were treated with CM, as in Figure 2, along with 20 μM MT. IDO activity (A) and kynurenines in cells measured by HPLC (B) were higher in the presence of CM and increased further in the presence of HG, but inhibited by MT (open bars). Hsp27 levels, determined by Western blotting, were restored to control levels by MT treatment in cells treated with CM (C) and in cells co-treated with HG (D). CM generated ROS in HREC, which was increased by HG (E). The ROS production (measured as fluorescent derivatives) by CM was largely inhibited by MT (open bars). In E, control values (cells alone or cells + MT) are subtracted from the values shown. Exogenous kynurenines (100 μM, Kyn) had no effect on Hsp27 levels (F). β-actin was used as the loading control in Western blotting. The bars represent means ± SD of three independent experiments. Representative Western blots are shown at the top of each figure. * p < 0.05 and *** p < 0.0005 relative to control in A, and between MT treated and untreated in B and E.
3.5. CM upregulates NOS2 and increases nitric oxide in HREC
Treatment of HREC with the CM resulted in the upregulation of NOS2 (Fig. 5A). While CM1 increased NOS2 levels by ~14% over controls, CM2 increased NOS2 levels by 21% (p < 0.05). The increase in NOS2 levels resulted in higher levels of NO, as indicated by the higher nitrite levels in the culture medium (Fig. 5B). Whether the increased production of NO was responsible for the downregulation of Hsp27 was then tested. The addition of an NO donor, DETA NONOate (in the absence of cytokines), to the culture medium increased NO levels significantly (Fig. 5C, p <0.0005), but this increase in NO (in the absence of CM) had no effect on Hsp27 levels (Fig. 5D). However, the addition of the NOS inhibitor, L-NAME along with CM and HG prevented Hsp27 downregulation (Fig. 5E, and F). These data suggest that CM increase NO production in HREC during downregulation of Hsp27, but NO alone is not responsible for such downregulation.
Figure 5. CM upregulates NOS2 and increases NO production in HREC.

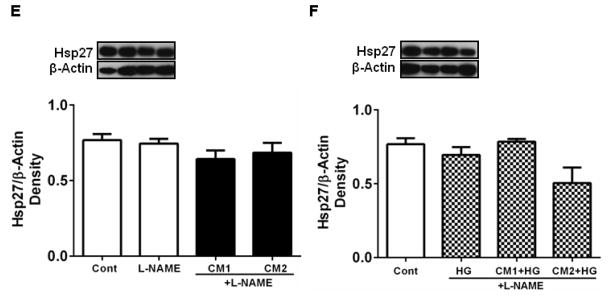
HREC were treated with CM as in Fig. 2. Treatment with CM upregulated NOS2 (A) and consequently NO (nitric oxide) in the culture medium (B). However, treatment with the NO donor, DETA NONOate increased NO levels (C) but had no effect on Hsp27 (D). Hsp27 levels in HREC treated with CM + L-NAME, CM + HG + L-NAME are shown in E and F, respectively, and are similar, though L-NAME treatment blocked the downregulation of Hsp27 in CM2 + HG samples. Representative Western blots are shown at the top of each figure. β-actin was used as the loading control in Western blotting. Data are means ± SD of three independent experiments.
3.6. Peroxynitrite downregulates Hsp27 in HREC
From the above findings, it is clear that neither ROS nor NO alone is responsible for the cytokine-mediated downregulation of Hsp27 in HREC. We hypothesized that the downregulation of Hsp27 could be due to peroxynitrite radical (ONOO·) produced from the reaction of NO and superoxide. We tested this by adding a ONOO· donor, SIN-1, to the culture media. We found a slight but statistically insignificant drop in the Hsp27 levels by this treatment (Fig. 6A). However, direct treatment with peroxynitrite had a significant inhibitory effect on Hsp27 expression, which was time-dependent (p < 0.05, Fig. 6B). We then checked the possibility that ONOO· downregulated HSF-1 and thus reduced Hsp27 levels. Treatment with peroxynitrite had no effect on cytosolic (Fig. 6C) or nuclear levels (Fig. 6D) of Hsp27. We then tested whether ONOO· affected the binding of HSF-1 to DNA using an EMSA. The results in Fig. 6E showed that direct addition of peroxynitrite had no effect on the binding of HSF-1 to DNA.
Figure 6. Peroxynitrite downregulates Hsp27 in HREC.

HREC were treated with a peroxynitrite donor (SIN-1, 1 and 2 mM) for 3 h (A) or peroxynitrite (200 μM) for 20 min, followed by incubation for 0, 24 and 48 h (B). Peroxynitrite (200 μM) had no effect on HSF-1 level (determined by Western blotting) in the cytoplasm (C) or nucleus (D). EMSA (E) showed that peroxynitrite treatment did not alter the nuclear binding of HSF-1. Lane 1, control, lanes 2 and 3 are from cells treated with ONOO· for 20 min and incubated 24 and 48 h, respectively. Arrow indicates DNA-bound HSF-1. FP=free probe. Representative Western blots are shown at the top of figures. Data are means ± SD of three independent experiments. * p < 0.05
To further examine the role of ONOO·, we used L-NAME together with TEMPOL (to block NO production and ROS sequestration, respectively) in the culture medium. The cytokine-mediated reduction in Hsp27 level (seen in Fig. 2) was prevented by these two reagents (Fig. 7A). Addition of TEMPOL and L-NAME resulted in inhibition of ROS and NO in CM ± HG-treated cells (Fig. 7B, C, compare results in Fig. 4E and 5B). In fact, the treatment of L-NAME + TEMPOL resulted in ROS levels falling below the basal levels found in untreated (control) cells (7B). These results confirm the causal role of ONOO· in the Hsp27 downregulation by CM in HREC.
Figure 7. Inhibition of ROS and NO production normalizes Hsp27 levels CM-treated HREC.

To determine whether inhibition of NO and ROS products would restore Hsp27 to control levels in CM treated cells (in the presence or absence of HG), cells were treated with 500 μM of a NOS inhibitor, L-NAME and 100 μM of a ROS scavenger, TEMPOL for 48 h (A). Hsp27 was measured by Western blotting and the densitometric data are shown. Data are means ± SD of three independent experiments. ROS levels were inhibited significantly, and the levels were similar among CM with or without HG samples (B). Nitrite levels in the culture media are shown in C. L-NAME and TEMPOL inhibited NO production with CM. *** p < 0.0005 between (L-NAME + TEMPOL) and untreated cells.
3.7. Downregulation of Hsp27 leads to apoptosis of HREC
Hsp27 is an anti-apoptotic protein, and thus its downregulation by cytokines could lead to apoptosis of HREC. To test this, we measured apoptotic cells in the cytokine (± HG)- treated cells. Treatment with CM1 resulted in ~14% apoptotic cells (p < 0.05), which increased to ~24% with CM2 (Fig. 8A). While HG treatment alone showed ~16% apoptotic cells (7% more than controls, p < 0.005), the combination of LC and HG showed 2.5-fold more apoptotic cells than seen with LC alone (Fig. 8B, p < 0.0005). These treatments resulted in significant increases in caspase-3 activity (p < 0.0005, Fig. 8C). To further assess the role of Hsp27, we treated HREC with an siRNA for Hsp27. siRNA treatment resulted in a significant (p < 0.0005) ~90% downregulation of Hsp27 (Fig. 8D). Scrambled siRNA did not show a similar effect. This siRNA treatment resulted in a significant increase in apoptosis of HRECs (Fig. 8E, p < 0.005). The role of ROS and NO in cytokine-induced apoptosis of HREC was investigated further. The CM-induced apoptosis was largely prevented by the MT and L-NAME treatments (Fig. 8F, G). Furthermore, the addition of SIN-1 at 2 mM caused a significant increase in apoptosis of HRECs (p < 0.0005, Fig. 8H). Together, these data suggest that downregulation of Hsp27 by ONOO· results in apoptosis of HRECs.
Figure 8. CM induces apoptosis of HREC through downregulation of Hsp27.


HREC cells were treated with CM as in Figure 2. Apoptosis was measured using Hoechst reagent. CM (A) and in combination with HG (B) induced apoptosis of HREC. Caspase-3 activity was measured in cell lysates (= 10 μg protein) using a fluorogenic substrate, Ac-DEVD-AFC (C). To determine the specific role of Hsp27, cells were treated with an siRNA or a scrambled siRNA for Hsp27 for 48 h; targeted siRNA treatment reduced Hsp27 levels significantly, as determined by Western blotting (D) and induced apoptosis (E). Treatment with 20 μM MT for 48 h (F) or 500 μM L-NAME for 48 h (G) inhibited apoptosis in CM ± HG treated cells. Treatment of cells with a peroxynitrite donor, SIN-1, for 3 h also induced apoptosis in HREC (H). Data are means ± SD of three independent experiments. * p < 0.05, ** p < 0.005 and *** p < 0.0005, versus controls.
To test whether overexpression of Hsp27 offered protection against cytokine-induced apoptosis, we transfected HREC with human Hsp27. Transfected cells showed 15 and 20% increase in Hsp27 after 24 and 48 h (Fig. 9A). After 24 h of transfection, cells were treated with CM1 and CM2 ± HG for 48 h. Unlike non-transfected cells that showed 13 and 22% apoptotic cells with CM1 and CM2 treatments (Fig. 8), Hsp27 overexpressing cells showed only 8 and 9% apoptotic cells (Fig. 9B). Hsp27 overexpression also mitigated the effect of cytokines + HG. These results further support the notion that a reduction in Hsp27 is central to cytokine-mediated apoptosis of HREC.
Figure 9. Hsp27 overexpression reduces CM-induced apoptosis in HREC.
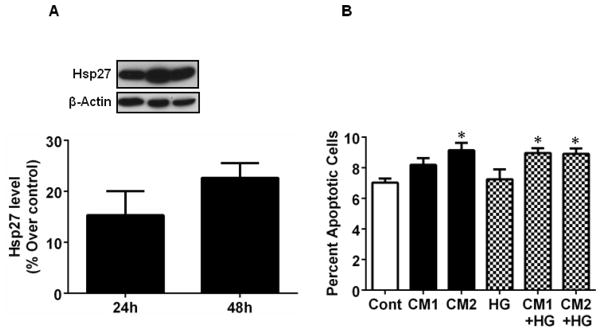
HREC were transfected with Hsp27 using Superfect Transfection Reagent. Overexpression of Hsp27 after 24 and 48 h of transfection was confirmed by Western blotting (A). 24 h post-transfection cells were treated with CM1 and CM2 and HG for 48 h as in Figure 2. Apoptotic cells were counted after staining with Hoechst reagent. Data are means ± SD of three independent experiments. * p < 0.05.
4. Discussion
Our initial experiments showed that HREC contained several small heat shock proteins, including Hsp27 (data not shown). We expected Hsp27 to be upregulated by the cytokine and HG treatments because they can impose significant cellular stress. However, we found that a combination of pro-inflammatory cytokines downregulated Hsp27 and HG exacerbated such an effect. Interestingly, individual cytokines, when tested separately, did not elicit a similar response. That the response occurred only when all three cytokines (TNF-α, IL-1β, IFN-γ) were present simultaneously suggests a synergistic cooperative mechanism in the Hsp27 downregulation. Similar synergy has been observed during cytokine-mediated apoptosis of corneal endothelial cells [32], although the mechanism(s) of such synergistic actions remain unknown. Additionally, our data showed that S82 phosphorylation in Hsp27 was reduced by treatment of HREC with CM and HG. S82 is a major site for phosphorylation in Hsp27, mediated by p38MAPK/MAPKAK-2. Phosphorylation of Hsp27 changes its tertiary structure from a polymeric to predominantly dimeric [33]. Phosphorylation is required for the anti-apoptotic function of Hsp27 [reviewed in [34]]. Whether the downregulation of S82 phosphorylation is due to adverse effects of cytokines on the upstream kinases need to be determined. That CM neither reduced the total or nuclear HSF-1 levels nor its binding to DNA points to other mechanisms in Hsp27 downregulation (see below).
Our data showed that the CM induced IDO in HREC and that this induction resulted in the activation of the kynurenine pathway and, consequently, the formation of ROS. Previous studies have also shown that kynurenines can produce ROS in cells [29, 35]. The ability of MT to inhibit ROS production and simultaneously block cytokine-mediated downregulation of Hsp27 points to an important role for ROS. However, addition of kynurenine to culture media, which can increase intracellular ROS [35], failed to downregulate Hsp27, indicating that ROS produced by CM treatment is necessary but insufficient to downregulate Hsp27.
Our data showed that CM upregulated NOS2 and increased NO levels in HREC. To test whether NO was involved in Hsp27 downregulation, we tested the effect of a NO donor on HREC. Although it did result in high NO levels, this treatment failed to downregulate Hsp27, suggesting the possibility that ONOO·, formed from the reaction of NO with superoxide, is responsible for the downregulation of Hsp27. In fact, addition of ONOO· to the culture medium decreased Hsp27 levels, similar to CM, confirming that ONOO· downregulated Hsp27. ONOO· has been linked to inflammation-mediated damage in cells and tissues and apoptosis [36]. However, the mechanism of such apoptosis is not clear. Protein nitration, which has been shown to be increased in the diabetic retina, is a consequence of increased ONOO [37]. Whether protein nitration contributes to the cytokine-mediated Hsp27 downregulation needs to be investigated further.
The CM brought about apoptosis of HREC, and Hsp27 downregulation appeared to be a key mechanism in this apoptosis. The requirement of Hsp27 for HREC viability was further reiterated by our data, showing that depletion of Hsp27 by siRNA lead to apoptosis in HREC. Previous studies have shown that hyperglycemia-mediated inflammatory cytokines cause apoptosis in retinal capillary cells [38, 39], although the mechanisms are still unclear. The present study provides a mechanistic link between inflammatory cytokines and apoptosis, through ONOO· formation and downregulation of Hsp27. Hsp27 inhibits apoptosis by several mechanisms, including binding to cytochrome c, activating Akt and blocking caspase-3 activation [40]. Whether a decrease in these functions as a consequence of a reduction in Hsp27 activity leads to apoptosis of HREC remains to be shown. Moreover, whether Hsp27 is downregulated in endothelial cells of diabetic retina is unknown. There is evidence for upregulation of heat shock proteins in general and the specific upregulation of Hsp27 in the diabetic retina [31, 41, 42]. It is possible that there may be an overall increase in these proteins in diabetic retina, but at the same time locally within capillary endothelial cells, there may be a decrease. Further studies are needed to examine this. A schematic representation of the possible mechanisms by which CM may bring about apoptosis of HREC is shown in Figure 10.
Figure 10. A conceptual view of the mechanisms by which CM downregulates Hsp27 in HREC and bring about apoptosis in diabetes.

Inflammatory cytokines are upregulated in the diabetic eye. Inflammatory cytokines activate NOS2 and IDO to generate NO and ROS, respectively, which react to form ONOO·. The ONOO·, by an unknown mechanism, downregulates Hsp27 and brings about apoptosis of HREC. IFGNR=IFN-γ receptor1, ILR=IL-1β receptor, and TLR, TNF-α receptor.
In summary, we showed that Hsp27 is required for HREC viability and its decrease through an upregulation of inflammatory cytokines and ONOO· formation could be a mechanism in acellular capillary formation in the early phase of diabetic retinopathy, reinforcing the idea that anti-inflammatory agents could be of therapeutic benefit in diabetic retinopathy.
Supplementary Material
Highlights.
Pro-inflammatory cytokines downregulate Hsp27 in human retinal endothelial cells.
High glucose exacerbates the Hsp27 downregulation by pro-inflammatory cytokines.
Pro-inflammatory cytokines upregulate IDO and NOS2.
Peroxynitrite mediates the downregulation of Hsp27.
Downregulation of Hsp27 results in apoptosis of human retinal endothelial cells
Acknowledgments
We thank Dawn Smith in the Visual Sciences Research Center, CWRU for her help with the isolation and culture of human retinal endothelial cells. This research was supported by the NIH grants EYP30-11373 (to CWRU) and EYR01-020895 (PEF), The International Retinal Research Foundation, AL (RHN), W. R. Bryan Diabetic Eye Disease Research Fund from the Ohio Lions Eye Research Foundation (RHN) and Research to Prevent Blindness, NY (CWRU).
Abbreviations
- HREC
human retinal endothelial cells
- IDO
indoleamine 2,3-dioxygenase
- MT
1-methyl-DL-tryptophan
- NOS
nitric oxide synthase
- ROS
reactive oxygen species
- TNF-α
tumor necrosis factor-α
- IL-1β
interleukin-1β
- IFN-γ
interferon-γ
- VCAM-1
vascular cell adhesion molecule-1
- ICAM-1
intracellular adhesion molecule-1
- MCP-1
monocyte chemotactic protein-1
- COX2
cyclooxygenase-2
- L-NAME
Nω-nitro-L-arginine methyl ester hydrochloride
- TEMPOL
1-oxyl-2,2,6,6-tetramethyl-4-hydroxypiperidine
- HG
high glucose
- CM
cytokine mixture
- CM-H2DCFDA
5-(and-6)-chloromethyl-2′,7′-dichlorodihydro-fluorescein diacetate
- Ac-DEVD-AFC
N-Acetyl-Asp-Glu-Val-Asp-7-amido-4-trifluoromethylcoumarin
- DETA NONOate
(Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate
- NO
nitric oxide
- ONOO·
peroxynitrite
- HSF-1
heat shock factor-1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.I.D. Federation. IDF Diabetes Atlas: The Global Burden. 2011 http://www.idf.org/diabetesatlas/5e/the-global-burden.
- 2.WHO. Prevention of blindness from diabetes mellitus. 2005 http://www.who.int/blindness/PreventionofBlindnessfromDiabetesMellitus-with-cover-small.pdf.
- 3.Bronson-Castain KW, Bearse MA, Jr, Neuville J, Jonasdottir S, King-Hooper B, Barez S, Schneck ME, Adams AJ. Early neural and vascular changes in the adolescent type 1 and type 2 diabetic retina. Retina. 2012;32:92–102. doi: 10.1097/IAE.0b013e318219deac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kern TS, Barber AJ. Retinal ganglion cells in diabetes. J Physiol. 2008;586:4401–4408. doi: 10.1113/jphysiol.2008.156695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammes HP, Porta M, editors. Frontiers in Diabetes. Vol. 20. S. Krager AG; Switzerland: 2010. Experimental Approaches to Diabetic Retinopathy; pp. 42–60. [Google Scholar]
- 6.Tang J, Kern TS. Inflammation in diabetic retinopathy. Prog Retin Eye Res. 2011;30:343–358. doi: 10.1016/j.preteyeres.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kowluru RA, Zhong Q, Kanwar M. Metabolic memory and diabetic retinopathy: role of inflammatory mediators in retinal pericytes. Exp Eye Res. 2010;90:617–623. doi: 10.1016/j.exer.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang W, Liu H, Al-Shabrawey M, Caldwell RW, Caldwell RB. Inflammation and diabetic retinal microvascular complications. J Cardiovasc Dis Res. 2011;2:96–103. doi: 10.4103/0975-3583.83035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnsen-Soriano S, Sancho-Tello M, Arnal E, Navea A, Cervera E, Bosch-Morell F, Miranda M, Javier Romero F. IL-2 and IFN-gamma in the retina of diabetic rats. Graefes Arch Clin Exp Ophthalmol. 2010;248:985–990. doi: 10.1007/s00417-009-1289-x. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, Biarnes Costa M, Gerhardinger C. IL-1beta is upregulated in the diabetic retina and retinal vessels: cell-specific effect of high glucose and IL-1beta autostimulation. PLoS One. 2012;7:e36949. doi: 10.1371/journal.pone.0036949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Behl Y, Krothapalli P, Desta T, DiPiazza A, Roy S, Graves DT. Diabetes-enhanced tumor necrosis factor-alpha production promotes apoptosis and the loss of retinal microvascular cells in type 1 and type 2 models of diabetic retinopathy. Am J Pathol. 2008;172:1411–1418. doi: 10.2353/ajpath.2008.071070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang H, Gandhi JK, Zhong X, Wei Y, Gong J, Duh EJ, Vinores SA. TNFalpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Invest Ophthalmol Vis Sci. 2011;52:1336–1344. doi: 10.1167/iovs.10-5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joussen AM, Poulaki V, Mitsiades N, Kirchhof B, Koizumi K, Dohmen S, Adamis AP. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. Faseb J. 2002;16:438–440. doi: 10.1096/fj.01-0707fje. [DOI] [PubMed] [Google Scholar]
- 15.Vincent JA, Mohr S. Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes. 2007;56:224–230. doi: 10.2337/db06-0427. [DOI] [PubMed] [Google Scholar]
- 16.Nagai N, Izumi-Nagai K, Oike Y, Koto T, Satofuka S, Ozawa Y, Yamashiro K, Inoue M, Tsubota K, Umezawa K, Ishida S. Suppression of diabetes-induced retinal inflammation by blocking the angiotensin II type 1 receptor or its downstream nuclear factor-kappaB pathway. Invest Ophthalmol Vis Sci. 2007;48:4342–4350. doi: 10.1167/iovs.06-1473. [DOI] [PubMed] [Google Scholar]
- 17.Du Y, Sarthy VP, Kern TS. Interaction between NO and COX pathways in retinal cells exposed to elevated glucose and retina of diabetic rats. Am J Physiol Regul Integr Comp Physiol. 2004;287:R735–741. doi: 10.1152/ajpregu.00080.2003. [DOI] [PubMed] [Google Scholar]
- 18.Leal EC, Manivannan A, Hosoya K, Terasaki T, Cunha-Vaz J, Ambrosio AF, Forrester JV. Inducible nitric oxide synthase isoform is a key mediator of leukostasis and blood-retinal barrier breakdown in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2007;48:5257–5265. doi: 10.1167/iovs.07-0112. [DOI] [PubMed] [Google Scholar]
- 19.Li J, Wang JJ, Yu Q, Wang M, Zhang SX. Endoplasmic reticulum stress is implicated in retinal inflammation and diabetic retinopathy. FEBS Lett. 2009;583:1521–1527. doi: 10.1016/j.febslet.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagaraj RH, Mailankot M. High glucose sensitizes human retinal endothelial cells for IFN-g-mediated apoptosis. Acta Ophthalmol. 2010;88 Abstract. [Google Scholar]
- 21.Sun Y, MacRae TH. Small heat shock proteins: molecular structure and chaperone function. Cell Mol Life Sci. 2005;62:2460–2476. doi: 10.1007/s00018-005-5190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fort PE, Freeman WM, Losiewicz MK, Singh RS, Gardner TW. The retinal proteome in experimental diabetic retinopathy: up-regulation of crystallins and reversal by systemic and periocular insulin. Mol Cell Proteomics. 2009;8:767–779. doi: 10.1074/mcp.M800326-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kannan R, Sreekumar PG, Hinton DR. Novel roles for alpha-crystallins in retinal function and disease. Prog Retin Eye Res. 2012;31:576–604. doi: 10.1016/j.preteyeres.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strunnikova N, Baffi J, Gonzalez A, Silk W, Cousins SW, Csaky KG. Regulated heat shock protein 27 expression in human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2001;42:2130–2138. [PubMed] [Google Scholar]
- 25.Gangalum RK, Atanasov IC, Zhou ZH, Bhat SP. AlphaB-crystallin is found in detergent-resistant membrane microdomains and is secreted via exosomes from human retinal pigment epithelial cells. J Biol Chem. 2011;286:3261–3269. doi: 10.1074/jbc.M110.160135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Straume O, Shimamura T, Lampa MJ, Carretero J, Oyan AM, Jia D, Borgman CL, Soucheray M, Downing SR, Short SM, Kang SY, Wang S, Chen L, Collett K, Bachmann I, Wong KK, Shapiro GI, Kalland KH, Folkman J, Watnick RS, Akslen LA, Naumov GN. Suppression of heat shock protein 27 induces long-term dormancy in human breast cancer. Proc Natl Acad Sci U S A. 2012;109:8699–8704. doi: 10.1073/pnas.1017909109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dimberg A, Rylova S, Dieterich LC, Olsson AK, Schiller P, Wikner C, Bohman S, Botling J, Lukinius A, Wawrousek EF, Claesson-Welsh L. alphaB-crystallin promotes tumor angiogenesis by increasing vascular survival during tube morphogenesis. Blood. 2008;111:2015–2023. doi: 10.1182/blood-2007-04-087841. [DOI] [PubMed] [Google Scholar]
- 28.Busik JV, Mohr S, Grant MB. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes. 2008;57:1952–1965. doi: 10.2337/db07-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mailankot M, Nagaraj RH. Induction of indoleamine 2,3-dioxygenase by interferon-gamma in human lens epithelial cells: apoptosis through the formation of 3-hydroxykynurenine. Int J Biochem Cell Biol. 2010;42:1446–1454. doi: 10.1016/j.biocel.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pasupuleti N, Gangadhariah M, Padmanabha S, Santhoshkumar P, Nagaraj RH. The role of the cysteine residue in the chaperone and anti-apoptotic functions of human Hsp27. J Cell Biochem. 2010;110:408–419. doi: 10.1002/jcb.22552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heise EA, Fort PE. Impact of diabetes on alpha-crystallins and other heat shock proteins in the eye. J Ocul Biol Dis Infor. 2011;4:62–69. doi: 10.1007/s12177-011-9073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sagoo P, Chan G, Larkin DF, George AJ. Inflammatory cytokines induce apoptosis of corneal endothelium through nitric oxide. Invest Ophthalmol Vis Sci. 2004;45:3964–3973. doi: 10.1167/iovs.04-0439. [DOI] [PubMed] [Google Scholar]
- 33.Rogalla T, Ehrnsperger M, Preville X, Kotlyarov A, Lutsch G, Ducasse C, Paul C, Wieske M, Arrigo AP, Buchner J, Gaestel M. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. J Biol Chem. 1999;274:18947–18956. doi: 10.1074/jbc.274.27.18947. [DOI] [PubMed] [Google Scholar]
- 34.Vidyasagar A, Wilson NA, Djamali A. Heat shock protein 27 (HSP27): biomarker of disease and therapeutic target. Fibrogenesis Tissue Repair. 2012;5:7. doi: 10.1186/1755-1536-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song H, Park H, Kim YS, Kim KD, Lee HK, Cho DH, Yang JW, Hur DY. L-kynurenine-induced apoptosis in human NK cells is mediated by reactive oxygen species. Int Immunopharmacol. 2011;11:932–938. doi: 10.1016/j.intimp.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 36.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 37.Zhan X, Du Y, Crabb JS, Gu X, Kern TS, Crabb JW. Targets of tyrosine nitration in diabetic rat retina. Mol Cell Proteomics. 2008;7:864–874. doi: 10.1074/mcp.M700417-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Joussen AM, Doehmen S, Le ML, Koizumi K, Radetzky S, Krohne TU, Poulaki V, Semkova I, Kociok N. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol Vis. 2009;15:1418–1428. [PMC free article] [PubMed] [Google Scholar]
- 39.Kowluru RA, Odenbach S. Role of interleukin-1beta in the pathogenesis of diabetic retinopathy. Br J Ophthalmol. 2004;88:1343–1347. doi: 10.1136/bjo.2003.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arrigo AP. The cellular “networking” of mammalian Hsp27 and its functions in the control of protein folding, redox state and apoptosis. Adv Exp Med Biol. 2007;594:14–26. doi: 10.1007/978-0-387-39975-1_2. [DOI] [PubMed] [Google Scholar]
- 41.Du Y, Tang J, Li G, Berti-Mattera L, Lee CA, Bartkowski D, Gale D, Monahan J, Niesman MR, Alton G, Kern TS. Effects of p38 MAPK inhibition on early stages of diabetic retinopathy and sensory nerve function. Invest Ophthalmol Vis Sci. 2010;51:2158–2164. doi: 10.1167/iovs.09-3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pinach S, Burt D, Berrone E, Barutta F, Bruno G, Porta M, Perin PC, Gruden G. Retinal heat shock protein 25 in early experimental diabetes. Acta Diabetol. 2013;50:579–585. doi: 10.1007/s00592-011-0346-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.