

Abstract
Nedd4 family interacting protein 1 (Ndfip1) is an adaptor protein that regulates Itch, an E3 ubiquitin ligase that ubiquitylates JunB, thereby preventing interleukin (IL)-4 and IL-5 production. Mice lacking Ndfip1 or Itch develop T helper type 2 (TH2)-mediated inflammation in the skin and lungs and die prematurely. In this study we show that Ndfip1 − / − mice also develop inflammation in the gastrointestinal tract. Inflammation is characterized by infiltration of eosinophils and T cells and is accompanied by a failure to gain weight. T cells are both necessary and sufficient for eosinophil recruitment and inflammation. This is because Ndfip1 − / − T cells become activated and produce IL-5. Itch mutant mice develop much less severe gastrointestinal inflammation, suggesting that Ndfip1 regulation of Itch cannot entirely account for this phenotype and that Ndfip1 has both Itch-dependent and Itch-independent roles. Ndfip1 may also regulate human disease. We found single-nucleotide polymorphisms in the Ndfip1 locus that associate with inflammatory bowel disease. Taken together, our data support a role for Ndfip1 in gastrointestinal inflammation in both mice and humans.
INTRODUCTION
T-cell receptor stimulation, in the presence of costimulatory receptor signaling, leads to proliferation, effector differentiation, and adaptive immunity. These pathways are tightly regulated to prevent the deleterious effects of excessive immune activation and inflammation, and are often found to be defective in autoimmune and allergy-mediated diseases. There is increasing evidence that ubiquitin-mediated protein degradation has a role in suppressing the immune response through the targeted destruction of signaling proteins and proinflammatory transcription factors.1 E3 ubiquitin ligases bring specificity to this degradation process by selecting proteins for ubiquitin-mediated destruction. 2 Supporting this, several E3 ubiquitin ligases have been shown to regulate T-cell activation, most notably Itch, Roquin, and Cbl-b.3–6 In the absence of these E3 ligases, mechanisms of immunological tolerance fail, and mice lacking some of these proteins develop overt inflammation and/or auto-immune-like symptoms.7
Nedd4 family interacting protein 1(Ndfip1) was originally identified because of its ability to bind the WW domains of Nedd4, the prototypic member of the Nedd4 family of E3 ubiquitin ligases.8 In vitro , Ndfip1 was shown to bind most of the E3 ligases in this family;8–11 however, its role as an adaptor protein was only recently revealed. In T cells, we showed that Ndfip1 promotes the function of Itch. 12 Mice that are deficient in Ndfip1 develop inflammation in the skin and lungs and die prematurely. Inflammation in these mice is characterized by T helper type 2 (TH2)-polarized T cells and high levels of circulating IgE,12 the hallmarks of atopy. The TH2 bias of Ndfip1−/− T cells can be explained by the role of Ndfip1 in the regulation of Itch. Itch ubiquitylates and causes the destruction of JunB,13 a transcription factor that promotes the expression of the TH2 cytokines interleukin (IL)-4 and IL-5. In the absence of Ndfip1, Itch is unable to initiate the destruction of JunB.12
The extent to which the inflammation in Ndfip1−/− mice is initiated by defects in T cells vs. cells of the innate immune system is not known. It is also not known why the inflammation in mice lacking Ndfip1 preferentially occurs in the skin and lung, the known sites of environmental antigen exposure. One possibility is that the immune system of these mice responds to environmental antigens as though they are pathogenic. If this was the case, one might also expect TH2-mediated inflammation to be evident within the gastrointestinal (GI) tract, the major site of environmental antigen encounter.
In this report, we show that mice that lack Ndfip1 develop GI inflammation at a very young age. GI inflammation is characterized by an influx of high numbers of T cells and eosinophils. GI inflammation is dependent on the presence of T cells. Furthermore, Ndfip1−/− T cells are sufficient to drive disease in the GI tract. This is because Ndfip1−/− T cells become activated in vivo and produce high levels of IL-5. Importantly, a less severe GI phenotype is seen in Itch mutant mice. This is because Ndfip1 has both Itch-dependent and Itch-independent roles. This may have relevance for human disease as we provide evidence that polymorphisms in Ndfip1 are associated with the development of inflammatory bowel disease (IBD). Taken together, our data suggest that Ndfip1 regulates multiple E3 ubiquitin ligases to prevent T cell-mediated GI inflammation in both mice and humans.
RESULTS
Ndfip1-deficient mice develop inflammation along the GI tract
The skin and lung inflammation in Ndfip1−/− mice occurs in the absence of known pathogen exposure, suggesting that immune activation may result from inappropriate immune responses to environmental antigens. The major site of environmental antigen exposure is the GI tract. Thus, we tested whether Ndfip1−/− mice show evidence of inflammation in the GI tract. On gross inspection of the different regions of the GI tract, we found that the small bowel was thicker than that of wild-type (WT) mice (Figure 1a). Histological analysis of Ndfip1−/− mice showed an infiltration of inflammatory cells in the esophagus, small bowel, and colon (Figure 1b ). In the small bowel and colon, the infiltrating cells were characteristic of eosinophils, based on their granularity and red pigment when stained with eosin. These cells were evident in both the lamina propria and between the cells of the GI epithelium. In Figure 1, the ileum is shown; however, a similar level of eosinophil infiltration was seen along the entire small bowel. Analysis of the esophagus showed severe epithelial thickening, with an increase in the number of eosinophils as well as in cells that resembled lymphocytes. Eosinophils were also evident in the stomach (Supplementary Figure S1 online) and cecum (data not shown).
Figure 1.

Ndfip1 − / − mice develop gastrointestinal (GI) inflammation. (a) Gross anatomy of the small bowel of Ndfip1 − / − vs. Ndfip1 + / + littermate controls. (b) Histological analysis of the esophagus (×20), small bowel (×40), and colon (×40) of Ndfip1 − / − vs. Ndfip1 + / + littermate controls. (c) Weights of Ndfip1 − / − or Ndfip1 + / + littermates at different ages as indicated. Each point is the average weight of 3–5 different mice; *P < 0.01. (d) Flow cytometric analysis of cells from different sections of the GI tract. Cells from these organs were analyzed for their expression of CD4 + (T cells) and Siglec-F + (eosinophils). The data in a, b, and d are representative of > 5 independent experiments. (e) The percentages of eosinophils observed in d were compiled and analyzed. *P < 0.05, **P < 0.005. Each dot represents an individual mouse. Mice were analyzed at 6–12 weeks of age.
Inflammation of the GI tract in Ndfip1−/− mice is accompanied by a failure to gain weight. Mice lacking Ndfip1 do not gain as much weight as their Ndfip1+/+ littermates (Figure 1c), and this is more evident in males; perhaps because WT female mice do not gain as much weight at this age. The difference in weights becomes more apparent by 6 weeks of age. This time point coincides with the onset of skin lesions. Their failure to gain weight might be because of the presence of eosinophils in the GI tract, which are known to induce tissue destruction.14
To confirm that the infiltrating cells were eosinophils and T cells, tissue homogenates were prepared from esophagus, small bowel, and colon. Isolated cells were stained with antibodies for Siglec-F, a marker of eosinophils, or CD4. Flow cytometric analysis revealed that the esophagus and colon of WT (Ndfip1+/+) mice contain very few eosinophils, whereas the small bowel has measurable numbers of these cells. In contrast, Ndfip1−/− mice showed a significant increase in the percentages of eosinophils in the esophagus (Figure 1d,e) as well as in the small bowel.
The percentages of eosinophils were only modestly increased in the colon. CD4 T cells were also increased in the GI tract of Ndfip1−/− mice (Figure 1d), although this was mostly evident in the esophagus. The percentages of leukocytes in the blood of Ndfip1−/− and Ndfip1+/+ littermates were also analyzed. Ndfip1−/− mice contained increased percentages of circulating eosinophils compared with littermate controls (Supplementary Figure S2 online); however, there was no difference in the percentage of CD4 + T cells in the blood. Taken together, these data suggest that in the absence of Ndfip1, immune regulatory mechanisms fail, leading to inflammation along the GI tract. Although the inflammation seen in Ndfip1−/− mice involves both T cells and eosinophils, it does not reveal whether GI inflammation is initiated by a defect in T cells or eosinophils.
T-cell activation and infiltration into the GI tract precedes that of eosinophils
We next wanted to determine whether T cells or eosinophils were initiating inflammation. At 4 weeks of age, there were no overt signs of inflammation in Ndfip1−/− mice. Interestingly, at this time point, Ndfip1−/− mice already showed increased percentages of activated CD4 T (CD44hi and CD62Llo) cells in peripheral lymphoid organs (Figure 2a ). To test whether T-cell movement into the esophagus preceded GI eosinophilia, we looked at the percentages of eosinophils and CD4 T cells in the esophagus of Ndfip1−/− mice at this early age. At this time point, the percentage of eosinophils in the esophagus was not different from that seen in WT mice (Figure 2b,c). In contrast, about half of the 4-week-old Ndfip1−/− mice already showed increased percentages of CD4 T cells in their esophagus. Thus, T-cell activation occurs prior to, and thus may trigger, eosinophil recruitment into the GI tract.
Figure 2.

T-cell activation and migration precedes eosinophilic infiltration into the gastrointestinal (GI) tract. (a) CD4 T cells from the spleen and lymph nodes of 4-week-old Ndfip1 − / − and Ndfip1 + / + littermates were analyzed for markers of activation (CD44 and CD62L). Activated T cells (CD44hi) are indicated by the gate and percentages of these cells are noted. (b) The percentages of eosinophils (Siglec-F + ) and T cells (CD4 + ) isolated from the esophagus of Ndfip1 − / − vs. Ndfip1 + / + littermate controls are shown. Data in a and b are representative of three different experiments. (c) The percentages of eosinophils in the esophagus of Nfip1 − / − and Ndfip1 + / + littermate control mice from all three experiments are graphed with each circle representing an individual mouse.
T cells are required for the development of GI inflammation in the Ndfip1 − / − mice
Several publications have described eosinophils as antigen-presenting cells capable of activating T cells and initiating tissue inflammation.15,16 However, in Ndfip1−/− mice, CD4 T-cell activation and migration into the esophagus occurs prior to the infiltration of eosinophils, suggesting that activated CD4 T cells could be recruiting eosinophils into this tissue. To test whether GI inflammation results from defective T cells, we crossed Ndfip1−/− mice to mice that lack T cells, namely Rag1−/−mice.17 Mice deficient in both Ndfip1 and Rag1 showed no signs of inflammation along the GI tract and had a similar body weight compared with their Ndfip1+/+ Rag−/− littermates (Figure 3a,b). These data suggest that T cells are required for the GI inflammation in Ndfip1−/− mice. Given that Rag1−/−mice also lack B cells, we further tested the role of T cells in the induction of GI inflammation through a transfer experiment described below.
Figure 3.
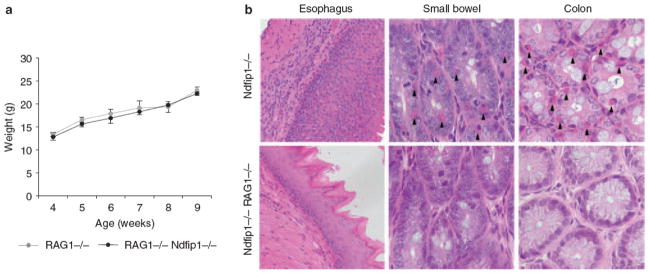
T cells are required for gastrointestinal (GI) inflammation in Ndfip1 − / − mice. (a) Weights of Ndfip1 + / + Rag1 − / − and Ndfip1 − / − Rag1 − / − mice are shown. Each data point includes weights from 3 to 5 different mice. (b) Histological analysis of the esophagus (×20), small bowel (×40), and colon (×40) from Rag1 − / − Ndfip1 − / − vs. Ndfip1 − / − animals. Data shown are representative of at least three mice of each genotype.
Ndfip1-deficient mice have elevated levels of serum IL-5 and IL-5-producing effector T cells
Under normal conditions, a small number of eosinophils are released from the bone marrow and these home to the small bowel and colon because of expression of eotaxin.18 Overexpression of IL-5 leads to an increased release of eosinophils from the bone marrow and promotes eosinophil recruitment into the GI tract.19 Thus, we reasoned that IL-5, produced by activated CD4 T cells, could drive eosinophil recruitment into the GI tract of Ndfip1−/− mice. Thus, we first measured IL-5 levels in the serum of Ndfip1−/− and Ndfip1+/+ animals. We found that IL-5 was significantly increased in the serum of Ndfip1−/− mice (Figure 4a). Furthermore, Ndfip1−/−Rag1−/− mice did not show measurable levels of IL-5 in the serum. These data suggested that Ndfip1−/− T cells might produce IL-5 and initiate the recruitment of eosinophils into the GI tract.
Figure 4.

T cells in Ndfip1 − / − mice are more likely to make interleukin (IL-5). (a) Serum levels of IL-5 measured by enzyme-linked immunosorbent assay (ELISA). Each circle represents an individual mouse. The data include 12 Ndfip1 − / − , 12 Ndfip1 + / + littermates, and 3 Ndfip1 − / − RAG − / − mice. (b) IL-5 from spleen or lymph node cells isolated from Ndfip1 − / − (black) or Ndfip1 + / + (white) littermates. The graph shows the average and s.d. from 3 to 4 different experiments. (c) Intracellular stain of IL-5 in gated CD4 T cells isolated from the spleen of Ndfip1 − / − or Ndfip1 + / + littermates. *P < 0.05, **P < 0.0005.
To test whether Ndfip1−/− mice have effector T cells in the peripheral lymphoid organs that make IL-5, total spleen and lymph node cells from Ndfip1−/− or Ndfip1+/+ littermates were activated in the presence of anti-CD3 for 3 days and the culture supernatants were analyzed for the presence of IL-5. We found that IL-5 was significantly higher in the supernatants of cells from Ndfip1−/− mice than in those from Ndfip1+/+ animals (Figure 4b). We also detected a significant increase in IL-4 production in spleen cultures from Ndfip1−/− mice, but very low levels of interferon-γ (Supplementary Figure S3 online), which is consistent with the previously observed bias of Ndfip1−/− T cells toward the TH2 lineage.12 To test whether the T cells in these cultures were producing IL-5, we measured intracellular IL-5 by flow cytometry. We found that Ndfip1−/−spleens contained increased percentages of IL-5 + CD4 T cells than their Ndfip1+/+ littermates (Figure 4c). These data show that Ndfip1−/− T cells produce significant quantities of IL-5 and may account for the high levels of IL-5 in the serum of Ndfip1−/− mice.
Overproduction of IL-5 by CD4 T cells is sufficient to induce GI eosinophilia.19 Thus, we sought to test whether IL-5 was responsible for the GI eosinophilia in Ndfip1−/− mice. To do this, we treated Ndfip1−/− bone marrow chimeras with either an IL-5blocking antibody, or with an isotype control antibody. At 4 weeks after bone marrow reconstitution, and prior to onset of disease, mice were treated for 2 weeks and then analyzed for the number of eosinophils in their GI tract. We found that mice treated with the anti-IL-5 blocking antibody had a lower percentage of eosinophils in the esophagus and small bowel compared with those treated with an isotype control (Supplementary Figure S4 online). These data show that IL-5 promotes the recruitment of eosinophils into the GI tract of Ndfip1−/− mice.
Ndfip1-deficient CD4 T cells are sufficient to drive GI inflammation
So far, our results indicate that Ndfip1−/− CD4 T cells become activated in vivo and make IL-5, and this promotes eosinophil recruitment into the GI tract. This suggests that Ndfip1−/−CD4 T cells would be sufficient to drive GI inflammation. To test this, we sorted naive (CD44low CD62Lhi) CD4 T cells from spleen and lymph nodes of Ndfip1−/− or Ndfip1+/+ mice and transferred these cells into Rag1−/− recipients. When sorting for naive cells we did not gate away from CD25+ cells; therefore, there were regulatory T cells present in the naive CD4 T-cell population, which we have corroborated by FoxP3 (forkhead box P3) expression analysis through flow cytometry (data not shown). Importantly, Ndfip1−/− mice have normal percentages of FoxP3 + regulatory T cells in spleen and lymph nodes. Furthermore, we know that the inflammation seen in Ndfp1−/−mice is because of a defect in the effector T-cell population and not the regulatory T-cell population, given that mixed chimeras using WT and Ndfip1−/− bone marrow, which have WT regulatory T cells, still develop inflammation.12 Between 4 and 5 weeks after transfer, Rag−/− mice that received Ndfip1−/−T cells began to loose weight (Figure 5a ). At 6 weeks after transfer, histological analysis revealed eosinophils in the GI tract and extensive inflammation, particularly in the esophagus (Figure 5b). On the other hand, Rag−/− mice that received Ndfip1+/+ cells were devoid of inflammatory infiltrate and showed a healthy appearance. This indicates that Ndfip1−/−CD4 T cells are sufficient to drive eosinophil recruitment and inflammation in the GI tract.
Figure 5.

Ndfip1 − / − CD4 T cells are sufficient to induce gastrointestinal (GI) inflammation. Naive CD4 T cells were sorted from spleen and lymph nodes of Ndfip1 − / − or Ndfip1 + / + littermates and transferred to Rag1 − / − mice. (a) The percent weight change in mice that received either naive Ndfip1 − / − (black) or Ndfip1 + / + (white) CD4 T cells is shown. Each point in the graph represents the weight of an individual mouse as a percentage of its initial weight at the time of transfer. (b) Histological analysis of GI sections from mice that received naive Ndfip1 − / − or Ndfip1 + / + CD4 + T cells. (c) Serum interleukin-5 (IL-5) levels were analyzed by enzyme-linked immunosorbent assay (ELISA) at 5 to 6 weeks after transfer of Ndfip1 − / − or Ndfip1 + / + CD4 T cells. Each point in the graph represents one mouse. (d) Levels of IL-5 produced by anti-CD3-stimulated spleen and lymph node cultures from mice that received Ndfip1 − / − (black) or Ndfip1 + / + (white) cells. *P<0.05.
To test whether IL-5 was being produced by the transferred CD4 T cells, we measured IL-5 production in cells from lymph nodes and spleen as described in Figure 4b. Consistent with our previous results, we found that spleen and lymph node cells isolated from recipients that received Ndfip1 − / − T cells produced a significantly higher amount of IL-5 compared with cells isolated from Ndfip1 + / + T-cell recipients (Figure 5c). There was also an increase in IL-4 in Ndfip1 − / − spleen cultures (data not shown), as would be expected, given the TH2-biased phenotype of Ndfip1 − / − T cells. Furthermore, IL-5 was detected only in the serum of mice that received Ndfip1 − / −T cells (Figure 5d). Taken together, these data indicate that the loss of Ndfip1 in CD4 T cells leads to activation, IL-5 production, and consequent recruitment of eosinophils into the GI tract.
The GI inflammation seen in Ndfip1-deficient mice involves Itch-dependent as well as Itch-independent pathways
We have previously shown that Ndfip1 binds Itch and promotes the ubiquitylation and degradation of JunB, a transcription factor involved in TH2 differentiation.20 Both Ndfip1−/− and Itch mutant mice develop a TH2-mediated pathology in the skin and lung. Ndfip1−/− mice develop inflammation with much faster kinetics, showing signs of inflammation as early as at 6 weeks of age compared with 5 to 6 months for Itch-deficient mice. 5,12 Interestingly, GI inflammation in Itch-deficient mice has not been described. Thus, we tested whether Itch mutant mice also develop GI inflammation. Sections along the GI tract were analyzed from Itch mutant and WT control mice at 5 to 6 months of age, given that this is the time point at which Itch mutant mice show inflammation in the skin and lungs. The esophagus showed an increase in the percentage of eosinophils, although the inflammation seen histologicaly was not as profound as that seen in Ndfip1−/− mice (Figure 6a,b). Furthermore, even at 5 months of age, the percentages of eosinophils in the esophagus of Itch-deficient mice are not as high as those seen in 5–8-week-old Ndfip1−/− mice. Supporting this, the small bowel and colon in Itch-deficient mice showed eosinophilia but to a much lower degree compared with Ndfip1−/− mice (Figures 1b and 6a). The percentage of eosinophils in the small bowel of Itch mutant mice was not significantly different from that of WT controls (Figure 6b).
Figure 6.

Itch mutant mice have a less severe gastrointestinal (GI) phenotype. (a) Histological analysis of the GI tract from Itch-deficient mice and wild-type (WT) mice at 4 to 5 months of age. (b) The percentage of eosinophils determined by flow cytometric analysis of cells isolated from different GI sections from 4 to 5 months old Itch-deficient mice and age-matched controls. (c) Serum levels of interleukin-5 (IL-5) from Ndfip1 − / − , WT, or Itch-deficient mice at different ages measured by enzyme-linked immunosorbent assay (ELISA). Each point in the graph represents an individual mouse. The data include 12 Ndfip1 − / − , 3 Itch-deficient mice, and 3 WT mice for each age indicated. (d) Levels of IL-5 produced from anti-CD3 stimulated spleen cultures measured by ELISA. Intracellular cytokine staining for IL-4 and IL-5 in T helper type 2 (TH2)-differentiated CD4 T cells from WT, Itch mutant, or Ndfip1 − / − mice. Activation status of gated spleen CD4 + T cells from mice that are WT, Itch mutant, or Ndfip1 − / − at 5–7 weeks of age. Gate represents CD44 high cells. Same as c but including five mice of each genotype. *P < 0.01.
We next measured IL-5 in the serum of young (6 weeks of age) and old (5 to 6 months) Itch mutant mice. Old Itch mutant mice showed elevated levels of IL-5 in the serum, although to a lesser degree than in Ndfip1−/− mice (Figure 6c). Young Itch mutant mice, on the contrary, did not show detectable IL-5 in the serum. IL-5 was detected in total spleen cell cultures from old Itch mutant mice, and to a lesser extent in young Itch mutant mice, after anti-CD3 treatment (Figure 6d). However, these levels were lower that those observed in Ndfip1−/− mice. Taken together, these data indicate that only part of the phenotype seen in Ndfip1−/− mice can be explained by the role of this protein in the regulation of Itch, and that Ndfip1 therefore might also regulate other E3 ligases.
SNPs within the Ndfip1 locus associate with IBD
IBD is a group of inflammatory diseases with unknown etiology, which can affect the small bowel and colon; this includes ulcerative colitis (UC) and Crohn’s disease (CD). Identification of genetic abnormalities that contribute to IBD is of considerable importance as it could identify proteins with therapeutic potential. One way through which genetic abnormalities are identified is through a comparative analysis of single-nucleotide polymorphisms (SNPs) in patients and controls. To test whether SNPs in the locus encoding Ndfip1 associate with IBD, we examined polymorphisms within a 130-kilobase region of chromosome 5q31.3. Of 17 SNPs covering the Ndfip1 gene, 7 were found to associate with IBD, when combining the P -values for the discovery and replication (Wellcome Trust Case Control Consortium (WTCCC)) cohorts (Table 1 ); on the contrary, no SNPs in this study associated with celiac disease, rheumatoid arthritis, or type 1 diabetes (data not shown). Three of these SNPs (SNP11) sit upstream of the first coding exon, whereas the other four lie within the Ndfip1 introns (Figure 7a). Pairwise linkage disequilibrium for the SNPs analyzed showed a strong linkage disequilibrium for SNPs that lie within the Ndfip1-coding region (Figure 7b ), suggesting that the SNPs identified may be inherited with other polymorphisms that have yet to be identified. Using our discovery cohort, we analyzed the association of Ndfip1 with UC vs. CD, and found the significant P-values to associate with UC not CD (data not shown); however, we are not able to corroborate these data using the replication (WTCCC) cohort, given that they do not have a specific UC data set. Together, these data support a role for Ndfip1 in the susceptibility to IBD.
Table 1.
SNP association analysis
| Discovery
|
Replication
|
Combined
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| SNP no. | SNP name | Allele | P-value | OR 1 | Allele | P-value | OR | P-value | Corrected P |
| 11 | rs2338874 | G | 0.0122 | 0.8642 | A | 0.0171 | 1.1199 | 0.0020 | 0.0340 |
| 12 | rs891988 | G | 0.3978 | 0.9538 | G | 0.1191 | 1.0775 | NA | NA |
| 13 | rs12655443 | C | 0.0007 | 0.8436 | A | 0.0725 | 1.0799 | 0.0005 | 0.0085 |
| 14 | rs1821263 | C | 0.0404 | 0.902 | C | 0.6949 | 0.9833 | 0.1284 | 1.0000 |
| 15 | rs891989 | C | 0.8732 | 0.9821 | C | 0.6256 | 1.0429 | NA | NA |
| 16 | rs2879271 | C | 0.9206 | 1.005 | C | 0.1062 | 1.0744 | 0.3251 | 5.5267 |
| 27 | rs11167764 | A | 0.0131 | 0.8531 | A | 0.0015 | 0.8469 | 0.0002 | 0.0034 |
| 28 | rs11745990 | A | 0.3912 | 0.9434 | A | 0.8858 | 1.0085 | NA | NA |
| 29 | rs735683 | T | 0.3681 | 0.9482 | G | 0.4345 | 0.9612 | NA | NA |
| 20 | rs4912622 | G | 0.0065 | 0.8672 | A | 0.0081 | 1.1244 | 0.0006 | 0.0102 |
| 21 | rs11749731 | A | 0.0058 | 0.8656 | A | 0.0080 | 0.8892 | 0.0005 | 0.0085 |
| 22 | rs7709361 | T | 0.0084 | 0.871 | C | 0.0079 | 1.1249 | 0.0007 | 0.0119 |
| 3 | rs12655675 | C | 0.2788 | 1.058 | C | 0.1561 | 1.0632 | 0.1799 | 1.0000 |
| 24 | rs1062158 | C | 0.0069 | 0.868 | C | 0.0079 | 0.8890 | 0.0006 | 0.0102 |
| 25 | rs3850577 | A | 0.4958 | 1.055 | C | 0.2606 | 0.9213 | 0.3936 | 1.0000 |
Abbreviations: NA, not applicable; OR, odds ratio; SNP, single-nucleotide polymorphism.
Bold indicates a significant P-value for combined data sets.
Figure 7.

Single-nucleotide polymorphism (SNP) analysis. In all, 37 different SNPs that span the region including 50 kb before the start site of Nedd4 family interacting protein 1(Ndfip1) and 50 kb downstream of the coding region were analyzed. (a) Each SNP is represented by a dot at the indicated locations on human chromosome 5. Five of the analyzed SNPs showed significant differences between a population of healthy individuals and inflammatory bowel disease (IBD) patients and are represented by arrows, which show their location with respect to the Ndfip1 gene. (b) A linkage disequilibrium plot of all 37 SNPs was analyzed. The locations of the SNPs analyzed are indicated. Each square shows the linkage disequilibrium between two SNPs. Darker squares indicate a higher linkage disequilibrium.
DISCUSSION
Ndfip1 was recently identified as an adaptor for the Nedd4 family E3 ubiquitin ligase known as Itch. Little is known as to which pathways this adaptor regulates or whether Itch is the only E3 ligase regulated by Ndfip1. Although Ndfip1−/− and Itch-deficient mice have similar phenotypes, there are also differences, such as the timing of disease onset. In this study we show that mice lacking Ndfip1 have activated effector CD4 T cells that produce IL-5, and that these cells are required for eosinophil infiltration and GI inflammation. The aberrant cytokine production can be explained by the role of Ndfip1 regulating the ubiquitylation of JunB by promoting Itch function. In fact, we have previously shown that Ndfip1−/− T cells have elevated levels of JunB.12 However, overexpression of JunB and a bias toward TH2 differentiation does not entirely account for the phenotype of Ndfip1−/− mice. Supporting this, JunB-overexpressing T cells have been shown to express higher levels of IL-4 and IL-5 after in vitro activation.20 However, mice that overexpress JunB in their CD4 + T cells have not been shown to develop inflammatory disease. 20 We therefore believe that Ndfip1 might regulate two aspects of T-cell function, TH2 differentiation (through Itch and JunB) and T-cell activation, the second of which is currently being examined in our laboratory. Interestingly, mice that are Ndfip1−/− Rag−/− OTII do not develop disease (data not shown), indicating that Ndfip1−/− T cells must see antigen to become activated and cause disease. Given that the inflammation in Ndfip1−/− mice occurs in the skin, lung, and GI tract, the known sites of environmental antigen exposure, we believe that T cells are responding to environmental antigens rather than to self. This would imply that Ndfip1 regulates tolerance to environmental antigens.
Ndfip1 has been shown to bind to the WW domains of several members of the Nedd4 family of E3 ubiquitin ligases in vitro.9–11 Based on these data, it seems likely that Ndfip1 regulates the function of other Nedd4 family E3 ubiquitin ligases in vivo. In T cells, under physiologic conditions, there is only evidence supporting its role in the regulation of Itch. 12 Ndfip1 was originally identified based on its ability to bind to Nedd4, and Nedd4 is expressed in T cells, and hence it seems obvious to speculate that Ndfip1 might also regulate Nedd4. However, Nedd4 promotes, rather than inhibits, T-cell activation.21 Thus, it seems unlikely that Ndfip1 would regulate Nedd4 function in a similar manner to that of Itch; instead, it might regulate the function of another E3 ligase.
Loss of Ndfip1 in mice leads to the development GI inflammation; furthermore, our SNP data show a possible link between Ndfip1 and IBD in humans. We have found that the frequency of several SNPs in the Ndfip1 locus differ significantly between patients with IBD and healthy controls. This suggests that Ndfip1 may have a role in the susceptibility to this disease. Interestingly, the differences in allele frequencies were found in populations with IBD but not in patients with celiac disease, rheumatoid arthritis, or type 1 diabetes. Eosinophilia, such as we observed in Ndfip1−/− mice, is frequently observed in patients in IBD,22,23 especially in the earlier phases of disease development. However, other aspects of the pathology associated with IBD are not reproduced in our mice. One possible reason is that Ndfip1 deficiency causes GI pathology that manifests itself differently between mice and humans. The GI disease in Ndfip1−/− mice resembles certain eosinophilic disorders, namely eosinophilic esophagitis, eosinophilic gastrointestinal disorder, or food allergy. Future experiments will be needed to determine whether SNPs in Ndfip1 associate with these diseases. Nevertheless, it is clear that Ndfip1 prevents GI disease in mice and might also prevent the development of GI diseases in humans. There has been a major effort in trying to identify genes that increase the risk of developing IBD,24–26 given that this disease has an obvious genetic component.27 Several genes associated with IBD have been identified, including the IL-23R, CCR6, TL1A, and others.26 Here, we have identified Ndfip1 as an additional gene that may influence the risk of developing IBD in humans.
METHODS
Mice
The Ndfip1−/− and Itch mutant mice have been previously described,5,12 and have been backcrossed to C57BL6 for > 9 generations. All mice were bred and maintained in a semi-barrier facility at the Children ’ s Hospital of Philadelphia. All experimentation was approved and followed guidelines established by the institutional animal care and use committee of the Children ’ s Hospital of Philadelphia. Itch mutant mice were bred from homozygous mutant parents. Given that Ndfip1−/− mice die prematurely, they were bred from heterozygous parents and their WT littermates were used as controls. Ndfip1−/− mice were genotyped using purified genomic DNA from tails and the following PCR primers: Ndfip1 wild-type forward 5′-TAGGCCAAGGTGAAAACTGG-3′, Ndfip1 wild-type reverse 5′-AGAGGTGGGTTCAACAGTGG-3′, Ndfip1 knockout forward 5′-CGACTTCCAGTTCAACATCAGC-3′,andNdfip1knockoutreverse5′-GTCTGTTGTGCCCAGTCATAGC-3′.TheRag1−/−micewereobtained form Jackson Laboratories (Bar Harbor, ME) and genotyped by PCR as previously described.
Histological analysis of GI sections
Sections of the esophagus, stomach, small bowel, and colon were dissected and flushed with cold phosphate-buffered saline (PBS). All sections were then fixed in 10 % formalin for at least 24 h. Tissues were then paraffin-embedded, sectioned, and stained with hematoxylin and eosin. Stained sections were analyzed using a Leica microscope (Bannockburn, IL) with a bright field objective at ×5, ×20, or ×40 magnifications.
Flow cytometric analysis of digested GI sections
The esophagus, small bowel, and colon were dissected. The lumen of the small bowel and colon were flushed with cold PBS. The entire esophagus and colon and 1to 2 inches of the small bowel were minced in Dulbecco’s modied Eagle’s medium media with 0.9 mg ml −1 of collagenase A (Sigma), 0.8mgml−1 collagenase 1A (Sigma), and 20 μgml−1 of DNase I (Sigma, St Louis, MO). Minced tissues were then incubated for 1 h at room temperature, with end-over-end mixing. The resulting cell suspension was passed through 100μm filters and then 40 μm filters, after which 10 % fetal bovine serum was added. Cells were washed with fluorescence-activated cell sorting buffer (3% fetal bovine serum in PBS) and stained with anti-Siglec-F-PE (BD Pharmingen, San Diego, CA) and anti-CD4-APC (Biolegend, San Diego, CA). Data were collected using a FACSCalibur (BD Biosciences, San Jose, CA) and analyzed using FlowJo (TreeStar, Ashland, OR).
Enzyme-linked immunosorbent assay (ELISA)
ELISA was performed using serum samples or using supernatants from cultured cells. For serum, immediately after animals were killed, blood samples were collected by heart puncture and centrifuged in serum separation tubes (Greiner Bio-One, Monroe, NC). The serum was stored at - 80 °C until used for ELISA. For cell supernatants, spleen or lymph node cells were isolated and cultured in complete media with soluble anti-CD3 (5 μg ml−1 ). Cells were plated in 96-well plates, in quadruplets, at 2× 105 per well. Supernatants were collected on days 1 or 3 and kept at − 80 °C until used for ELISA. ELISAs were performed using the BD Biosciences Kit for detection of IL-4 or IL-5, according to the manufacturer ’ s instructions. ELISA plates were analyzed using a Synergy HT Microplate Reader (BioTek, Winooski, VT).
Intracellular IL-5 staining
Total splenocytes from Ndfip1−/− or Ndfip1+/+ littermates were cultured at 1×106cells per well in 24-well plates with 30 μgml−1 of phorbol myristate acetate, 1 × 3 ionomycin, and gologistop (BD Biosciences) for 4.5 h. Cells were collected and stained with anti-CD4-APC. Cells were fixed and analyzed for intracellular IL-5 using the BD Biosciences Kit. The staining was performed using anti-IL-5-PE (BD Pharmingen).
Transfer of naive CD4 T cells
Naive CD4 T cells (CD62Lhigh/CD44low) were sorted using florescence activated cell sorting. A total of 3–4×106 cells in PBS were injected into Rag1−/− mice intravenously.
Generation and anti-IL-5 treatment of bone marrow chimeras
Bone marrow from Ndfip1−/− mice was injected into irradiated B6 mice. The resulting chimeras were treated with anti-IL-5 (TRFK5) blocking antibody (Biolegend) or a rat IgG1 isotype control (eBioscience). Injections were performed using 100 μg of antibody in 200 μl of PBS. Mice were injected every 3 days starting at week 4 after bone marrow reconstitution, and analyzed 6 weeks after reconstitution.
SNP analysis
Cases and controls for the discovery cohort were genotyped at the Center for Applied Genomics, Children’s Hospital of Philadelphia using the Illumina HumanHap550 Beadchip (San Diego, CA) as previously described.28 The replication cohort consists of patients from the WTCCC who had been genotyped on the Affymetrix 500K set (Affymetrix, Santa Clara, CA). 29 The discovery cohort includes a mixture of 605 CD, 293 UC, and 123 undetermined colitis patients. In all, 3,737 controls were recruited and genotyped at the Children’s Hospital of Philadelphia, and were free of a diagnosis of an inflammatory or autoimmune disease. The replication cohort utilized the WTCCC CD cohort of 1,749 patients as cases, along with the national blood service and 1958 birth cohorts of 2,938 patients as controls. All patients who were used in both the discovery and replication sets were of European ancestry. To test for association with the SNP and disease status, we used an allelic χ2 test. The analysis of the discovery cohort was done using plink (http://pngu.mgh.harvard.edu/~purcell/plink/) and the analysis of the replication set was done using summary stats reported by the WTCCC. The P-values for the discovery and WTCCC were combined using Haploview (Cambridge, MA). Multiple testing was corrected for 17 independent signals found through the linkage disequilibrium analysis using Plink.
Statistics
All statistical analyses were performed using Student’s t-tests. A P-value of ≤ 0.05 was considered to determine statistical significance. Error bars represent s.d. of the mean. For SNP analysis we used a more stringent P<0.01.
Supplementary Material
Acknowledgments
We thank Dr Janis K. Burkhardt and Dr Edwin de Zoeten for helpful discussions and Amy Laroche for technical assistance, as well as the staff of the flow cytometry core at the University of Pennsylvania. This work was supported by the NIH grants RO3 AR057144 and 5-T32-AI055428.
Footnotes
DISCLOSURE
The authors declared no conflict of interest.
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at http://www.nature.com/mi
References
- 1.Mueller DL. E3 ubiquitin ligases as T cell anergy factors. Nat Immunol. 2004;9:883–890. doi: 10.1038/ni1106. [DOI] [PubMed] [Google Scholar]
- 2.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 3.Bachmaier K, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000;403:211–216. doi: 10.1038/35003228. [DOI] [PubMed] [Google Scholar]
- 4.Chiang YJ, et al. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403:216–220. doi: 10.1038/35003235. [DOI] [PubMed] [Google Scholar]
- 5.Fang D, et al. Dysregulation of T lymphocyte function in itchy mice: a role for Itch in TH2 differentiation. Nat Immunol. 2002;3:281–287. doi: 10.1038/ni763. [DOI] [PubMed] [Google Scholar]
- 6.Vinuesa CG, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005;435:452–458. doi: 10.1038/nature03555. [DOI] [PubMed] [Google Scholar]
- 7.Lin AE, Mak TW. The role of E3 ligases in autoimmunity and the regulation of autoreactive T cells. Curr Opin Immunol. 2007;6:665–673. doi: 10.1016/j.coi.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Harvey KF, Shearwin-Whyatt LM, Fotia A, Parton RG, Kumar S. N4WBP5, a potential target for ubiquitination by the Nedd4 family of proteins, is a novel Golgi-associated protein. J Biol Chem. 2002;277:9307–9317. doi: 10.1074/jbc.M110443200. [DOI] [PubMed] [Google Scholar]
- 9.Foot NJ, et al. Regulation of the divalent metal ion transporter DMT1 and iron homeostasis by a ubiquitin-dependent mechanism involving Ndfips and WWP2. Blood. 2008;112:4268–4275. doi: 10.1182/blood-2008-04-150953. [DOI] [PubMed] [Google Scholar]
- 10.Howitt J, et al. Divalent metal transporter 1 (DMT1) regulation by Ndfip1 prevents metal toxicity in human neurons. Proc Natl Acad Sci USA. 2009;106:15489–15494. doi: 10.1073/pnas.0904880106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mund T, Pelham HR. Control of the activity of WW-HECT domain E3 ubiquitin ligases by NDFIP proteins. EMBO Rep. 2009;10:501–507. doi: 10.1038/embor.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliver PM, et al. Ndfip1 protein promotes the function of itch ubiquitin ligase to prevent T cell activation and T helper 2 cell-mediated inflammation. Immunity. 2006;25:929–940. doi: 10.1016/j.immuni.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao M, et al. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science. 2004;306:271–275. doi: 10.1126/science.1099414. [DOI] [PubMed] [Google Scholar]
- 14.Rothenberg ME, Mishra A, Brandt EB, Hogan SP. Gastrointestinal eosinophils. Immunol Rev. 2001;179:139–155. doi: 10.1034/j.1600-065x.2001.790114.x. [DOI] [PubMed] [Google Scholar]
- 15.Padigel UM, et al. Eosinophils act as antigen-presenting cells to induce immunity to Strongyloides stercoralis in mice. J Infect Dis. 2007;196:1844–1851. doi: 10.1086/522968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi HZ. Eosinophils function as antigen-presenting cells. J Leukoc Biol. 2004;76:520–527. doi: 10.1189/jlb.0404228. [DOI] [PubMed] [Google Scholar]
- 17.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1 deficient mice have no mature T and B lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 18.Rothenberg ME, Hogan SP. The eosinophil. Annu Rev Immunol. 2006;24:147–174. doi: 10.1146/annurev.immunol.24.021605.090720. [DOI] [PubMed] [Google Scholar]
- 19.Mishra A, Hogan SP, Brandt EB, Rothenberg ME. IL-5 promotes eosinophil trafficking to the esophagus. J Immunol. 2002;168:2464–2469. doi: 10.4049/jimmunol.168.5.2464. [DOI] [PubMed] [Google Scholar]
- 20.Li B, Tournier C, Davis RJ, Flavell RA. Regulation of IL-4 expression by the transcription factor JunB during T helper cell differentiation. EMBO J. 1999;18:420–432. doi: 10.1093/emboj/18.2.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang B, et al. Nedd4 augments the adaptive immune response by promoting ubiquitin-mediated degradation of Cbl-b in activated T cells. Nat Immunol. 2008;9:1356–1363. doi: 10.1038/ni.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Al-Haddad S, Riddell RH. The role of eosinophils in inflammatory bowel disease. Gut. 2005;54:1674–1675. doi: 10.1136/gut.2005.072595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogan SP, et al. Eosinophils: biological properties and role in health and disease. Clin Exp Allergy. 2008;38:709–750. doi: 10.1111/j.1365-2222.2008.02958.x. [DOI] [PubMed] [Google Scholar]
- 24.Barrett JC, et al. Genome-wide association defines more than 30 distinct loci for Crohn’s disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kugathasan S, et al. Loci on 20q13 and 21q22 are associated with pediatric-onset inflammatory bowel disease. Nat Genet. 2008;40:1211–1215. doi: 10.1038/ng.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shih DQ, Targan SR, McGovern D. Recent advances in IBD pathogenesis: genetics and immunobiology. Curr Gastroenterol Rep. 2008;10:568–575. doi: 10.1007/s11894-008-0104-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xavier RJ, Podolsky DK. Unraveling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 28.Hakonarson H, et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature. 2007;448:591–594. doi: 10.1038/nature06010. [DOI] [PubMed] [Google Scholar]
- 29.Welcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 common controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.