

Abstract
Hepatitis C virus (HCV) is a significant health problem facing the world. This virus infects more than 170 million people worldwide and is considered the major cause of both acute and chronic hepatitis. Persons become infected mainly through parenteral exposure to infected material by blood transfusions or injections with nonsterile needles. Although the sexual behavior is considered as a high risk factor for HCV infection, the transmission of HCV infection through sexual means, is less frequently. Currently, the available treatment for patients with chronic HCV infection is interferon based therapies alone or in combination with ribavirin and protease inhibitors. Although a sustained virological response of patients to the applied therapy, a great portion of patients did not show any response. HCV infection is mostly associated with progressive liver diseases including fibrosis, cirrhosis and hepatocellular carcinoma. Although the focus of many patients and clinicians is sometimes limited to that problem, the natural history of HCV infection (HCV) is also associated with the development of several extrahepatic manifestations including dermatologic, rheumatologic, neurologic, and nephrologic complications, diabetes, arterial hypertension, autoantibodies and cryglobulins. Despite the notion that HCV-mediated extrahepatic manifestations are credible, the mechanism of their modulation is not fully described in detail. Therefore, the understanding of the molecular mechanisms of HCV-induced alteration of intracellular signal transduction pathways, during the course of HCV infection, may offer novel therapeutic targets for HCV-associated both hepatic and extrahepatic manifestations. This review will elaborate the etiopathogenesis of HCV-host interactions and summarize the current knowledge of HCV-associated diseases and their possible therapeutic strategies.
Keywords: Hepatitis C virus, Hepatocellular carcinoma, Extrahepatic, Signalling, Therapy
INTRODUCTION
Hepatitis C virus (HCV) infects more than 170 million people worldwide[1-3]. This virus is considered one of the major causes of both acute and chronic hepatitis. Persons become infected mainly through parenteral exposure to infected material by blood transfusions or injections with nonsterile needles. Although the sexual behavior is considered as a high risk factor for HCV infection, the transmission of HCV infection through sexual means, is less frequently[4,5]. Besides the cause of liver disease, the natural history of HCV infection (HCV) is also associated with the development of several extrahepatic manifestations[6,7]. Patients (40%-74%) infected with HCV might develop at least one extrahepatic manifestation during the course of the infection[6,7]. HCV-associated liver diseases range from chronic hepatitis to fibrosis, cirrhosis and hepatocellular carcinoma (HCC)[8]. Whereas, the extrahepatic manifestations include dermatologic, rheumatologic, neurologic, and nephrologic complications; and diabetes; arterial hypertension; autoantibodies and cryoglobulins[9,10].
Currently, the available treatment for patients with chronic HCV infection is interferon based therapies alone or in combination with ribavirin. Although a sustained virological response of patients to the applied therapy, a great portion of patients did not show any response[11]. Despite the mechanisms underlying the failure of interferon therapy are not well understood, several studies revealed that host response to interferon therapy is controlled by both viral and host factors[12]. Therefore, the current knowledge obtained from the functional analysis of both viral- and host factors, during the infection might help for the development of a novel therapeutic strategy in the future.
HCV GENOME AND ITS FUNCTIONAL ORGANIZATION
HCV belongs to the family Flaviviridae and is a single positive strand of RNA (about 9.6 kb) and contains a long open reading frame flanked by both 5’ and 3’ untranslated regions that are important for both translation and replication processes of the viral RNA genome[13,14]. Based on the sequence variation of HCV genome, six genotypes and more than 50 subtypes have been identified[15,16]. The RNA genome of the virus encodes for a single polyprotein that is mainly processed by cellular and viral proteases into at least 10 structural (Core, E1, E2/p7) and nonstructural (NS2, NS3, NS4A, NS4B, NS5A and NS5B) proteins (Figure 1).
Figure 1.

Hepatitis C virus genomic organization,replication,translation and generation of functional proteins. Proteins encoded by the hepatitis C virus (HCV) genome. HCV is formed by an enveloped particle harboring a plus-strand RNA of about 9.6 kb. The genome carries a long open-reading frame (ORF) encoding a polyprotein precursor of 3010 amino acids. Translation of the HCV ORF is directed via a 5’ nontranslated region (NTR) functioning as an internal ribosome entry site; it permits the direct binding of ribosomes in close proximity to the start codon of the ORF. The HCV polyprotein is cleaved co- and post-translationally by cellular and viral proteases into ten different products, with the structural proteins core (C), envelop 1 (E1) and envelop 2 (E2) located in the N-terminal third, whereas, the nonstructural (NS2, NS3, NS4A, NS4B, NS5A, NS5B) replicative proteins are located in the remainder. Putative functions of the cleavage products are shown.
Proteins that are derived from the amino-terminal of the viral polyprotein are called viral structural proteins, these include core and two envelope glycoproteins, E1 and E2. HCV core is a basic protein with variable molecular weights (17-21 kDa) and is characterized by its RNA-binding activity that is thought to be responsible for the comprising of the viral nucleocapsid[17-19]. However, the localization of HCV core protein in various subcellular compartments, including cytosol, lipid droplets, endoplasmic reticulum (ER), golgi apparatus, mitochondria, and nuclei suggests the contribution of HCV core protein in the modulation of different cellular processes[20].
The two HCV envelope glycoproteins E1 and E2 interact with cell surface molecules including CD81, claudin-1, scavenger receptor class B type I, and thereby facilitate the virus entry into the mammalian cells[21,22]. HCV p7 protein is a small transmembrane protein that is characterized by its functional activity as an ion channel protein[23]. In addition to the mentioned structural proteins, the non-structural protein NS2 is recognized to play a central role in polyprotein processing and virus assembly[24].
HCV non-structural proteins (NS3, NS4A, NS4B, NS5A, and NS5B) are essential for both viral RNA replication and polyprotein processing[25]. Besides its serine protease activity that is responsible for the cleavage of HCV polyprotein, and subsequently the generation of the amino termini of NS4A, NS4B, NS5A, and NS5B[26], NS3 serves as an RNA helicase and NTPase, and is considered an essential component of the RNA replicase complex[27,28]. NS4A, a small 54-amino-acid protein that forms a stable complex with the amino-terminal third of NS3, protease domain, and is required for a complete serine protease activity[29]. NS4B, an integral membrane protein that is mostly localized on the cytoplasmic side of the ER membrane and is implicated in assembly of the replicase complex on lipid rafts[30,31]. NS5A, a phosphoprotein that plays a role in viral resistance to interferon[32,33]. NS5A also plays a role in RNA replication, and virus assembly[34]. NS5B is the RNA-dependent RNA polymerase, and acts as the catalytic core of the macromolecular replicase complex essential for HCV RNA replication[25,35].
The functional analysis of HCV genome using cloned HCV gene expression in mammalian cells, the development of subgenomic or full-length replicon derived from HCV, and the generation of infectious HCV genotypes 1a and 2a in human hepatocyte derived cell lines have significantly contributed to the advancement of HCV research[36-39]. Recently, autophagy has gained importance as it plays an important role in HCV life cycle. Also, the role of HCV in the modulation of autophagy in hepatocytes has been reported[40-43]. HCV may induce accumulation of autophagosomes via the induction of ER stress and the unfolded protein response[40-43]. Similar to several viruses including poliovirus or coxsackie viruses, the induction of autophagosomes seems to play an important role in HCV replication[40-43]. Taken together, the knowledge obtained from the functional analysis of the molecular mechanisms of HCV-induced autophagy in hepatocytes may help for the development of a therapeutic strategy for treatment of HCV infection.
INTERFERENCE OF HCV PROTEINS WITH INTRACELLULAR SIGNAL TRANSDUCTION PROCESSES
The most studied transmembrane and intracellular signal transduction pathways, in the liver, are the mitogen-activated protein kinases (MAPKs), the transforming growth factor (TGF)-β, and the Janus kinase (JAK), tumor necrosis factor (TNF)-α and sphingolipid (SP). However, the activation of these signaling pathways by either cytokines or growth factors leads to the regulation of specific cellular processes including proliferation, growth, differentiation, adhesion, migration, apoptosis, and both synthesis and degradation of the extracellular matrix[44-47].
As recognized, the replication cycle of HCV is an intracellular mechanism that requires the intracellular signal transduction processes of the host cell to ensure genome replication, transcription and translation[48,49]. A proposed model for the interference of HCV with cellular signal transduction processes is demonstrated in Figure 2.
Figure 2.

A proposed model for the consequences resulting from the interference of hepatitis C virus with signal transduction processes in host cells. HCV: Hepatitis C virus; TNFR: Tumor necrosis factor receptor; JAK: Janus kinase; EGFR: Endothelial growth factor receptor; IFN: Interferon; IFNR: IFN receptor; TRAF: Tumor necrosis factor associated factor; TRADD: TNFR-associated protein with death domain; JAK: Janus kinase; JNK: c-Jun N-terminal kinase; SOCS: Suppressor of cytokine signaling; PI3K: Phosphatidylinositol-3-kinase; ERK: Extracellular regulated protein kinase; RIP: Receptor-interacting protein; ROS: Reactive oxygen species; STAT: Signal transducers and activators of transcription; NF-κB: Nuclear factor κB; IL: Interleukin; PPAR: Peroxisome proliferator-activated receptor.
MAPK signal transduction pathway
The legation of endothelial growth factor (EGF), hepatocyte growth factor (HGF) and TGF-α to their corresponding membrane receptors results mainly in the activation of the intrinsic tyrosine kinase that leads to ligand-receptor complex formation and subsequently autophosphorylation[50-52]. As a consequence, the formation of a transient complex from Ras proteins and GTP that subsequently mediates the activation of RAF and MAPKK kinases that, in turn, enhance the activation of MAPK by dual phosphorylation of threonine and tyrosine. Activated MAPK results in the phosphorylation of transcription factors such as cAMP response element-binding protein and Ets-related transcription factor 1 (ELK-1)[47,53-56]. Evolutionary, MAPK signal transduction pathway is considered as one of the oldest signal transduction pathways in eukaryotic cells. This kinase contains three different signal pathways: the extracellular regulated protein kinase (ERK, p42/44 MAPK), the stress activated protein kinase [stress activated protein kinases (SAPK), p38 MAPK, p38-RK or p38], and the c-Jun N-terminal kinase (JNK, p64/54 SAPK). All of these pathways are implicated in the regulation of cellular processes including cell growth, differentiation, maturation, proliferation and apoptosis[53-63]. In mammalian cells every single pathway is activated by two mitogen-activated protein kinase kinase (MKK), e.g., JNK is activated by MKK4 and MKK7, ERK is activated by MKK1 and MKK2, whereas p38 is activated by MKK3 and MKK6[54,55,64]. However, the dual role of MKK in the activation of JNK, ERK or p38 signal transduction pathways still remains to be investigated in detail. Although ERK has been shown to play a key role in the regeneration of liver cells[65,66], the role of p38, especially in hepatocytes regeneration, so far, is not clear[67,68]. Physiologically, the activity of JNK in the liver is minimal, however, its increase during liver regeneration or HCV infection may result from the direct effect of the elevated level of hepatic TNF-α[60,69]. Also, the promotion of cell proliferation in liver and non liver cells by HCV proteins is associated with the activation of MAP kinase signaling pathways JNK, p38 and ERK[47,54,55,70-72]. An overview of HCV-induced alteration of MAP kinase signaling pathways is summarized in Figure 3.
Figure 3.
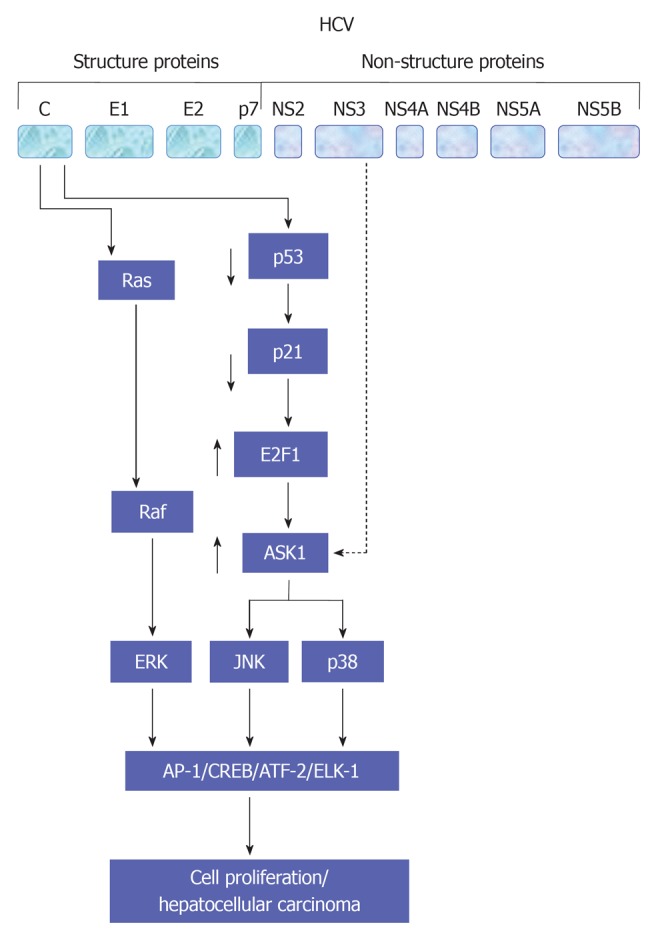
A schematic view for the molecular mechanisms,which are involved in the regulation of hepatitis C virus-induced associated cell proliferation. A central role for mitogen-activated protein kinase signaling pathways in the modulation of hepatitis C virus-associated hepatocellular carcinoma is also demonstrated. HCV: Hepatitis C virus; AP-1: Activator protein-1; JNK: c-Jun N-terminal kinase; CREB: cAMP response element-binding protein; ERK: Extracellular regulated protein kinase.
TGF-β signal transduction pathway
TGF-β is a cytokine family member that plays a key role in the regulation of different cellular events including growth, differentiation, adhesion, apoptosis, and synthesis and degradation of the extracellular matrix[47,73,74]. Although the elevation of TGF-β concentration is observed, during liver regeneration, no marked apoptosis was noted in hepatocytes[75]. However, the inhibition of TGF-β-mediated apoptosis in hepatocytes may be linked with parallel augmentation of Smads, and other antiapoptotic proteins such as Bcl-2 and Bcl-X in hepatocytes[76,77].
In the liver, TGF-β is responsible for hepatocytes regeneration, the development of fibrosis, and HCC, as well as for the proliferation and differentiation of epithelial cells[47,78]. Moreover, the elevation of TGF-β2 expression, in liver cells, during the course of HCV infection is associated with the development of neoangiogenesis[47,79]. Also, the higher the concentration of both TGF-β1 and TGF-β2 in the sera of patients with chronic liver diseases, such as chronic HCV infection, the more severe the liver failure; an evidence for the association between the level of these cytokines and the development of liver diseases including hepatic fibrosis, cirrhosis and HCC[47,80,81]. Simultaneously, in patients with chronic HCV, TGF-β1 serum concentration decreases and normalizes after successful antiviral therapy[82]. However, the inhibition of TGF-β pathway during the infection with HCV, hepatitis B virus (HBV), adenoviruses or human papilloma virus (HPV) has been reported[83]. Also, the mechanism whereby HCV protein NS5A inhibits the activity of TGF-β signal transduction pathway is reported[83]. The inhibition of TGF-β pathway by HCV protein NS5A results from its direct reaction with its specific receptor[84].
TGF-β signaling pathway appears to be most prominent at the interface between development and cancer both in liver and gut epithelial cells[85]. Since this signaling pathway is considered to play a pivotal role in the proliferation of embryogenic hepatocyte as well as in the formation of gastrointestinal cancers[86,87]. In addition, many studies have reported a reduction of TGF-β receptors in up to 70% of HCC[88,89]. Although Smad proteins have been shown to be impaired in different cancers, it appears to play a minor role in HCC[90,91]. Yet, TGF-β levels in serum and urine are increased in HCC patients[92,93]. However, High TGF-β levels have been noted during the course of advanced clinical stage of HCC[94,95]. This dual role of TGF-β signaling in HCC was explained by its effect on the tumor tissue microenvironment and on selective loss of the TGF-β-induced antiproliferative pathway[88]. However, the role of TGF-β signaling pathway in the development of both hepatic angiogenesis (Figure 4A) and HCC (Figure 4B) during the course of HCV infection is demonstrated in detail.
Figure 4.

A proposed model for hepatitis C virus - mediated effects in liver cells and the modulatory role of transforming growth factor β in the regulation of both angiogenesis (A) and hepatocellular carcinoma (B). PKC: Protein kinase C; HCV: Hepatitis C virus; AP-1: Activator protein-1; CREB: cAMP response element-binding protein; JNK: c-Jun N-terminal kinase; ERK: Extracellular regulated protein kinase; TGF: Transforming growth factor; VEGF: Vascular endothelial growth factor; ROS: Reactive oxygen species; PPAR: Peroxisome proliferator-activated receptor.
TNF-α signal transduction pathway
As recognized, macrophages, monocytes, mast cells and NK cells are the main source of TNF-α production that is considered to be one of the major mediators of the antiviral inflammatory response, which results in the enhancement of lymphocytes proliferation and differentiation, production of acute phase proteins and cell apoptosis[60,96,97].
The two essential TNF-α membrane receptors are the TNF-R1 and TNF-R2[98]. TNF-R1 is an extracellular transmembrane receptor that consists of extracellular, transmembrane and intracellular [death domain (DD)] domains. TNFR1 is considered to play a key role in liver, and is expressed in hepatocytes as well as in kupffer cells and hepatic sinusoidal endothelial cells[99-101].
Activated TNF-R1 binds, via the DD, to an adaptor protein TNFR-associated protein with DD, which afterwards activates Fas associated DD proteins, TNF associated factor 2 and receptor-interacting protein. All of these proteins influence different signal transduction pathways, which are involved in the regulation of apoptosis[102], and anti-apoptotic effects of TNF-α as the activation of nuclear factor κB (NF-κB) factor, as well as JNK and ERK from MAPK signal transduction pathway[103].
Although the main target cells of HCV infection are the hepatocytes, the infection of B lymphocytes by HCV virus is documented[104]. Therefore, the participation of both innate and adoptive immune system at the etiopathogenesis of HCV during the course of HCV infection is expected.
The induction of TNF-α, during the infection with chronic HCV, results mainly in the activation of NF-κB pathway that subsequently stimulates transcription of genes encode for cytokines, acute phase proteins, immunoglobulins (Ig) and adhesion factors[60,103,105]. The ligation of TNF-α to TNF-R1, depending on the activated cellular proteins, leads to either cell proliferation or apoptosis[106].
TNF-α plays a diverse role in HCV infection. The activation of TNF-α has a pivotal role in the inflammatory process of chronic hepatitis C, and TNF-α levels correlate with the degree of inflammation[107,108].
HCV viral proteins including core, NS3 and NS4B protein have been reported to be involved in the modulation of cell proliferation[54,55,60,109,110] and production of proinflammatory cytokines TNF-α through NF-κB (Hassan et al[47,54,60], 2007), activator protein-1 (AP-1) and serum response element (SRE)[55]. AP-1 is a complex of homo- or heterodimers encoded by c-jun and c-fos family genes[55]. However, the ability of AP-1 to stimulate proliferation seems to be growth factors[47,111], oncogenes and inflammatory peptides- dependent mechanism[112]. SRE regulates the promoters of immediate early genes such as c-fos and PIP92. MAPK cascade activation phosphorylates Elk-1 factor binding with SRE and serum response factor[113]. Thus, the created complexes affect transcription of genes taking part in cell proliferation. A schematic view demonstrates HCV-mediated pathways leading to TNF-α production is outlined in Figure 5.
Figure 5.
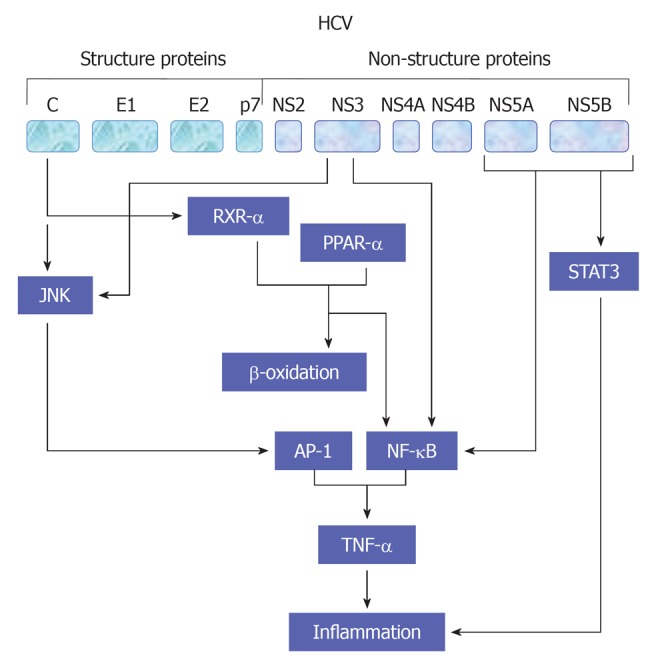
A suggested model for hepatitis C virus -associated inflammation and the role of tumor necrosis factor α in the regulation of this mechanism. HCV: Hepatitis C virus; JNK: c-Jun N-terminal kinase; AP-1: Activator protein-1; NF-κB: Nuclear factor κB; PPAR: Peroxisome proliferator-activated receptor; TNF: Tumor necrosis factor; STAT: Signal transducers and activators of transcription.
JAK signal transduction pathway
JAK signal transduction pathway can be activated by different cytokines and growth factors. This intracellular pathway operates in hepatocytes[114,115] as well as in immune[116], hematopoietic[117,118] and neural system cells[119]. After extracellular ligand-receptor interaction, receptor multimerization and the activation of JAK1, JAK2, JAK3 and tyrosine kinase 2 (Tyk2) is observed[120]. The receptor-kinase complex phosphorylates cytoplasmic SH-2-containing transcription factors: signal transducers and activators of transcription (STAT)1-6. STATs are specifically inhibited by protein inhibitors of activated STAT and by suppressor of cytokine signaling (SOCS) through negative feedback control[121]. SOCS proteins include SOCS 1, 2, 3 and cytokine-induced Src homology 2 protein, which bind to JAK kinase inhibiting its enzymatic activity[122]. STATs perform different, often opposing functions in the liver. STAT1 is mainly activated by interferon (IFN) type I (IFN-α/β) and IFN type II (IFN-γ). Its essential function in liver is the participation in antiviral immune defense, as well as in the development of inflammation and apoptosis. IFN-α/β and IFN-γ are ligands for STAT2, whose major function is antiviral defense. Membrane the IFN-α/β receptor (IFNAR) is a complex of two subunits: IFNAR1 and IFNAR2. IFNAR2 presents three diverse forms: full-length IFNAR2c is responsible for signal transduction and transcription process, whereas short form IFNAR2b and soluble form IFNAR2a inhibit these processes[123]. The complex IFN-α/β - IFNAR activates JAK1 and Tyk2 kinases. IFN-γ takes effect by IFN-γ receptor (IFNGR): IFNGR1 and IFNGR2. STAT3 function is especially regulated by interleukin (IL)-6 and its family members such as cardiotrophin-1, oncostatin M, IL-11, leukemia inhibitory factor or ciliary neurotrophic factor, by IL-10, IL-22, EGF and HCV proteins. STAT3 participates in the acute phase response, stimulates hepatocytes regeneration and regulates lipid and carbohydrate metabolism in the liver[124]. Moreover STAT3 is one of the main anti-HCV defense elements that act by increasing the IFN-α antiviral effect and by its direct cytoprotective and anti-inflammatory influence on hepatocytes[125]. IL-6 and its related cytokines bind gp130 receptor protein, which plays a key role in liver regeneration.
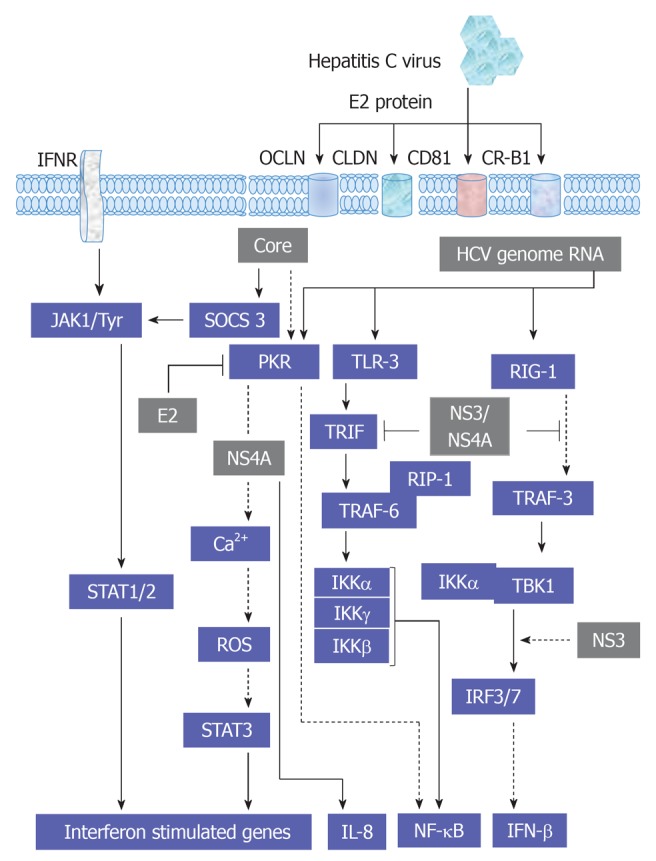
Furthermore, the activation of gp130 is independent of the activation of other kinases, such as MAKP[71]. The ligand-gp130 complex activates JAK1, JAK2 and Tyk2 and subsequently leads to the activation of STAT1-3. However, the modulation of JAK1, JAK2 and Tyk2-mediated activation of STAT1-3 factors by HCV infection or by HCV structural and non-structural proteins has been demonstrated. Thus, the inhibition of the transmembrane and intracellular signal transduction pathways could be a new therapeutic target in chronic HCV treatment. HCV structural proteins Core, E2 and non-structural protein NS5A were reported to reduce the number of IFN-α receptors (IFN-α R1 and IFN-αR2c) and subsequently inhibit IFN-α-induced activation of STAT1-3[126-128]. As a result, viral replication, as well as inflammation and fibrosis in the liver, is augmented and has a negative effect on IFN-α treatment response among patients with severe liver damage. However, HCV does not affect IFN-γ function, and in consequence, STAT1 activation[129]. Moreover, the production of IFN-γ by NK cells during HCV infection is associated with the inhibition of hepatocytes regeneration[124]. Also, STAT4 has been shown to be activated by IL-12 and to play a critical role in hepatocytes damage during hepatic ischemia/reperfusion injury[130]. Whereas, STAT5 that is mainly activated by growth factors and involved in the regulation of the expression of genes encoding cytochrome P450, HGF and insulin growth factor 1, which are essential for hepatocytes metabolism, growth and differentiation[131,132]. STAT6 that is mainly regulated by IL-4, IL-12 and IL-13 and contributes in Th2 lymphocytes response during viral hepatitis and reduces hepatocytes damage during hepatic ischemia/reperfusion injury[133]. An overview demonstrating the interference of HCV with JAK/STAT, cytokines and IFN-associated pathways is outlined in Figure 6.
Figure 6.

Proposed model for the molecular mechanisms, which are involved in the regulation of hepatitis C virus-associated steatosis. HCV: Hepatitis C virus; ROS: Reactive oxygen species; PPAR: Peroxisome proliferator-activated receptor.
SP signal transduction pathway
Although SPs are considered to be the major components of eukaryotic plasma membranes and mediators of cell-to-cell interactions, their role as second messengers in transmembrane and intracellular signal transduction is documented[134]. The main function of the SP signal transduction pathway is the modulation of specific cell reactions including proliferation, growth arrest, differentiation, apoptosis and calcium homeostasis (Boya et al[135], 2005). SP pathways can be activated by many pro-apoptotic and promitotic factors[136-138].
Ceramide is the most intensively studied second messenger of SP signal transduction pathway. Ceramide can mediate its antiproliferative effect by the activation of JNK, SAPK, cathepsin D, methionine adenosyl transferase 1A and caspase 3, leading to the destruction of the cytoskeleton, nuclear and plasma membranes[139]. In addition to its antiproliferative effects, ceramide has the ability to trigger mitochondrial dysfunction by the enhancement of reactive oxygen species (ROS) accumulation and cytochrome c release[140]. However, the antiapoptotic function of ceramide depends on its ability to decreases the intracellular level of anti-apoptotic proteins of the Bcl-2 family as well as the activity of anti-apoptotic enzymes such as Ca2+-kinases including protein kinase C (PKC), PKCα and PKCβα/Akt[141]. Besides its role in the regulation of both cell survival and death, ceramide can also inhibit autophagocytosis by a mechanism based on the enhancement of apoptotic pathway[135,142]. As known, autophagocytosis is an intracellular process that relies on degradation of damaged, dead or used cell structures to prolong cell life[142].
Sphingosine (SFO) that is synthesized mainly from the hydrolysis of Ceramide by ceramidases is a member of the second messengers of SP signal transduction pathway. SFO plays a key role in promotion of apoptosis by the enhancement of ROS production in mitochondria and activation of caspase 3, 7 and 8[137]. Also, the inhibition of AKT by SFO leads to the augmentation of the cellular effects of both cytochrome c and caspase-3[137].
Besides its negative effect on DNA synthesis, methylation and replication, SFO reduces the activity of protein kinases including, PKC, calmodulin-dependent protein kinase and insulin receptor kinase[137], and thereby leads to disturbances of nuclear proteins phosphorylation including RNA polymerase, topoisomerase II, histones and matrix proteins[143].
Some studies underline the proliferative character of SFO suggesting that low cellular concentrations of SFO leads to stimulation of cell proliferation and DNA synthesis, whereas the high concentrations is associated with the induction of apoptosis. SFO-1-phosphate (S1P) that mainly synthesized from SFO, has been reported to have an anti-apoptotic potential[135]. An increase in the intracellular level of S1P can activate cell proliferation and its passing from G1 phase to S phase, augment the general number of cells resting in S phase, shorten the time needed for cell division, enhance survival rate of cells subjected to pro-apoptotic factors, mobilize calcium ions from intracellular compartments, influence cytoskeletal architecture and the processes of cell migration and adhesion[144]. S1P modulates cell functions in two different ways: as an intracellular messenger and as a ligand of G protein-coupled receptors, known as endothelial differentiation genes (Edg) - Edg-1, -3, -5, -6 and -8[144]. Cer can be phosphorylated by ceramide kinase to ceramide-1-phosphate (C1P), which can be dephosphorylated back to ceramide by C1P phosphatase[145]. Similarly to S1P, C1P promotes cell proliferation[145]. Recently, some studies have shown that the inhibition of SP metabolism can be a new therapeutic target for HCV infection[146].
HCV-ASSOCIATED STEATOSIS
Fatty liver (liver steatosis) is recognized as a histological phenotype of HCV infection that is occurring in all patients independent from the genotype[147,148]. Although the development of steatosis seems to be a direct consequence of viral protein expression, the molecular mechanism of its occurring appears to be genotype-specific[149].
The promotion of lipid homeostasis by HCV is mediated by the increasing of lipogenesis via a process including the activation of ER membrane bound transcription factors (SREBPs), reducing oxidation and lipid export[149]. As recognized, the main function of SREBPs is the regulation of the transcription of genes that encode for the enzymes which are essential for the biosynthesis of both cholesterol and fatty acid[150]. However, the suppression of both HCV replication and release in response to the inhibition of SREBP[151], or by the fatty acid synthase, an enzyme that is primarily involved in the biosynthesis of fatty acids[151,152], suggest that the host lipid metabolic pathways is considered a potential target for the treatment of HCV infection. A schematic view suggests HCV-mediated pathways leading to the development of steatosis are outlined in Figure 7.
Figure 7.
A schematic overview for the interference of hepatitis C virus with cytokines-associated Janus kinase/signal transducers and activators of transcription and cytokines-associated pathways. HCV: Hepatitis C virus; JAK: Janus kinase; EGFR: Endothelial growth factor receptor; IFN: Interferon; IFNR: IFN receptor; TRAF: Tumor necrosis factor associated factor; JAK: Janus kinase; SOCS: Suppressor of cytokine signaling; RIP: Receptor-interacting protein; ROS: Reactive oxygen species; STAT: Signal transducers and activators of transcription; NF-κB: Nuclear factor κB; IL: Interleukin; PPAR: Peroxisome proliferator-activated receptor.
HCV-RELATED CRYOGLOBULINEMIC
Cryoglobulinemia is defined as the presence of circulating Ig that precipitate at temperatures below 37 °C and redissolve on rewarming[153]. Such an in vitro phenomenon is detectable in a wide number of chronic infectious and immunological disorders, as well as in some hematological malignancies[153-155].
Cryoglobulinemia is usually classified into serological subsets namely type I or monoclonal cryoimmunoglobulinemia that is composed by single monoclonal Ig, mixed cryoglobulinemia (MC) that contains a mixture of polyclonal IgG and monoclonal (type II) or polyclonal (type III) IgM rheumatoid factor (RF)[156]. Type I cryoglobulinemia is frequently associated with hematological disorder including multiple myeloma, immunocytoma or Waldenstrom’s macroglobulinaemia, and is mostly asymptomatic except in the case of hyperviscosity syndrome[157-159]. Whereas, MC is characterized by a typical triad purpura, weakness, arthralgias as well as by multisystem organ involvement including chronic hepatitis, membranoproliferative glomerulonephritis, peripheral neuropathy, skin ulcers, widespread vasculitis, and less frequently lymphatic and hepatic malignancies[154-156,160].
The pathological feature of MC is a leucocytoclastic vasculitis, including both small and medium sized vessels, which are responsible for cutaneous and visceral organ involvement[154-156,160,161]. However, based on clinico-serological and pathological alterations, the terms MC and cryoglobulinemic vasculitis (CV) are referred to the same clinical syndrome[160-162].
CV is considered to be a relatively rare disorder; its prevalence among different countries show a great geographical heterogeneity[155,156,160].
Currently, there are no available classification/diagnostic criteria for CV. However, in the clinical practice, the main diagnostic parameters include serum mixed cryoglobulins with RF activity, low C4, orthostatic skin purpura, and leukocytoclastic vasculitis of small/medium-sized blood vessels secondary to the deposition of circulating immune-complexes and complement[154-156,160,161].
The causative role of hepatotropic viruses in the development of CV had been hypothesized previously[163-165], when a role of HBV in another systemic vasculitis - the polyarteritis nodosa had been demonstrated[166]. However, soon after the identification of CV in patients with HBV infection[167], the role of HCV in the development of CV has been considered[168,169], the majority HCV patients was characterized by the presence of CV[170], an evidence for the association between the chronic infection with HCV and the development of CV.
Currently, the role of HCV infection in the modulation of CV has been consequently established in several studies[160,169-172]. Therefore, a direct role for HCV in the formation of immune-complex-mediated vasculitis is considered. Besides its role as a main triggering factor of CV, HCV infection is thought to play an important role in the underlying lymphoproliferative disorder[160,169-172]. However, the detection of both active and latent HCV viral replication in the peripheral lymphocytes of patients with HCV infection and/or CV suggesting further a dual feature for HCV as both hepato- and lymphotropic virus[169,171]. Also, the affinity of lymphoid tissue to the infection with HCV supports further the occurring of both autoimmune and lymphoproliferative disorders in patients with chronic HCV infection[160,170-174].
Although the HCV infection presents homogenous distribution worldwide, the geographical heterogeneity in the context of HCV-associated immune disorders is common[174]. Thus, the involvement of particular HCV genotypes, environmental and/or host genetic factors may play a central role in the development of HCV-associated CV.
Although the strong affinity of the HCV envelop protein E2 to CD81 and subsequently the modulation of immunological disorders[175], the HCV, based on its biological features and several laboratory studies, seems to be insufficient to drive the different autoimmune-lymphoproliferative disorders in infected patients[173,174,176]. CD81 is a cell surface protein that expressed in both hepatocytes and B-lymphocytes[177], and is recognized to play an important role by the entry of HCV particles[178], in addition to the modulation of HCV-induced autoimmunity[179]. Therefore, the elevation of HCV-associated autoimmune diseases including CV during the course of infection[180,181], may be a consequence of the interaction between HCV-E2 and CD81 leading the increase of the frequency of VDJ rearrangement in antigen-reactive B-cell[169]. Thus, the expansion of B-lymphocyte may be the main actor that is responsible for the observed autoantibody production during the course of HCV infection[174,182-184]. Also, another mechanisms including the molecular mimicry such as HCV antigens or host autoantigens may be involved in the activation of B lymphocyte and thereby increase the production of autoantibodies[160].
In addition to its immunopathogeneicity, HCV is reported to exert oncogenic potential that is mainly involved in the development of HCC[47,54,60,109,185], and in the lymphomagenesis and, possibly, in other malignancies including thyroid cancer[184-186]. However, the modulation of TNF signaling of the host cells by HCV NS5A protein and the inhibition of JAK-STAT pathway by HCV core following the treatment with either IL-6 or IFN-γ stimuli[126,186,187], suggesting an important role for HCV in the dysregulation of the immune system.
The outcome and the severity of CV among patients are largely variable, also the behavior of the disease is mostly unpredictable and patient show usually a relatively benign clinical course. Thus, based on its complicated etiopathogenesis the treatment of CV syndrome must deal with the total clinical picture of the conflicting conditions including: HCV infection, autoimmune, and lymphoproliferative alterations[160]. Thus, according to the pathogenetic process leading to HCV infection and subsequently to the appearance of CV, the treatment of the disease may be applied at three different levels by means of etiologic, pathogenetic, and/ or symptomatic therapies.
DERMATOLOGIC MANIFESTATIONS OF HCV
Dermatologic manifestations of HCV are classified, based on the disease to be proven, are either a suspected etiology or causation. The causal manifestation of the dermatologic diseases results from direct infection of HCV in the skin, lymphocytes, dendric antigen-presenting cells, and blood vessels[188]. Whereas, the etiological manifestation of dermatological diseases results indirectly when the disruption of another organ infected or affected by HCV is associated with skin manifestations, but not specific or typical of skin responses in relation to HCV-infected or affected organ[189,190]. The causal manifestation is directly mediated by HCV infection as evidenced by the detection of HCV-RNA particles in epidermal cells[191] as well as by the induction of epiphenomena, that results mainly from the disruption of immune responses, in the skin of HCV-infected patients[192,193]. Also, the leukocytoclastic vasculitis that is due to cryoglobulinemia is considered a good evidence for a specific skin manifestation that results in a great part from the production of Ig, that is associated with rheumatoid characteristics causing an immune complex-mediated vasculitis and thereby presents a good example for etiological skin manifestation[194,195]. Such skin responses result from a wide range of causes, for example the release of thyroid hormone in early HCV-linked autoimmune thyroiditis[196,197]. Also, chronic active hepatitis that mostly leads to fibrotic liver disease in patients with chronic hepatitis C infection can lead to the development of cutaneous vascular changes such as palmar erythema or spider nevus[197,198]. Moreover, arteriovenous hemangioma, a benign acquired cutaneous vascular lesion, has been reported, in patients with chronic active hepatitis associated with HCV infection[199].
Another category of dermatologic manifestations in HCV infections includes porphyria cutanea tarda (PCT), an example for HCV-related disease in which causation is either unexplained or undeniable[200].
HCV-ASSOCIATED LICHEN PLANUS
Oral lichen planus (LP) is a chronic inflammatory condition that affects the oral mucous membranes with a variety of clinical presentations, including reticular papular, plaque-like, atrophic, and ulcerative lesions[201]. Oral LP affects about 0.1% to 4% of the population, it is a middle-aged disease that is more common among women[202]. The occurring of LP is induced by a wide range of factors including both bacterial and viral infections that thought to trigger the regulation of cell-mediated mechanisms leading the formation of oral LP lesions[203,204]. The association of LP with chronic liver disease is reported[205], and seems to be geographical dependent disease[206]. However, the risk of chronic liver disorders in LP patients appears to be f age, sex, alcohol consumption and even hepatitis B infection (HBV)-independent[207]. Nevertheless, most patients with LP and chronic liver disease are not HBV[208,209] or hepatitis G virus-infected[210-213]. Although LP is rarely associated with various hepatic conditions such as Wilson’s disease, haemochromatosis, primary sclerosing choloangitis, and α-1-antitrypsin deficiency[214,215], the association of LP with primary biliary, and HCV infection is reported in several studies[209,216-224]. Thus, HCV- associated hepatic disease may precede LP onset or may be diagnosed together with it[225].
The geographic heterogeneity in the prevalence of HCV infection is reported in patients with other HCV-related extrahepatic conditions, such as serum autoantibodies, PCT or lymphoma[225], suggesting a genetic differences among the studied populations. Indeed, HCV-related oral LP seems to be associated mainly with the HLA-DR6 allele (Nagao et al[226], 1996) and this could partially explain the particularity of the geographic heterogeneity of HCV infection- associated LP. However, the pathogenetic link between LP and HCV is not fully understood, since the molecular mimicry between the virus and host epitopes is unexpected, and the viral factors including the genotype or the viral load[226-228]. Although the histological features of lesional tissue from HCV-positive or HCV-negative patients showed no substantial differences[229,230], the presence of HCV in oral LP lesional tissue becomes object of several investigations[222,230-233]. However, the presence of replicative intermediate HCV-RNA in LP specimens provide a strong evidence for the association between HCV infection and the development of LP in HCV infected patients[222,230-233]. Although, the compartmentalization of HCV in the oral mucosa, the infection with HCV does not seem to cause direct damage to epithelial cells in oral LP[234].
HCV AND TYPE 2 DIABETES
The epidemiological link between HCV and type 2 diabetes mellitus (T2DM) is common and widely reported[235-238], and the infection with HCV increase the risk of T2DM development[239]. Although the processes of T2DM development in patients with HCV infection is not fully described, the molecular mechanisms, which are involved in the regulation of HCV-induced insulin resistance is studied in details[240-243].
Chronic infection with HCV is mostly associated with insulin resistance that subsequently leads to the development of the metabolic disease T2DM. Thus, apart from the well-investigated complications of diabetes, the appearance of insulin resistance in patients with chronic HCV infection leads mostly to the enhancement of fibrosis, and cirrhosis, and in turn to the development of HCC[244]. Besides the liver complications, patients with insulin resistance show a poor response to antiviral therapy[245].
As known, insulin is an anabolic hormone that is secreted by pancreatic β-cells. This enzyme is essential for the maintenance of glucose homeostasis[246,247].
The pathway of insulin signaling pathway is involved mainly in the regulation of different cellular processes including the activation of insulin receptor (IR), IR substrates (IRS), phosphatidylinositol-3-kinase, Akt and PKC isoforms ζ and λ[248,249]. The activation of Akt promotes storage of excess glucose as glycogen by phosphorylating glycogen synthase kinase and subsequently suppresses gluconeogenesis by inhibition of phosphoenol-pyruvate carboxykinase and glucose-6 phosphatase. Whereas, the activation of Akt is involved in the translocation of the glucose transporter GLUT4 to the plasma membrane, and subsequently the enhancement of glucose uptake[248].
The direct interference of HCV with the insulin signaling cascade is experimentally documented in several studies[250]. Also, in patients with chronic hepatitis C, direct interactions between HCV and insulin signaling components occur and may result in insulin resistance, which in turn, may progress to T2D in at-risk individuals. In the transgenic mouse model[149], the core-encoding region of HCV is sufficient to induce IR. This effect was reversed by treatment with anti-TNF-antibodies, which suggested an increased level of serine phosphorylation of IRS-1 as induced by TNF-α. Thus, the core protein may induce IR indirectly via stimulation of the secretion of TNF-α. However, in vitro models suggest a direct interaction of the core protein with the insulin signaling pathway. An increased proteasome degradation of the IRS-1 and -2 via the activation of the SOCS-3[251]. Also, a genotype-specific mechanisms, in which down-regulation of peroxisome proliferator-activated receptor γ and up-regulation of SOCS-7 was observed in cells transfected with the core protein of genotype 3[252]. A schematic view suggests HCV-mediated pathways leading to insulin resistance during the course of HCV infection is shown in Figure 8.
Figure 8.
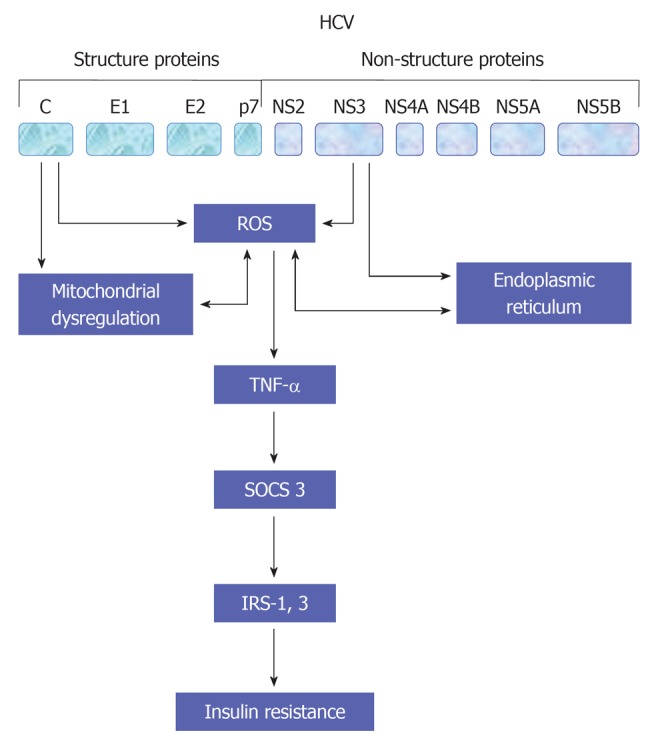
A proposed view for molecular mechanisms, which are involved in the modulation of hepatitis C virus-mediated insulin resistance during the course of hepatitis C virus infection. TNF: Tumor necrosis factor; HCV: Hepatitis C virus; SOCS: Suppressor of cytokine signaling; ROS: Reactive oxygen species; STAT: Signal transducers and activators of transcription; IRS: Insulin receptor substrates.
CONCLUSION
The replication cycle of HCV is host-dependent processes that require the intracellular signal transduction pathways of target cells to govern the nuclear factors, which are essential for the promotion of both transcriptional and translational mechanisms of the viral genome. Although the role of extracellular processes in the modulation of etiopathogenesis of HCV infection is not completely described, the role of intracellular signal transduction processes in the modulation of HCV-host interactions is established and seem to be the main actor in the regulation of HCV-associated both liver diseases and extrahepatic manifestations. The alteration of the physiological status of the intracellular signal transduction processes in response to their interaction with HCV viral proteins is thought to be responsible for the cause of the severe complications of chronic HCV infection including liver disease (e.g., hepatitis, fibrosis, cirrhosis and HCC) and extrahepatic manifestations (e.g., dermatologic, rheumatologic, neurologic, and nephrologic completions; and diabetes; arterial hypertension; autoantibodies and cryglobulins).
Thus, the knowledge obtained from the functional analysis of the intracellular signal transduction processes using an HCV artificial cellular systems may help to identify a new therapeutic target for the treatment of chronic HCV infection and diseases.
Alterations in cellular proteins and their regulation during HCV infection are clearly involved in the development and progression of HCV-associated both hepatic and extrahepatic diseases.
Both host lipid metabolic and very low-density lipoproteins (VLDL) pathways play a central role in the regulation of different viral processes including replication, assembly, secretion and entry. Thus, the association of HCV with VLDL is thought to be a virus strategy to evade host immune defense by masking the putative antigenic moieties from immune recognition. The understanding of the mechanistic details underlying the interactions between viral and host lipid metabolic pathways will help to identify potential host cell factors that may be required for HCV and the infectious processes and thereby gives the opportunity to design a potential therapeutic approach in order to eradicate HCV infection, and to decrease lipid metabolism-associated extrahepatic manifestation during the course of HCV.
The infection with HCV is more likely to favor IR in response to the accumulation of the viral proteins including core, NS3 and NS5A. However, the induction of IR by HCV infection is not merely because of glucose imbalances rather it involves upregulation of the gluconeogenic and lipogenic genes that promote glucose intolerance and progresses towards IR, a step towards HCC.
As recognized, the development of HCC is mostly associated with activation of different signaling pathways including Ras/Raf/MAP kinase, cyclin/cyclin-dependent kinase and wnt-1 pathways. The Constitutive expression of HCV viral proteins including core and NS3 results in a high basal activity of MAP kinase pathway and thereby potentiates hepatocyte transformation as well as the regulation of HGF, senescence and differentiation.
Also, HCV core protein can modulate the expression of cyclin-dependent inhibitor p21, which is considered the major target of p53 and triggers mainly the activities of cyclin/cyclin-dependent kinase complexes, which is implicated in cell-cycle control and tumor formation.
Moreover, the transcriptional upregulation of both Wnt-1 and its downstream target WISP-2 by HCV core protein suggested a possible role for Wnt-1 pathways in the modulation of HCV core and NS5A proteins in the development of HCC.
The functional analysis of JAK signal transduction pathway in the context of HCV-host interactions opened a new research option for a better understanding the mechanisms of HCV resistance to IFN-α therapy. JAK pathway is known to be the principal signaling pathway for IFN-α. Therefore, the inhibition of this pathway by viral proteins or by the reduction IFN-α receptors in response to the accumulation of both HCV structural (C and E2) and non-structural protein (NS5A) proteins may contribute to the resistant mechanisms of HCV to IFN therapy. Besides the negative effect on the virological response to IFN-α treatment, the reduction of IFN-α receptors plays a central role in the suppression of IFN-α-mediated activation of STAT1-3 and subsequently augments viral replication, inflammation and fibrosis during the course of HCV infection.
Therefore, the better understanding of the molecular mechanisms, which are involved in the regulation of virus-host cell interactions may help to develop a new therapeutic strategies. These therapeutic strategies may help to decrease or even to inhibit HCV-associated both hepatic and extrahepatic diseases, and make IFN-α therapy more effective in HCV-infected patients.
Footnotes
Peer reviewer: Chih-Ping Hsu, Assistant Professor, Department of Medical Laboratory Science and Biotechnology, Yuanpei University, No. 306 Yuanpei Street, Hsinchu 30015, Taiwan, China
S- Editor Li JY L- Editor A E- Editor Zheng XM
References
- 1.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–567. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 2.Reed KE, Rice CM. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr Top Microbiol Immunol. 2000;242:55–84. doi: 10.1007/978-3-642-59605-6_4. [DOI] [PubMed] [Google Scholar]
- 3.Alter MJ, Kruszon-Moran D, Nainan OV, McQuillan GM, Gao F, Moyer LA, Kaslow RA, Margolis HS. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med. 1999;341:556–562. doi: 10.1056/NEJM199908193410802. [DOI] [PubMed] [Google Scholar]
- 4.Nasir A, Todd CS, Stanekzai MR, Bautista CT, Botros BA, Scott PT, Strathdee SA, Tjaden J. Prevalence of HIV, hepatitis B and hepatitis C and associated risk behaviours amongst injecting drug users in three Afghan cities. Int J Drug Policy. 2011;22:145–152. doi: 10.1016/j.drugpo.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quer J, Murillo P, Esteban JI, Martell M, Esteban R, Guardia J. Sexual transmission of hepatitis C virus from a patient with chronic disease to his sex partner after removal of an intrauterine device. Sex Transm Dis. 2003;30:470–471. doi: 10.1097/00007435-200305000-00015. [DOI] [PubMed] [Google Scholar]
- 6.Cacoub P, Poynard T, Ghillani P, Charlotte F, Olivi M, Piette JC, Opolon P. Extrahepatic manifestations of chronic hepatitis C. MULTIVIRC Group. Multidepartment Virus C. Arthritis Rheum. 1999;42:2204–2212. doi: 10.1002/1529-0131(199910)42:10<2204::AID-ANR24>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 7.Cacoub P, Renou C, Rosenthal E, Cohen P, Loury I, Loustaud-Ratti V, Yamamoto AM, Camproux AC, Hausfater P, Musset L, et al. Extrahepatic manifestations associated with hepatitis C virus infection. A prospective multicenter study of 321 patients. The GERMIVIC. Groupe d'Etude et de Recherche en Medecine Interne et Maladies Infectieuses sur le Virus de l'Hepatite C. Medicine (Baltimore) 2000;79:47–56. doi: 10.1097/00005792-200001000-00005. [DOI] [PubMed] [Google Scholar]
- 8.Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann Intern Med. 2000;132:296–305. doi: 10.7326/0003-4819-132-4-200002150-00008. [DOI] [PubMed] [Google Scholar]
- 9.Andreone P, Gramenzi A, Cursaro C, Bernardi M, Zignego AL. Monoclonal gammopathy in patients with chronic hepatitis C virus infection. Blood. 1996;88:1122. [PubMed] [Google Scholar]
- 10.Zignego AL, Macchia D, Monti M, Thiers V, Mazzetti M, Foschi M, Maggi E, Romagnani S, Gentilini P, Bréchot C. Infection of peripheral mononuclear blood cells by hepatitis C virus. J Hepatol. 1992;15:382–386. doi: 10.1016/0168-8278(92)90073-x. [DOI] [PubMed] [Google Scholar]
- 11.Pawlotsky JM. Therapy of hepatitis C: from empiricism to eradication. Hepatology. 2006;43:S207–S220. doi: 10.1002/hep.21064. [DOI] [PubMed] [Google Scholar]
- 12.Gao L, Aizaki H, He JW, Lai MM. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J Virol. 2004;78:3480–3488. doi: 10.1128/JVI.78.7.3480-3488.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yi M, Lemon SM. 3' nontranslated RNA signals required for replication of hepatitis C virus RNA. J Virol. 2003;77:3557–3568. doi: 10.1128/JVI.77.6.3557-3568.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friebe P, Lohmann V, Krieger N, Bartenschlager R. Sequences in the 5' nontranslated region of hepatitis C virus required for RNA replication. J Virol. 2001;75:12047–12057. doi: 10.1128/JVI.75.24.12047-12057.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simmonds P. The origin and evolution of hepatitis viruses in humans. J Gen Virol. 2001;82:693–712. doi: 10.1099/0022-1317-82-4-693. [DOI] [PubMed] [Google Scholar]
- 16.Simmonds P. Virology of hepatitis C virus. Clin Ther. 1996;18 Suppl B:9–36. doi: 10.1016/S0149-2918(96)80193-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yasui K, Wakita T, Tsukiyama-Kohara K, Funahashi SI, Ichikawa M, Kajita T, Moradpour D, Wands JR, Kohara M. The native form and maturation process of hepatitis C virus core protein. J Virol. 1998;72:6048–6055. doi: 10.1128/jvi.72.7.6048-6055.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ray RB, Lagging LM, Meyer K, Steele R, Ray R. Transcriptional regulation of cellular and viral promoters by the hepatitis C virus core protein. Virus Res. 1995;37:209–220. doi: 10.1016/0168-1702(95)00034-n. [DOI] [PubMed] [Google Scholar]
- 19.Harada S, Watanabe Y, Takeuchi K, Suzuki T, Katayama T, Takebe Y, Saito I, Miyamura T. Expression of processed core protein of hepatitis C virus in mammalian cells. J Virol. 1991;65:3015–3021. doi: 10.1128/jvi.65.6.3015-3021.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ray RB, Ray R. Hepatitis C virus core protein: intriguing properties and functional relevance. FEMS Microbiol Lett. 2001;202:149–156. doi: 10.1111/j.1574-6968.2001.tb10796.x. [DOI] [PubMed] [Google Scholar]
- 21.Meyer K, Beyene A, Bowlin TL, Basu A, Ray R. Coexpression of hepatitis C virus E1 and E2 chimeric envelope glycoproteins displays separable ligand sensitivity and increases pseudotype infectious titer. J Virol. 2004;78:12838–12847. doi: 10.1128/JVI.78.23.12838-12847.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer K, Basu A, Ray R. Functional features of hepatitis C virus glycoproteins for pseudotype virus entry into mammalian cells. Virology. 2000;276:214–226. doi: 10.1006/viro.2000.0547. [DOI] [PubMed] [Google Scholar]
- 23.Griffin SD, Beales LP, Clarke DS, Worsfold O, Evans SD, Jaeger J, Harris MP, Rowlands DJ. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 2003;535:34–38. doi: 10.1016/s0014-5793(02)03851-6. [DOI] [PubMed] [Google Scholar]
- 24.Yi M, Ma Y, Yates J, Lemon SM. Trans-complementation of an NS2 defect in a late step in hepatitis C virus (HCV) particle assembly and maturation. PLoS Pathog. 2009;5:e1000403. doi: 10.1371/journal.ppat.1000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lohmann V, Roos A, Körner F, Koch JO, Bartenschlager R. Biochemical and kinetic analyses of NS5B RNA-dependent RNA polymerase of the hepatitis C virus. Virology. 1998;249:108–118. doi: 10.1006/viro.1998.9311. [DOI] [PubMed] [Google Scholar]
- 26.Tomei L, Failla C, Vitale RL, Bianchi E, De Francesco R. A central hydrophobic domain of the hepatitis C virus NS4A protein is necessary and sufficient for the activation of the NS3 protease. J Gen Virol. 1996;77(Pt 5):1065–1070. doi: 10.1099/0022-1317-77-5-1065. [DOI] [PubMed] [Google Scholar]
- 27.Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, Moradpour D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol. 2003;77:5487–5492. doi: 10.1128/JVI.77.9.5487-5492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzich JA, Tamura JK, Palmer-Hill F, Warrener P, Grakoui A, Rice CM, Feinstone SM, Collett MS. Hepatitis C virus NS3 protein polynucleotide-stimulated nucleoside triphosphatase and comparison with the related pestivirus and flavivirus enzymes. J Virol. 1993;67:6152–6158. doi: 10.1128/jvi.67.10.6152-6158.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhattacherjee V, Prescott LE, Pike I, Rodgers B, Bell H, El-Zayadi AR, Kew MC, Conradie J, Lin CK, Marsden H. Use of NS-4 peptides to identify type-specific antibody to hepatitis C virus genotypes 1, 2, 3, 4, 5 and 6. J Gen Virol. 1995;76(Pt 7):1737–1748. doi: 10.1099/0022-1317-76-7-1737. [DOI] [PubMed] [Google Scholar]
- 30.Hügle T, Fehrmann F, Bieck E, Kohara M, Kräusslich HG, Rice CM, Blum HE, Moradpour D. The hepatitis C virus nonstructural protein 4B is an integral endoplasmic reticulum membrane protein. Virology. 2001;284:70–81. doi: 10.1006/viro.2001.0873. [DOI] [PubMed] [Google Scholar]
- 31.Gao B, Hong F, Radaeva S. Host factors and failure of interferon-alpha treatment in hepatitis C virus. Hepatology. 2004;39:880–890. doi: 10.1002/hep.20139. [DOI] [PubMed] [Google Scholar]
- 32.Izumi N, Enomoto N, Uchihara M, Murakami T, Ono K, Noguchi O, Miyake S, Nouchi T, Fujisawa K, Marumo F, et al. Hepatic iron contents and response to interferon-alpha in patients with chronic hepatitis C. Relationship to genotypes of hepatitis C virus. Dig Dis Sci. 1996;41:989–994. doi: 10.1007/BF02091542. [DOI] [PubMed] [Google Scholar]
- 33.Gale MJ, Korth MJ, Tang NM, Tan SL, Hopkins DA, Dever TE, Polyak SJ, Gretch DR, Katze MG. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 34.Appel N, Zayas M, Miller S, Krijnse-Locker J, Schaller T, Friebe P, Kallis S, Engel U, Bartenschlager R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008;4:e1000035. doi: 10.1371/journal.ppat.1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Behrens SE, Tomei L, De Francesco R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1996;15:12–22. [PMC free article] [PubMed] [Google Scholar]
- 36.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 38.Cai Z, Zhang C, Chang KS, Jiang J, Ahn BC, Wakita T, Liang TJ, Luo G. Robust production of infectious hepatitis C virus (HCV) from stably HCV cDNA-transfected human hepatoma cells. J Virol. 2005;79:13963–13973. doi: 10.1128/JVI.79.22.13963-13973.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heller T, Saito S, Auerbach J, Williams T, Moreen TR, Jazwinski A, Cruz B, Jeurkar N, Sapp R, Luo G, et al. An in vitro model of hepatitis C virion production. Proc Natl Acad Sci USA. 2005;102:2579–2583. doi: 10.1073/pnas.0409666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ait-Goughoulte M, Kanda T, Meyer K, Ryerse JS, Ray RB, Ray R. Hepatitis C virus genotype 1a growth and induction of autophagy. J Virol. 2008;82:2241–2249. doi: 10.1128/JVI.02093-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sir D, Liang C, Chen WL, Jung JU, Ou JH. Perturbation of autophagic pathway by hepatitis C virus. Autophagy. 2008;4:830–831. doi: 10.4161/auto.6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dreux M, Chisari FV. Autophagy proteins promote hepatitis C virus replication. Autophagy. 2009;5:1224–1225. doi: 10.4161/auto.5.8.10219. [DOI] [PubMed] [Google Scholar]
- 43.Tanida I, Fukasawa M, Ueno T, Kominami E, Wakita T, Hanada K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy. 2009;5:937–945. doi: 10.4161/auto.5.7.9243. [DOI] [PubMed] [Google Scholar]
- 44.Cornelissen C, Lüscher-Firzlaff J, Baron JM, Lüscher B. Signaling by IL-31 and functional consequences. Eur J Cell Biol. 2012;91:552–566. doi: 10.1016/j.ejcb.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 45.Bauvois B. New facets of matrix metalloproteinases MMP-2 and MMP-9 as cell surface transducers: outside-in signaling and relationship to tumor progression. Biochim Biophys Acta. 2012;1825:29–36. doi: 10.1016/j.bbcan.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 46.Jin J, Samuvel DJ, Zhang X, Li Y, Lu Z, Lopes-Virella MF, Huang Y. Coactivation of TLR4 and TLR2/6 coordinates an additive augmentation on IL-6 gene transcription via p38MAPK pathway in U937 mononuclear cells. Mol Immunol. 2011;49:423–432. doi: 10.1016/j.molimm.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hassan M, Selimovic D, Ghozlan H, Abdel-kader O. Hepatitis C virus core protein triggers hepatic angiogenesis by a mechanism including multiple pathways. Hepatology. 2009;49:1469–1482. doi: 10.1002/hep.22849. [DOI] [PubMed] [Google Scholar]
- 48.Ashfaq UA, Javed T, Rehman S, Nawaz Z, Riazuddin S. An overview of HCV molecular biology, replication and immune responses. Virol J. 2011;8:161. doi: 10.1186/1743-422X-8-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pei R, Chen H, Lu L, Zhu W, Beckebaum S, Cicinnati V, Lu M, Chen X. Hepatitis C virus infection induces the expression of amphiregulin, a factor related to the activation of cellular survival pathways and required for efficient viral assembly. J Gen Virol. 2011;92:2237–2248. doi: 10.1099/vir.0.032581-0. [DOI] [PubMed] [Google Scholar]
- 50.Martínez-Palacián A, del Castillo G, Herrera B, Fernández M, Roncero C, Fabregat I, Sánchez A. EGFR is dispensable for c-Met-mediated proliferation and survival activities in mouse adult liver oval cells. Cell Signal. 2012;24:505–513. doi: 10.1016/j.cellsig.2011.09.031. [DOI] [PubMed] [Google Scholar]
- 51.Minjgee M, Toulany M, Kehlbach R, Giehl K, Rodemann HP. K-RAS(V12) induces autocrine production of EGFR ligands and mediates radioresistance through EGFR-dependent Akt signaling and activation of DNA-PKcs. Int J Radiat Oncol Biol Phys. 2011;81:1506–1514. doi: 10.1016/j.ijrobp.2011.05.057. [DOI] [PubMed] [Google Scholar]
- 52.Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signalling pathway in cancer. Eur J Cancer. 2010;46:1260–1270. doi: 10.1016/j.ejca.2010.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selimovic D, Hassan M, Haikel Y, Hengge UR. Taxol-induced mitochondrial stress in melanoma cells is mediated by activation of c-Jun N-terminal kinase (JNK) and p38 pathways via uncoupling protein 2. Cell Signal. 2008;20:311–322. doi: 10.1016/j.cellsig.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 54.Hassan M, Ghozlan H, Abdel-Kader O. Activation of c-Jun NH2-terminal kinase (JNK) signaling pathway is essential for the stimulation of hepatitis C virus (HCV) non-structural protein 3 (NS3)-mediated cell growth. Virology. 2005;333:324–336. doi: 10.1016/j.virol.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 55.Erhardt A, Hassan M, Heintges T, Häussinger D. Hepatitis C virus core protein induces cell proliferation and activates ERK, JNK, and p38 MAP kinases together with the MAP kinase phosphatase MKP-1 in a HepG2 Tet-Off cell line. Virology. 2002;292:272–284. doi: 10.1006/viro.2001.1227. [DOI] [PubMed] [Google Scholar]
- 56.Stuart KA, Riordan SM, Lidder S, Crostella L, Williams R, Skouteris GG. Hepatocyte growth factor/scatter factor-induced intracellular signalling. Int J Exp Pathol. 2000;81:17–30. doi: 10.1046/j.1365-2613.2000.00138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cetindere T, Nambiar S, Santourlidis S, Essmann F, Hassan M. Induction of indoleamine 2, 3-dioxygenase by death receptor activation contributes to apoptosis of melanoma cells via mitochondrial damage-dependent ROS accumulation. Cell Signal. 2010;22:197–211. doi: 10.1016/j.cellsig.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 58.Hassan M, Alaoui A, Feyen O, Mirmohammadsadegh A, Essmann F, Tannapfel A, Gulbins E, Schulze-Osthoff K, Hengge UR. The BH3-only member Noxa causes apoptosis in melanoma cells by multiple pathways. Oncogene. 2008;27:4557–4568. doi: 10.1038/onc.2008.90. [DOI] [PubMed] [Google Scholar]
- 59.Hassan M, Feyen O, Grinstein E. Fas-induced apoptosis of renal cell carcinoma is mediated by apoptosis signal-regulating kinase 1 via mitochondrial damage-dependent caspase-8 activation. Cell Oncol. 2009;31:437–456. doi: 10.3233/CLO-2009-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hassan M, Selimovic D, Ghozlan H, Abdel-Kader O. Induction of high-molecular-weight (HMW) tumor necrosis factor(TNF) alpha by hepatitis C virus (HCV) non-structural protein 3 (NS3) in liver cells is AP-1 and NF-kappaB-dependent activation. Cell Signal. 2007;19:301–311. doi: 10.1016/j.cellsig.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 61.Selimovic D, Ahmad M, El-Khattouti A, Hannig M, Haïkel Y, Hassan M. Apoptosis-related protein-2 triggers melanoma cell death by a mechanism including both endoplasmic reticulum stress and mitochondrial dysregulation. Carcinogenesis. 2011;32:1268–1278. doi: 10.1093/carcin/bgr112. [DOI] [PubMed] [Google Scholar]
- 62.Selimovic D, Sprenger A, Hannig M, Haïkel Y, Hassan M. Apoptosis related protein-1 triggers melanoma cell death via interaction with the juxtamembrane region of p75 neurotrophin receptor. J Cell Mol Med. 2012;16:349–361. doi: 10.1111/j.1582-4934.2011.01304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou X, Fang Y, Jing H, Zhong L, Luo P, Song H, Yang B, He Q. Involvement of mitogen-activated protein kinase in signal transducer and activator of transcription-1 mediated differentiation induced by bortezomib in acute myeloid leukemia cells. Mol Carcinog. 2011:Epub ahead of print. doi: 10.1002/mc.20873. [DOI] [PubMed] [Google Scholar]
- 64.Tournier C, Dong C, Turner TK, Jones SN, Flavell RA, Davis RJ. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 2001;15:1419–1426. doi: 10.1101/gad.888501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Won M, Park KA, Byun HS, Kim YR, Choi BL, Hong JH, Park J, Seok JH, Lee YH, Cho CH, et al. Protein kinase SGK1 enhances MEK/ERK complex formation through the phosphorylation of ERK2: implication for the positive regulatory role of SGK1 on the ERK function during liver regeneration. J Hepatol. 2009;51:67–76. doi: 10.1016/j.jhep.2009.02.027. [DOI] [PubMed] [Google Scholar]
- 66.Talarmin H, Rescan C, Cariou S, Glaise D, Zanninelli G, Bilodeau M, Loyer P, Guguen-Guillouzo C, Baffet G. The mitogen-activated protein kinase kinase/extracellular signal-regulated kinase cascade activation is a key signalling pathway involved in the regulation of G(1) phase progression in proliferating hepatocytes. Mol Cell Biol. 1999;19:6003–6011. doi: 10.1128/mcb.19.9.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Campbell JS, Argast GM, Yuen SY, Hayes B, Fausto N. Inactivation of p38 MAPK during liver regeneration. Int J Biochem Cell Biol. 2011;43:180–188. doi: 10.1016/j.biocel.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stepniak E, Ricci R, Eferl R, Sumara G, Sumara I, Rath M, Hui L, Wagner EF. c-Jun/AP-1 controls liver regeneration by repressing p53/p21 and p38 MAPK activity. Genes Dev. 2006;20:2306–2314. doi: 10.1101/gad.390506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Diehl AM, Yin M, Fleckenstein J, Yang SQ, Lin HZ, Brenner DA, Westwick J, Bagby G, Nelson S. Tumor necrosis factor-alpha induces c-jun during the regenerative response to liver injury. Am J Physiol. 1994;267:G552–G561. doi: 10.1152/ajpgi.1994.267.4.G552. [DOI] [PubMed] [Google Scholar]
- 70.Spaziani A, Alisi A, Sanna D, Balsano C. Role of p38 MAPK and RNA-dependent protein kinase (PKR) in hepatitis C virus core-dependent nuclear delocalization of cyclin B1. J Biol Chem. 2006;281:10983–10989. doi: 10.1074/jbc.M512536200. [DOI] [PubMed] [Google Scholar]
- 71.Li W, Liang X, Kellendonk C, Poli V, Taub R. STAT3 contributes to the mitogenic response of hepatocytes during liver regeneration. J Biol Chem. 2002;277:28411–28417. doi: 10.1074/jbc.M202807200. [DOI] [PubMed] [Google Scholar]
- 72.Feng DY, Sun Y, Cheng RX, Ouyang XM, Zheng H. Effect of hepatitis C virus nonstructural protein NS3 on proliferation and MAPK phosphorylation of normal hepatocyte line. World J Gastroenterol. 2005;11:2157–2161. doi: 10.3748/wjg.v11.i14.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Binamé F, Lassus P, Hibner U. Transforming growth factor beta controls the directional migration of hepatocyte cohorts by modulating their adhesion to fibronectin. Mol Biol Cell. 2008;19:945–956. doi: 10.1091/mbc.E07-09-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schuster N, Krieglstein K. Mechanisms of TGF-beta-mediated apoptosis. Cell Tissue Res. 2002;307:1–14. doi: 10.1007/s00441-001-0479-6. [DOI] [PubMed] [Google Scholar]
- 75.Thenappan A, Shukla V, Abdul Khalek FJ, Li Y, Shetty K, Liu P, Li L, Johnson RL, Johnson L, Mishra L. Loss of transforming growth factor β adaptor protein β-2 spectrin leads to delayed liver regeneration in mice. Hepatology. 2011;53:1641–1650. doi: 10.1002/hep.24111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Torre C, Benhamouche S, Mitchell C, Godard C, Veber P, Letourneur F, Cagnard N, Jacques S, Finzi L, Perret C, et al. The transforming growth factor-α and cyclin D1 genes are direct targets of β-catenin signaling in hepatocyte proliferation. J Hepatol. 2011;55:86–95. doi: 10.1016/j.jhep.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 77.Herrera B, Alvarez AM, Beltrán J, Valdés F, Fabregat I, Fernández M. Resistance to TGF-beta-induced apoptosis in regenerating hepatocytes. J Cell Physiol. 2004;201:385–392. doi: 10.1002/jcp.20078. [DOI] [PubMed] [Google Scholar]
- 78.Weidenaar AC, ter Elst A, Kampen KR, Meeuwsen-de Boer TG, de Jonge HJ, Scherpen FJ, den Dunnen WF, Kamps WA, de Bont ES. Stromal interaction essential for vascular endothelial growth factor A-induced tumour growth via transforming growth factor-β signalling. Br J Cancer. 2011;105:1856–1863. doi: 10.1038/bjc.2011.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fransvea E, Mazzocca A, Santamato A, Azzariti A, Antonaci S, Giannelli G. Kinase activation profile associated with TGF-β-dependent migration of HCC cells: a preclinical study. Cancer Chemother Pharmacol. 2011;68:79–86. doi: 10.1007/s00280-010-1459-x. [DOI] [PubMed] [Google Scholar]
- 80.Flisiak R, Pytel-Krolczuk B, Prokopowicz D. Circulating transforming growth factor beta(1) as an indicator of hepatic function impairment in liver cirrhosis. Cytokine. 2000;12:677–681. doi: 10.1006/cyto.1999.0660. [DOI] [PubMed] [Google Scholar]
- 81.Talaat RM. Soluble angiogenesis factors in sera of Egyptian patients with hepatitis C virus infection: correlation with disease severity. Viral Immunol. 2010;23:151–157. doi: 10.1089/vim.2009.0089. [DOI] [PubMed] [Google Scholar]
- 82.Flisiak R, Jaroszewicz J, Lapinski TW, Flisiak I, Prokopowiczi D. Effect of pegylated interferon alpha 2b plus ribavirin treatment on plasma transforming growth factor-beta1, metalloproteinase-1, and tissue metalloproteinase inhibitor-1 in patients with chronic hepatitis C. World J Gastroenterol. 2005;11:6833–6838. doi: 10.3748/wjg.v11.i43.6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Choi SH, Hwang SB. Modulation of the transforming growth factor-beta signal transduction pathway by hepatitis C virus nonstructural 5A protein. J Biol Chem. 2006;281:7468–7478. doi: 10.1074/jbc.M512438200. [DOI] [PubMed] [Google Scholar]
- 84.Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science. 2003;299:574–577. doi: 10.1126/science.1075994. [DOI] [PubMed] [Google Scholar]
- 85.Mishra L, Derynck R, Mishra B. Transforming growth factor-beta signaling in stem cells and cancer. Science. 2005;310:68–71. doi: 10.1126/science.1118389. [DOI] [PubMed] [Google Scholar]
- 86.Massagué J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 87.Chang H, Brown CW, Matzuk MM. Genetic analysis of the mammalian transforming growth factor-beta superfamily. Endocr Rev. 2002;23:787–823. doi: 10.1210/er.2002-0003. [DOI] [PubMed] [Google Scholar]
- 88.Ikegami T. Transforming growth factor-beta signaling and liver cancer stem cell. Hepatol Res. 2009;39:847–849. doi: 10.1111/j.1872-034X.2009.00586.x. [DOI] [PubMed] [Google Scholar]
- 89.Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25:3787–3800. doi: 10.1038/sj.onc.1209556. [DOI] [PubMed] [Google Scholar]
- 90.Yakicier MC, Irmak MB, Romano A, Kew M, Ozturk M. Smad2 and Smad4 gene mutations in hepatocellular carcinoma. Oncogene. 1999;18:4879–4883. doi: 10.1038/sj.onc.1202866. [DOI] [PubMed] [Google Scholar]
- 91.Longerich T, Breuhahn K, Odenthal M, Petmecky K, Schirmacher P. Factors of transforming growth factor beta signalling are co-regulated in human hepatocellular carcinoma. Virchows Arch. 2004;445:589–596. doi: 10.1007/s00428-004-1118-x. [DOI] [PubMed] [Google Scholar]
- 92.Kim HG, Chung YH, Song BC, Kim J, Yang SH, Lee YS, Suh DJ. Expression of transforming growth factor beta-1 in chronic hepatitis and hepatocellular carcinoma associated with hepatitis C virus infection. Korean J Intern Med. 2000;15:165–170. doi: 10.3904/kjim.2000.15.3.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tsai JF, Jeng JE, Chuang LY, Yang ML, Ho MS, Chang WY, Hsieh MY, Lin ZY, Tsai JH. Elevated urinary transforming growth factor-beta1 level as a tumour marker and predictor of poor survival in cirrhotic hepatocellular carcinoma. Br J Cancer. 1997;76:244–250. doi: 10.1038/bjc.1997.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lu Y, Wu LQ, Li CS, Wang SG, Han B. Expression of transforming growth factors in hepatocellular carcinoma and its relations with clinicopathological parameters and prognosis. Hepatobiliary Pancreat Dis Int. 2008;7:174–178. [PubMed] [Google Scholar]
- 95.Ikeguchi M, Iwamoto A, Taniguchi K, Katano K, Hirooka Y. The gene expression level of transforming growth factor-beta (TGF-beta) as a biological prognostic marker of hepatocellular carcinoma. J Exp Clin Cancer Res. 2005;24:415–421. [PubMed] [Google Scholar]
- 96.Lgssiar A, Hassan M, Schott-Ohly P, Friesen N, Nicoletti F, Trepicchio WL, Gleichmann H. Interleukin-11 inhibits NF-kappaB and AP-1 activation in islets and prevents diabetes induced with streptozotocin in mice. Exp Biol Med (Maywood) 2004;229:425–436. doi: 10.1177/153537020422900511. [DOI] [PubMed] [Google Scholar]
- 97.Nagao Y, Tanaka J, Nakanishi T, Moriya T, Katayama K, Kumagai J, Komiya Y, Itoh Y, Myoken Y, Fujihara M, et al. High incidence of extrahepatic manifestations in an HCV hyperendemic area. Hepatol Res. 2002;22:27–36. doi: 10.1016/s1386-6346(01)00114-0. [DOI] [PubMed] [Google Scholar]
- 98.Dabrowska MM, Panasiuk A, Flisiak R. Signal transduction pathways in liver and the influence of hepatitis C virus infection on their activities. World J Gastroenterol. 2009;15:2184–2189. doi: 10.3748/wjg.15.2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Georgiadou M, Notas G, Xidakis C, Drygiannakis I, Sfakianaki O, Klironomos S, Valatas V, Kouroumalis E. TNF receptors in Kupffer cells. J Recept Signal Transduct Res. 2011;31:291–298. doi: 10.3109/10799893.2011.586354. [DOI] [PubMed] [Google Scholar]
- 100.Tarrats N, Moles A, Morales A, García-Ruiz C, Fernández-Checa JC, Marí M. Critical role of tumor necrosis factor receptor 1, but not 2, in hepatic stellate cell proliferation, extracellular matrix remodeling, and liver fibrogenesis. Hepatology. 2011;54:319–327. doi: 10.1002/hep.24388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Eum HA, Vallabhaneni R, Wang Y, Loughran PA, Stolz DB, Billiar TR. Characterization of DISC formation and TNFR1 translocation to mitochondria in TNF-α-treated hepatocytes. Am J Pathol. 2011;179:1221–1229. doi: 10.1016/j.ajpath.2011.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yoon JH, Gores GJ. Death receptor-mediated apoptosis and the liver. J Hepatol. 2002;37:400–410. doi: 10.1016/s0168-8278(02)00209-x. [DOI] [PubMed] [Google Scholar]
- 103.Wajant H, Scheurich P. Tumor necrosis factor receptor-associated factor (TRAF) 2 and its role in TNF signaling. Int J Biochem Cell Biol. 2001;33:19–32. doi: 10.1016/s1357-2725(00)00064-9. [DOI] [PubMed] [Google Scholar]
- 104.Stamataki Z. Hepatitis C infection of B lymphocytes: more tools to address pending questions. Expert Rev Anti Infect Ther. 2010;8:977–980. doi: 10.1586/eri.10.86. [DOI] [PubMed] [Google Scholar]
- 105.Beg AA, Finco TS, Nantermet PV, Baldwin AS. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of I kappa B alpha: a mechanism for NF-kappa B activation. Mol Cell Biol. 1993;13:3301–3310. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kato N, Yoshida H, Ono-Nita SK, Kato J, Goto T, Otsuka M, Lan K, Matsushima K, Shiratori Y, Omata M. Activation of intracellular signaling by hepatitis B and C viruses: C-viral core is the most potent signal inducer. Hepatology. 2000;32:405–412. doi: 10.1053/jhep.2000.9198. [DOI] [PubMed] [Google Scholar]
- 107.Ait-Goughoulte M, Banerjee A, Meyer K, Mazumdar B, Saito K, Ray RB, Ray R. Hepatitis C virus core protein interacts with fibrinogen-beta and attenuates cytokine stimulated acute-phase response. Hepatology. 2010;51:1505–1513. doi: 10.1002/hep.23502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tilg H, Wilmer A, Vogel W, Herold M, Nölchen B, Judmaier G, Huber C. Serum levels of cytokines in chronic liver diseases. Gastroenterology. 1992;103:264–274. doi: 10.1016/0016-5085(92)91122-k. [DOI] [PubMed] [Google Scholar]
- 109.Hassan M, Ghozlan H, Abdel-Kader O. Activation of RB/E2F signaling pathway is required for the modulation of hepatitis C virus core protein-induced cell growth in liver and non-liver cells. Cell Signal. 2004;16:1375–1385. doi: 10.1016/j.cellsig.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 110.Selimović D, Hassan M. Inhibition of hepatitis C virus (HCV) core protein- induced cell growth by non-structural protein 4A (NS4A) is mediated by mitochondrial dysregulation. Bosn J Basic Med Sci. 2008;8:4–11. doi: 10.17305/bjbms.2008.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hirota T, Irie K, Okamoto R, Ikeda W, Takai Y. Transcriptional activation of the mouse Necl-5/Tage4/PVR/CD155 gene by fibroblast growth factor or oncogenic Ras through the Raf-MEK-ERK-AP-1 pathway. Oncogene. 2005;24:2229–2235. doi: 10.1038/sj.onc.1208409. [DOI] [PubMed] [Google Scholar]
- 112.Andrade LJ, D'Oliveira A, Silva CA, Nunes P, França LS, Malta AM, Paraná R. A meta-analysis of patients with chronic hepatitis C treated with interferon-alpha to determine the risk of autoimmune thyroiditis. Acta Gastroenterol Latinoam. 2011;41:104–110. [PubMed] [Google Scholar]
- 113.Chung KC, Kim SM, Rhang S, Lau LF, Gomes I, Ahn YS. Expression of immediate early gene pip92 during anisomycin-induced cell death is mediated by the JNK- and p38-dependent activation of Elk1. Eur J Biochem. 2000;267:4676–4684. doi: 10.1046/j.1432-1327.2000.01517.x. [DOI] [PubMed] [Google Scholar]
- 114.Reindl KM, Kittilson JD, Bergan HE, Sheridan MA. Growth hormone-stimulated insulin-like growth factor-1 expression in rainbow trout (Oncorhynchus mykiss) hepatocytes is mediated by ERK, PI3K-AKT, and JAK-STAT. Am J Physiol Regul Integr Comp Physiol. 2011;301:R236–R243. doi: 10.1152/ajpregu.00414.2010. [DOI] [PubMed] [Google Scholar]
- 115.Hazari S, Chandra PK, Poat B, Datta S, Garry RF, Foster TP, Kousoulas G, Wakita T, Dash S. Impaired antiviral activity of interferon alpha against hepatitis C virus 2a in Huh-7 cells with a defective Jak-Stat pathway. Virol J. 2010;7:36. doi: 10.1186/1743-422X-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tamiya T, Kashiwagi I, Takahashi R, Yasukawa H, Yoshimura A. Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler Thromb Vasc Biol. 2011;31:980–985. doi: 10.1161/ATVBAHA.110.207464. [DOI] [PubMed] [Google Scholar]
- 117.Hamdorf M, Berger A, Schüle S, Reinhardt J, Flory E. PKCδ-induced PU.1 phosphorylation promotes hematopoietic stem cell differentiation to dendritic cells. Stem Cells. 2011;29:297–306. doi: 10.1002/stem.564. [DOI] [PubMed] [Google Scholar]
- 118.Medves S, Noël LA, Montano-Almendras CP, Albu RI, Schoemans H, Constantinescu SN, Demoulin JB. Multiple oligomerization domains of KANK1-PDGFRβ are required for JAK2-independent hematopoietic cell proliferation and signaling via STAT5 and ERK. Haematologica. 2011;96:1406–1414. doi: 10.3324/haematol.2011.040147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Imamura O, Satoh Y, Endo S, Takishima K. Analysis of extracellular signal-regulated kinase 2 function in neural stem/progenitor cells via nervous system-specific gene disruption. Stem Cells. 2008;26:3247–3256. doi: 10.1634/stemcells.2008-0578. [DOI] [PubMed] [Google Scholar]
- 120.Bauer JH, Liu KD, You Y, Lai SY, Goldsmith MA. Heteromerization of the gammac chain with the interleukin-9 receptor alpha subunit leads to STAT activation and prevention of apoptosis. J Biol Chem. 1998;273:9255–9260. doi: 10.1074/jbc.273.15.9255. [DOI] [PubMed] [Google Scholar]
- 121.Dimitriou ID, Clemenza L, Scotter AJ, Chen G, Guerra FM, Rottapel R. Putting out the fire: coordinated suppression of the innate and adaptive immune systems by SOCS1 and SOCS3 proteins. Immunol Rev. 2008;224:265–283. doi: 10.1111/j.1600-065X.2008.00659.x. [DOI] [PubMed] [Google Scholar]
- 122.Krebs DL, Hilton DJ. SOCS: physiological suppressors of cytokine signaling. J Cell Sci. 2000;113(Pt 16):2813–2819. doi: 10.1242/jcs.113.16.2813. [DOI] [PubMed] [Google Scholar]
- 123.Heim MH. Intracellular signalling and antiviral effects of interferons. Dig Liver Dis. 2000;32:257–263. doi: 10.1016/s1590-8658(00)80831-2. [DOI] [PubMed] [Google Scholar]
- 124.Sun R, Gao B. Negative regulation of liver regeneration by innate immunity (natural killer cells/interferon-gamma) Gastroenterology. 2004;127:1525–1539. doi: 10.1053/j.gastro.2004.08.055. [DOI] [PubMed] [Google Scholar]
- 125.Zhu H, Shang X, Terada N, Liu C. STAT3 induces anti-hepatitis C viral activity in liver cells. Biochem Biophys Res Commun. 2004;324:518–528. doi: 10.1016/j.bbrc.2004.09.081. [DOI] [PubMed] [Google Scholar]
- 126.Bode JG, Ludwig S, Ehrhardt C, Albrecht U, Erhardt A, Schaper F, Heinrich PC, Häussinger D. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J. 2003;17:488–490. doi: 10.1096/fj.02-0664fje. [DOI] [PubMed] [Google Scholar]
- 127.Basu A, Meyer K, Ray RB, Ray R. Hepatitis C virus core protein modulates the interferon-induced transacting factors of Jak/Stat signaling pathway but does not affect the activation of downstream IRF-1 or 561 gene. Virology. 2001;288:379–390. doi: 10.1006/viro.2001.1100. [DOI] [PubMed] [Google Scholar]
- 128.Polyak SJ, Khabar KS, Paschal DM, Ezelle HJ, Duverlie G, Barber GN, Levy DE, Mukaida N, Gretch DR. Hepatitis C virus nonstructural 5A protein induces interleukin-8, leading to partial inhibition of the interferon-induced antiviral response. J Virol. 2001;75:6095–6106. doi: 10.1128/JVI.75.13.6095-6106.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Larrea E, Aldabe R, Molano E, Fernandez-Rodriguez CM, Ametzazurra A, Civeira MP, Prieto J. Altered expression and activation of signal transducers and activators of transcription (STATs) in hepatitis C virus infection: in vivo and in vitro studies. Gut. 2006;55:1188–1196. doi: 10.1136/gut.2005.070060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wurster AL, Tanaka T, Grusby MJ. The biology of Stat4 and Stat6. Oncogene. 2000;19:2577–2584. doi: 10.1038/sj.onc.1203485. [DOI] [PubMed] [Google Scholar]
- 131.Blaas L, Kornfeld JW, Schramek D, Musteanu M, Zollner G, Gumhold J, van Zijl F, Schneller D, Esterbauer H, Egger G, et al. Disruption of the growth hormone--signal transducer and activator of transcription 5--insulinlike growth factor 1 axis severely aggravates liver fibrosis in a mouse model of cholestasis. Hepatology. 2010;51:1319–1326. doi: 10.1002/hep.23469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Eleswarapu S, Ge X, Wang Y, Yu J, Jiang H. Growth hormone-activated STAT5 may indirectly stimulate IGF-I gene transcription through HNF-3{gamma} Mol Endocrinol. 2009;23:2026–2037. doi: 10.1210/me.2009-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nofziger C, Vezzoli V, Dossena S, Schönherr T, Studnicka J, Nofziger J, Vanoni S, Stephan S, Silva ME, Meyer G, et al. STAT6 links IL-4/IL-13 stimulation with pendrin expression in asthma and chronic obstructive pulmonary disease. Clin Pharmacol Ther. 2011;90:399–405. doi: 10.1038/clpt.2011.128. [DOI] [PubMed] [Google Scholar]
- 134.Okazaki T, Bell RM, Hannun YA. Sphingomyelin turnover induced by vitamin D3 in HL-60 cells. Role in cell differentiation. J Biol Chem. 1989;264:19076–19080. [PubMed] [Google Scholar]
- 135.Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gulbins E, Li PL. Physiological and pathophysiological aspects of ceramide. Am J Physiol Regul Integr Comp Physiol. 2006;290:R11–R26. doi: 10.1152/ajpregu.00416.2005. [DOI] [PubMed] [Google Scholar]
- 137.Chang HC, Hsu C, Hsu HK, Yang RC. Functional role of caspases in sphingosine-induced apoptosis in human hepatoma cells. IUBMB Life. 2003;55:403–407. doi: 10.1080/15216540310001594184. [DOI] [PubMed] [Google Scholar]
- 138.Chang HC, Tsai LH, Chuang LY, Hung WC. Role of AKT kinase in sphingosine-induced apoptosis in human hepatoma cells. J Cell Physiol. 2001;188:188–193. doi: 10.1002/jcp.1108. [DOI] [PubMed] [Google Scholar]
- 139.Llacuna L, Marí M, Garcia-Ruiz C, Fernandez-Checa JC, Morales A. Critical role of acidic sphingomyelinase in murine hepatic ischemia-reperfusion injury. Hepatology. 2006;44:561–572. doi: 10.1002/hep.21285. [DOI] [PubMed] [Google Scholar]
- 140.Hearps AC, Burrows J, Connor CE, Woods GM, Lowenthal RM, Ragg SJ. Mitochondrial cytochrome c release precedes transmembrane depolarisation and caspase-3 activation during ceramide-induced apoptosis of Jurkat T cells. Apoptosis. 2002;7:387–394. doi: 10.1023/a:1020034906200. [DOI] [PubMed] [Google Scholar]
- 141.Osawa Y, Uchinami H, Bielawski J, Schwabe RF, Hannun YA, Brenner DA. Roles for C16-ceramide and sphingosine 1-phosphate in regulating hepatocyte apoptosis in response to tumor necrosis factor-alpha. J Biol Chem. 2005;280:27879–27887. doi: 10.1074/jbc.M503002200. [DOI] [PubMed] [Google Scholar]
- 142.Bollinger CR, Teichgräber V, Gulbins E. Ceramide-enriched membrane domains. Biochim Biophys Acta. 2005;1746:284–294. doi: 10.1016/j.bbamcr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 143.Musashi M, Ota S, Shiroshita N. The role of protein kinase C isoforms in cell proliferation and apoptosis. Int J Hematol. 2000;72:12–19. [PubMed] [Google Scholar]
- 144.Davaille J, Li L, Mallat A, Lotersztajn S. Sphingosine 1-phosphate triggers both apoptotic and survival signals for human hepatic myofibroblasts. J Biol Chem. 2002;277:37323–37330. doi: 10.1074/jbc.M202798200. [DOI] [PubMed] [Google Scholar]
- 145.Gómez-Muñoz A. Ceramide 1-phosphate/ceramide, a switch between life and death. Biochim Biophys Acta. 2006;1758:2049–2056. doi: 10.1016/j.bbamem.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 146.Sakamoto H, Okamoto K, Aoki M, Kato H, Katsume A, Ohta A, Tsukuda T, Shimma N, Aoki Y, Arisawa M, et al. Host sphingolipid biosynthesis as a target for hepatitis C virus therapy. Nat Chem Biol. 2005;1:333–337. doi: 10.1038/nchembio742. [DOI] [PubMed] [Google Scholar]
- 147.Negro F, Sanyal AJ. Hepatitis C virus, steatosis and lipid abnormalities: clinical and pathogenic data. Liver Int. 2009;29 Suppl 2:26–37. doi: 10.1111/j.1478-3231.2008.01950.x. [DOI] [PubMed] [Google Scholar]
- 148.Negro F. Hepatitis C virus and liver steatosis: when fat is not beautiful. J Hepatol. 2004;40:533–535. doi: 10.1016/j.jhep.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 149.Negro F, Alaei M. Hepatitis C virus and type 2 diabetes. World J Gastroenterol. 2009;15:1537–1547. doi: 10.3748/wjg.15.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, Bukh J, Purcell RH, Schultz PG, et al. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci USA. 2002;99:15669–15674. doi: 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yang W, Hood BL, Chadwick SL, Liu S, Watkins SC, Luo G, Conrads TP, Wang T. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology. 2008;48:1396–1403. doi: 10.1002/hep.22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lospalluto J, Dorward B, Miller W, Ziff M. Cryoglobulinemia based on interaction between a gamma macroglobulin and 7S gamma globulin. Am J Med. 1962;32:142–147. doi: 10.1016/0002-9343(62)90191-2. [DOI] [PubMed] [Google Scholar]
- 154.Gorevic PD, Kassab HJ, Levo Y, Kohn R, Meltzer M, Prose P, Franklin EC. Mixed cryoglobulinemia: clinical aspects and long-term follow-up of 40 patients. Am J Med. 1980;69:287–308. doi: 10.1016/0002-9343(80)90390-3. [DOI] [PubMed] [Google Scholar]
- 155.Gorevic PD, Frangione B. Mixed cryoglobulinemia cross-reactive idiotypes: implications for the relationship of MC to rheumatic and lymphoproliferative diseases. Semin Hematol. 1991;28:79–94. [PubMed] [Google Scholar]
- 156.Brouet JC, Clauvel JP, Danon F, Klein M, Seligmann M. Biologic and clinical significance of cryoglobulins. A report of 86 cases. Am J Med. 1974;57:775–788. doi: 10.1016/0002-9343(74)90852-3. [DOI] [PubMed] [Google Scholar]
- 157.Van De Donk N, De Weerdt O, Eurelings M, Bloem A, Lokhorst H. Malignant transformation of monoclonal gammopathy of undetermined significance: cumulative incidence and prognostic factors. Leuk Lymphoma. 2001;42:609–618. doi: 10.3109/10428190109099321. [DOI] [PubMed] [Google Scholar]
- 158.Gleissner B, Maurer J, Thiel E. Detection of immunoglobulin heavy chain genes rearrangements in B-cell leukemias, lymphomas, multiple myelomas, monoclonal and polyclonal gammopathies. Leuk Lymphoma. 2000;39:151–155. doi: 10.3109/10428190009053549. [DOI] [PubMed] [Google Scholar]
- 159.Garc-ia JF, Sánchez E, Lloret E, Martín J, Piris MA. Crystal-storing histiocytosis and immunocytoma associated with multifocal fibrosclerosis. Histopathology. 1998;33:459–464. doi: 10.1046/j.1365-2559.1998.00531.x. [DOI] [PubMed] [Google Scholar]
- 160.Ferri C, Zignego AL, Pileri SA. Cryoglobulins. J Clin Pathol. 2002;55:4–13. doi: 10.1136/jcp.55.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bombardieri S, Ferri C, Migliorini P, Pontrandolfo A, Puccetti A, Vitali C, Pasero G. Cryoglobulins and immune complexes in essential mixed cryoglobulinemia. Ric Clin Lab. 1986;16:281–288. doi: 10.1007/BF02909351. [DOI] [PubMed] [Google Scholar]
- 162.Meltzer M, Franklin EC, Elias K, McCluskey RT, Cooper N. Cryoglobulinemia--a clinical and laboratory study. II. Cryoglobulins with rheumatoid factor activity. Am J Med. 1966;40:837–856. doi: 10.1016/0002-9343(66)90200-2. [DOI] [PubMed] [Google Scholar]
- 163.Bombardieri S, Ferri C, Di Munno O, Pasero G. Liver involvement in essential mixed cryoglobulinemia. Ric Clin Lab. 1979;9:361–368. doi: 10.1007/BF02904572. [DOI] [PubMed] [Google Scholar]
- 164.Levo Y, Gorevic PD, Kassab HJ, Zucker-Franklin D, Franklin EC. Association between hepatitis B virus and essential mixed cryoglobulinemia. N Engl J Med. 1977;296:1501–1504. doi: 10.1056/NEJM197706302962605. [DOI] [PubMed] [Google Scholar]
- 165.Popp JW, Dienstag JL, Wands JR, Bloch KJ. Essential mixed cryoglobulinemia without evidence for hepatitis B virus infection. Ann Intern Med. 1980;92:379–383. doi: 10.7326/0003-4819-92-3-379. [DOI] [PubMed] [Google Scholar]
- 166.Gocke DJ. Extrahepatic manifestations of viral hepatitis. Am J Med Sci. 1975;270:49–52. doi: 10.1097/00000441-197507000-00007. [DOI] [PubMed] [Google Scholar]
- 167.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 168.Pascual M, Perrin L, Giostra E, Schifferli JA. Hepatitis C virus in patients with cryoglobulinemia type II. J Infect Dis. 1990;162:569–570. doi: 10.1093/infdis/162.2.569. [DOI] [PubMed] [Google Scholar]
- 169.Ferri C, La Civita L, Longombardo G, Greco F, Bombardieri S. Hepatitis C virus and mixed cryoglobulinaemia. Eur J Clin Invest. 1993;23:399–405. doi: 10.1111/j.1365-2362.1993.tb00782.x. [DOI] [PubMed] [Google Scholar]
- 170.Ferri C, Marzo E, Longombardo G, Lombardini F, Greco F, Bombardieri S. Alpha interferon in the treatment of mixed cryoglobulinaemia patients. Eur J Cancer. 1991;27 Suppl 4:S81–S82. doi: 10.1016/0277-5379(91)90583-y. [DOI] [PubMed] [Google Scholar]
- 171.Agnello V, Abel G. Localization of hepatitis C virus in cutaneous vasculitic lesions in patients with type II cryoglobulinemia. Arthritis Rheum. 1997;40:2007–2015. doi: 10.1002/art.1780401113. [DOI] [PubMed] [Google Scholar]
- 172.Sansonno D, De Vita S, Cornacchiulo V, Carbone A, Boiocchi M, Dammacco F. Detection and distribution of hepatitis C virus-related proteins in lymph nodes of patients with type II mixed cryoglobulinemia and neoplastic or non-neoplastic lymphoproliferation. Blood. 1996;88:4638–4645. [PubMed] [Google Scholar]
- 173.Zignego AL, Ferri C, Monti M, LaCivita L, Giannini C, Careccia G, Giannelli F, Pasero G, Bombardieri S, Gentilini P. Hepatitis C virus as a lymphotropic agent: evidence and pathogenetic implications. Clin Exp Rheumatol. 1995;13 Suppl 13:S33–S37. [PubMed] [Google Scholar]
- 174.Ferri C, Zignego AL. Relation between infection and autoimmunity in mixed cryoglobulinemia. Curr Opin Rheumatol. 2000;12:53–60. doi: 10.1097/00002281-200001000-00009. [DOI] [PubMed] [Google Scholar]
- 175.Keck ZY, Saha A, Xia J, Wang Y, Lau P, Krey T, Rey FA, Foung SK. Mapping a region of hepatitis C virus E2 that is responsible for escape from neutralizing antibodies and a core CD81-binding region that does not tolerate neutralization escape mutations. J Virol. 2011;85:10451–10463. doi: 10.1128/JVI.05259-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mascia MT, Ferrari D, Campioli D, Sandri G, Mussini C, Ferri C. Non HCV-related mixed cryoglobulinemia. Dig Liver Dis. 2007;39 Suppl 1:S61–S64. doi: 10.1016/s1590-8658(07)80013-2. [DOI] [PubMed] [Google Scholar]
- 177.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, et al. Binding of hepatitis C virus to CD81. Science. 1998;282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 178.Farquhar MJ, Harris HJ, McKeating JA. Hepatitis C virus entry and the tetraspanin CD81. Biochem Soc Trans. 2011;39:532–536. doi: 10.1042/BST0390532. [DOI] [PubMed] [Google Scholar]
- 179.Akeno N, Blackard JT, Tomer Y. HCV E2 protein binds directly to thyroid cells and induces IL-8 production: a new mechanism for HCV induced thyroid autoimmunity. J Autoimmun. 2008;31:339–344. doi: 10.1016/j.jaut.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zignego AL, Ferri C, Pileri SA, Caini P, Bianchi FB. Extrahepatic manifestations of Hepatitis C Virus infection: a general overview and guidelines for a clinical approach. Dig Liver Dis. 2007;39:2–17. doi: 10.1016/j.dld.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 181.Zignego AL, Giannelli F, Marrocchi ME, Mazzocca A, Ferri C, Giannini C, Monti M, Caini P, Villa GL, Laffi G, et al. T(14; 18) translocation in chronic hepatitis C virus infection. Hepatology. 2000;31:474–479. doi: 10.1002/hep.510310230. [DOI] [PubMed] [Google Scholar]
- 182.Lunel F, Musset L, Cacoub P, Frangeul L, Cresta P, Perrin M, Grippon P, Hoang C, Valla D, Piette JC. Cryoglobulinemia in chronic liver diseases: role of hepatitis C virus and liver damage. Gastroenterology. 1994;106:1291–1300. doi: 10.1016/0016-5085(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 183.Pawlotsky JM, Ben Yahia M, Andre C, Voisin MC, Intrator L, Roudot-Thoraval F, Deforges L, Duvoux C, Zafrani ES, Duval J. Immunological disorders in C virus chronic active hepatitis: a prospective case-control study. Hepatology. 1994;19:841–848. [PubMed] [Google Scholar]
- 184.Lenzi M, Johnson PJ, McFarlane IG, Ballardini G, Smith HM, McFarlane BM, Bridger C, Vergani D, Bianchi FB, Williams R. Antibodies to hepatitis C virus in autoimmune liver disease: evidence for geographical heterogeneity. Lancet. 1991;338:277–280. doi: 10.1016/0140-6736(91)90418-o. [DOI] [PubMed] [Google Scholar]
- 185.Antonelli A, Ferri C, Fallahi P, Nesti C, Zignego AL, Maccheroni M. Thyroid cancer in HCV-related mixed cryoglobulinemia patients. Clin Exp Rheumatol. 2002;20:693–696. [PubMed] [Google Scholar]
- 186.Park KJ, Choi SH, Choi DH, Park JM, Yie SW, Lee SY, Hwang SB. 1Hepatitis C virus NS5A protein modulates c-Jun N-terminal kinase through interaction with tumor necrosis factor receptor-associated factor 2. J Biol Chem. 2003;278:30711–30718. doi: 10.1074/jbc.M209623200. [DOI] [PubMed] [Google Scholar]
- 187.Hosui A, Ohkawa K, Ishida H, Sato A, Nakanishi F, Ueda K, Takehara T, Kasahara A, Sasaki Y, Hori M, et al. Hepatitis C virus core protein differently regulates the JAK-STAT signaling pathway under interleukin-6 and interferon-gamma stimuli. J Biol Chem. 2003;278:28562–28571. doi: 10.1074/jbc.M210485200. [DOI] [PubMed] [Google Scholar]
- 188.Rebora A. Skin diseases associated with hepatitis C virus: facts and controversies. Clin Dermatol. 2010;28:489–496. doi: 10.1016/j.clindermatol.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 189.Al-Ali J, Al-Mutari N, Ahmed el-SF. Hepatitis C virus and the skin. Hepatogastroenterology. 2011;58:880–886. [PubMed] [Google Scholar]
- 190.Márquez-Saavedra E, Artacho-Criado S, Morillo-Verdugo R, Grande-Santamaria L, Dorantes-Calderon B, Romero-Gómez M. Propranolol-induced angioedema in a patient with chronic hepatitis C virus infection. Am J Health Syst Pharm. 2010;67:1182–1184. doi: 10.2146/ajhp090120. [DOI] [PubMed] [Google Scholar]
- 191.Ilyin GP, Langouët S, Rissel M, Delcros JG, Guillouzo A, Guguen-Guillouzo C. Ribavirin inhibits protein synthesis and cell proliferation induced by mitogenic factors in primary human and rat hepatocytes. Hepatology. 1998;27:1687–1694. doi: 10.1002/hep.510270630. [DOI] [PubMed] [Google Scholar]
- 192.Terrier B, Sène D, Dechartres A, Saadoun D, Ortonne N, Rouvier P, Musset L, Rigon MR, Maisonobe T, Cacoub P. Systemic vasculitis in patients with hepatitis C virus infection with and without detectable mixed cryoglobulinemia. J Rheumatol. 2011;38:104–110. doi: 10.3899/jrheum.100191. [DOI] [PubMed] [Google Scholar]
- 193.Fernandes SS, Carvalho J, Leite S, Afonso M, Pinto J, Veloso R, Duarte R, Ferreira E, Fraga J. Erythema induratum and chronic hepatitis C infection. J Clin Virol. 2009;44:333–336. doi: 10.1016/j.jcv.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 194.Lidar M, Lipschitz N, Agmon-Levin N, Langevitz P, Barzilai O, Ram M, Porat-Katz BS, Bizzaro N, Damoiseaux J, Tervaert JW, et al. Infectious serologies and autoantibodies in hepatitis C and autoimmune disease-associated mixed cryoglobulinemia. Clin Rev Allergy Immunol. 2012;42:238–246. doi: 10.1007/s12016-011-8275-x. [DOI] [PubMed] [Google Scholar]
- 195.Vigani AG, Macedo de Oliveira A, Tozzo R, Pavan MH, Gonçales ES, Fais V, Gonçales NS, Gonçales FL. The association of cryoglobulinaemia with sustained virological response in patients with chronic hepatitis C. J Viral Hepat. 2011;18:e91–e98. doi: 10.1111/j.1365-2893.2010.01385.x. [DOI] [PubMed] [Google Scholar]
- 196.Andrade MV, Iwaki S, Ropert C, Gazzinelli RT, Cunha-Melo JR, Beaven MA. Amplification of cytokine production through synergistic activation of NFAT and AP-1 following stimulation of mast cells with antigen and IL-33. Eur J Immunol. 2011;41:760–772. doi: 10.1002/eji.201040718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Savvas SP, Papakostas N, Giannaris M, Malaktari S, Koskinas J, Archimandritis AJ. Interferon alpha-induced hashimoto thyroiditis followed by transient graves disease in a patient with chronic HCV infection. South Med J. 2010;103:585–588. doi: 10.1097/SMJ.0b013e3181ddd952. [DOI] [PubMed] [Google Scholar]
- 198.Cunha VS, Meotti C, Oliveira JH, Sprinz E, Alvares-da-Silva MR, Goldani LZ. Different patterns of dermatological presentations in patients co-infected with human immunodeficiency virus and hepatitis C virus (HCV), and those infected with HCV alone. Clin Exp Dermatol. 2012;37:122–127. doi: 10.1111/j.1365-2230.2011.04217.x. [DOI] [PubMed] [Google Scholar]
- 199.Civardi G, Vallisa D, Bertè R, Lazzaro A, Moroni CF, Cavanna L. Focal liver lesions in non-Hodgkin's lymphoma: investigation of their prevalence, clinical significance and the role of Hepatitis C virus infection. Eur J Cancer. 2002;38:2382–2387. doi: 10.1016/s0959-8049(02)00481-1. [DOI] [PubMed] [Google Scholar]
- 200.Cribier B, Chiaverini C, Dali-Youcef N, Schmitt M, Grima M, Hirth C, Lacour JP, Chosidow O. Porphyria cutanea tarda, hepatitis C, uroporphyrinogen decarboxylase and mutations of HFE gene. A case-control study. Dermatology. 2009;218:15–21. doi: 10.1159/000173696. [DOI] [PubMed] [Google Scholar]
- 201.Müller S. Oral manifestations of dermatologic disease: a focus on lichenoid lesions. Head Neck Pathol. 2011;5:36–40. doi: 10.1007/s12105-010-0237-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Nyirjesy P, Leigh RD, Mathew L, Lev-Sagie A, Culhane JF. Chronic vulvovaginitis in women older than 50 years: analysis of a prospective database. J Low Genit Tract Dis. 2012;16:24–29. doi: 10.1097/LGT.0b013e31822a198d. [DOI] [PubMed] [Google Scholar]
- 203.Shimoyama T, Horie N, Kato T, Kaneko T, Komiyama K. Helicobacter pylori in oral ulcerations. J Oral Sci. 2000;42:225–229. doi: 10.2334/josnusd.42.225. [DOI] [PubMed] [Google Scholar]
- 204.Riggio MP, Lennon A, Wray D. Detection of Helicobacter pylori DNA in recurrent aphthous stomatitis tissue by PCR. J Oral Pathol Med. 2000;29:507–513. doi: 10.1034/j.1600-0714.2000.291005.x. [DOI] [PubMed] [Google Scholar]
- 205.Birkenfeld S, Dreiher J, Weitzman D, Cohen AD. A study on the association with hepatitis B and hepatitis C in 1557 patients with lichen planus. J Eur Acad Dermatol Venereol. 2011;25:436–440. doi: 10.1111/j.1468-3083.2010.03809.x. [DOI] [PubMed] [Google Scholar]
- 206.Carrozzo M, Brancatello F, Dametto E, Arduino P, Pentenero M, Rendine S, Porter SR, Lodi G, Scully C, Gandolfo S. Hepatitis C virus-associated oral lichen planus: is the geographical heterogeneity related to HLA-DR6. J Oral Pathol Med. 2005;34:204–208. doi: 10.1111/j.1600-0714.2005.00303.x. [DOI] [PubMed] [Google Scholar]
- 207.Rebora A. Lichen planus and the liver. Int J Dermatol. 1992;31:392–395. doi: 10.1111/j.1365-4362.1992.tb02665.x. [DOI] [PubMed] [Google Scholar]
- 208.del Olmo JA, Llovet F, Rodrigo JM, Molina J, Aparisi L, Serra MA, Wassel A, Bixquert MA. Prevalence of liver disease and infection by hepatitis B, delta virus, and human immunodeficiency virus in two Spanish penitentiaries. Med Microbiol Immunol. 1990;179:43–48. doi: 10.1007/BF00190149. [DOI] [PubMed] [Google Scholar]
- 209.Carrozzo M, Gandolfo S, Carbone M, Colombatto P, Broccoletti R, Garzino-Demo P, Ghisetti V. Hepatitis C virus infection in Italian patients with oral lichen planus: a prospective case-control study. J Oral Pathol Med. 1996;25:527–533. doi: 10.1111/j.1600-0714.1996.tb01726.x. [DOI] [PubMed] [Google Scholar]
- 210.Nagao Y, Sata M, Noguchi S, Suzuki H, Mizokami M, Kameyama T, Tanikawa K. GB virus infection in patients with oral cancer and oral lichen planus. J Oral Pathol Med. 1997;26:138–141. doi: 10.1111/j.1600-0714.1997.tb00037.x. [DOI] [PubMed] [Google Scholar]
- 211.Lodi G, Carrozzo M, Harris K, Piattelli A, Teo CG, Gandolfo S, Scully C, Porter SR. Hepatitis C virus-associated oral lichen planus: no influence from hepatitis G virus co-infection. J Oral Pathol Med. 2000;29:39–42. doi: 10.1034/j.1600-0714.2000.290107.x. [DOI] [PubMed] [Google Scholar]
- 212.Rodríguez-Iñigo E, Arrieta JJ, Casqueiro M, Bartolomé J, López-Alcorocho JM, Ortiz-Movilla N, Manzarbeitia F, Pardo M, Carreño V. TT virus detection in oral lichen planus lesions. J Med Virol. 2001;64:183–189. doi: 10.1002/jmv.1034. [DOI] [PubMed] [Google Scholar]
- 213.Bez C, Hallett R, Carrozzo M, Lodi G, Gandolfo S, Carrassi A, Scully C, Porter SR. Lack of association between hepatotropic transfusion transmitted virus infection and oral lichen planus in British and Italian populations. Br J Dermatol. 2001;145:990–993. doi: 10.1046/j.1365-2133.2001.04518.x. [DOI] [PubMed] [Google Scholar]
- 214.Rebora A, Robert E, Rongioletti F. Clinical and laboratory presentation of lichen planus patients with chronic liver disease. J Dermatol Sci. 1992;4:38–41. doi: 10.1016/0923-1811(92)90054-f. [DOI] [PubMed] [Google Scholar]
- 215.Tong DC, Ferguson MM. Concurrent oral lichen planus and primary sclerosing cholangitis. Br J Dermatol. 2002;147:356–358. doi: 10.1046/j.1365-2133.2002.04773.x. [DOI] [PubMed] [Google Scholar]
- 216.Graham-Brown RA, Sarkany I, Sherlock S. Lichen planus and primary biliary cirrhosis. Br J Dermatol. 1982;106:699–703. doi: 10.1111/j.1365-2133.1982.tb11686.x. [DOI] [PubMed] [Google Scholar]
- 217.Oleaga JM, Gardeazabal J, Sanz de Galdeano C, Diaz PJ. Generalized lichen planus associated with primary biliar cirrhosis which resolved after liver transplantation. Acta Derm Venereol. 1995;75:87. doi: 10.2340/000155557587. [DOI] [PubMed] [Google Scholar]
- 218.Bagán JV, Ramón C, González L, Diago M, Milián MA, Cors R, Lloria E, Cardona F, Jiménez Y. Preliminary investigation of the association of oral lichen planus and hepatitis C. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;85:532–536. doi: 10.1016/s1079-2104(98)90286-4. [DOI] [PubMed] [Google Scholar]
- 219.Gimenez-García R, Pérez-Castrillón JL. Lichen planus and hepatitis C virus infection. J Eur Acad Dermatol Venereol. 2003;17:291–295. doi: 10.1046/j.1468-3083.2003.00631.x. [DOI] [PubMed] [Google Scholar]
- 220.Ilter N, Senol E, Gürer MA, Altay O. Lichen planus and hepatitis C-virus infection in Turkish patients. J Eur Acad Dermatol Venereol. 1998;10:192–193. doi: 10.1111/j.1468-3083.1998.tb00729.x. [DOI] [PubMed] [Google Scholar]
- 221.Kirtak N, Inalöz HS, Ozgöztasi Z. The prevalence of hepatitis C virus infection in patients with lichen planus in Gaziantep region of Turkey. Eur J Epidemiol. 2000;16:1159–1161. doi: 10.1023/a:1010968309956. [DOI] [PubMed] [Google Scholar]
- 222.Erkek E, Bozdogan O, Olut AI. Hepatitis C virus infection prevalence in lichen planus: examination of lesional and normal skin of hepatitis C virus-infected patients with lichen planus for the presence of hepatitis C virus RNA. Clin Exp Dermatol. 2001;26:540–544. doi: 10.1046/j.1365-2230.2001.00885.x. [DOI] [PubMed] [Google Scholar]
- 223.Ingafou M, Porter SR, Scully C, Teo CG. No evidence of HCV infection or liver disease in British patients with oral lichen planus. Int J Oral Maxillofac Surg. 1998;27:65–66. doi: 10.1016/s0901-5027(98)80101-x. [DOI] [PubMed] [Google Scholar]
- 224.Tucker SC, Coulson IH. Lichen planus is not associated with hepatitis C virus infection in patients from north west England. Acta Derm Venereol. 1999;79:378–379. doi: 10.1080/000155599750010328. [DOI] [PubMed] [Google Scholar]
- 225.Carrozzo M, Gandolfo S. Oral diseases possibly associated with hepatitis C virus. Crit Rev Oral Biol Med. 2003;14:115–127. doi: 10.1177/154411130301400205. [DOI] [PubMed] [Google Scholar]
- 226.Nagao Y, Sata M, Itoh K, Tanikawa K, Kameyama T. Quantitative analysis of HCV RNA and genotype in patients with chronic hepatitis C accompanied by oral lichen planus. Eur J Clin Invest. 1996;26:495–498. doi: 10.1046/j.1365-2362.1996.167314.x. [DOI] [PubMed] [Google Scholar]
- 227.Pawlotsky JM, Benchiki H, Pellet C, Duval J, Dhumeaux D, Revuz J, Bagot M. Lichen planus and hepatitis C virus (HCV)-related chronic hepatitis: evaluation of HCV genotypes. Br J Dermatol. 1995;133:666–667. doi: 10.1111/j.1365-2133.1995.tb02734.x. [DOI] [PubMed] [Google Scholar]
- 228.Lodi G, Carrozzo M, Hallett R, D'Amico E, Piattelli A, Teo CG, Gandolfo S, Carbone M, Porter SR. HCV genotypes in Italian patients with HCV-related oral lichen planus. J Oral Pathol Med. 1997;26:381–384. doi: 10.1111/j.1600-0714.1997.tb00235.x. [DOI] [PubMed] [Google Scholar]
- 229.Kirby AC, Lodi GL, Olsen I, Porter SR. Immunohistochemical and serological comparison of idiopathic and hepatitis C virus-associated forms of oral lichen planus. Eur J Oral Sci. 1998;106:853–862. doi: 10.1046/j.0909-8836.1998.eos106404.x. [DOI] [PubMed] [Google Scholar]
- 230.Nagao Y, Tsubone K, Kimura R, Hanada S, Kumashiro R, Ueno T, Sata M. High prevalence of anticardiolipin antibodies in patients with HCV-associated oral lichen planus. Int J Mol Med. 2002;9:293–297. doi: 10.3892/ijmm.9.3.293. [DOI] [PubMed] [Google Scholar]
- 231.Carrozzo M, Quadri R, Latorre P, Pentenero M, Paganin S, Bertolusso G, Gandolfo S, Negro F. Molecular evidence that the hepatitis C virus replicates in the oral mucosa. J Hepatol. 2002;37:364–369. doi: 10.1016/s0168-8278(02)00183-6. [DOI] [PubMed] [Google Scholar]
- 232.Mangia A, Andriulli A, Zenarola P, Lomuto M, Cascavilla I, Quadri R, Negro F. Lack of hepatitis C virus replication intermediate RNA in diseased skin tissue of chronic hepatitis C patients. J Med Virol. 1999;59:277–280. doi: 10.1002/(sici)1096-9071(199911)59:3<277::aid-jmv3>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 233.Pilli M, Penna A, Zerbini A, Vescovi P, Manfredi M, Negro F, Carrozzo M, Mori C, Giuberti T, Ferrari C, et al. Oral lichen planus pathogenesis: A role for the HCV-specific cellular immune response. Hepatology. 2002;36:1446–1452. doi: 10.1053/jhep.2002.37199. [DOI] [PubMed] [Google Scholar]
- 234.Arrieta JJ, Rodriguez-Inigo E, Casqueiro M, Bartolomé J, Manzarbeitia F, Herrero M, Pardo M, Carreno V. Detection of hepatitis C virus replication by In situ hybridization in epithelial cells of anti-hepatitis C virus-positive patients with and without oral lichen planus. Hepatology. 2000;32:97–103. doi: 10.1053/jhep.2000.8533. [DOI] [PubMed] [Google Scholar]
- 235.Mason AL, Lau JY, Hoang N, Qian K, Alexander GJ, Xu L, Guo L, Jacob S, Regenstein FG, Zimmerman R, et al. Association of diabetes mellitus and chronic hepatitis C virus infection. Hepatology. 1999;29:328–333. doi: 10.1002/hep.510290235. [DOI] [PubMed] [Google Scholar]
- 236.Qureshi H, Ahsan T, Mujeeb SA, Jawad F, Mehdi I, Ahmed W, Alam SE. Diabetes mellitus is equally frequent in chronic HCV and HBV infection. J Pak Med Assoc. 2002;52:280–283. [PubMed] [Google Scholar]
- 237.Okan V, Araz M, Aktaran S, Karsligil T, Meram I, Bayraktaroglu Z, Demirci F. Increased frequency of HCV but not HBV infection in type 2 diabetic patients in Turkey. Int J Clin Pract. 2002;56:175–177. [PubMed] [Google Scholar]
- 238.Chen HF, Li CY, Chen P, See TT, Lee HY. Seroprevalence of hepatitis B and C in type 2 diabetic patients. J Chin Med Assoc. 2006;69:146–152. doi: 10.1016/S1726-4901(09)70195-9. [DOI] [PubMed] [Google Scholar]
- 239.White DL, Ratziu V, El-Serag HB. Hepatitis C infection and risk of diabetes: a systematic review and meta-analysis. J Hepatol. 2008;49:831–844. doi: 10.1016/j.jhep.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Konrad T, Zeuzem S, Vicini P, Toffolo G, Briem D, Lormann J, Herrmann G, Berger A, Kusterer K, Teuber G, et al. Evaluation of factors controlling glucose tolerance in patients with HCV infection before and after 4 months therapy with interferon-alpha. Eur J Clin Invest. 2000;30:111–121. doi: 10.1046/j.1365-2362.2000.00608.x. [DOI] [PubMed] [Google Scholar]
- 241.Petit JM, Bour JB, Galland-Jos C, Minello A, Verges B, Guiguet M, Brun JM, Hillon P. Risk factors for diabetes mellitus and early insulin resistance in chronic hepatitis C. J Hepatol. 2001;35:279–283. doi: 10.1016/s0168-8278(01)00143-x. [DOI] [PubMed] [Google Scholar]
- 242.Chen LK, Hwang SJ, Tsai ST, Luo JC, Lee SD, Chang FY. Glucose intolerance in Chinese patients with chronic hepatitis C. World J Gastroenterol. 2003;9:505–508. doi: 10.3748/wjg.v9.i3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Mehta SH, Brancati FL, Sulkowski MS, Strathdee SA, Szklo M, Thomas DL. Prevalence of type 2 diabetes mellitus among persons with hepatitis C virus infection in the United States. Hepatology. 2001;33:1554. doi: 10.1053/jhep.2001.0103306le01. [DOI] [PubMed] [Google Scholar]
- 244.Balbi M, Donadon V, Ghersetti M, Grazioli S, Valentina GD, Gardenal R, Mas MD, Casarin P, Zanette G, Miranda C, et al. Alcohol and HCV chronic infection are risk cofactors of type 2 diabetes mellitus for hepatocellular carcinoma in Italy. Int J Environ Res Public Health. 2010;7:1366–1378. doi: 10.3390/ijerph7041366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Hung CH, Wang JH, Hu TH, Chen CH, Chang KC, Yen YH, Kuo YH, Tsai MC, Lu SN, Lee CM. Insulin resistance is associated with hepatocellular carcinoma in chronic hepatitis C infection. World J Gastroenterol. 2010;16:2265–2271. doi: 10.3748/wjg.v16.i18.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Ågren UM, Anttila M, Mäenpää-Liukko K, Rantala ML, Rautiainen H, Sommer WF, Mommers E. Effects of a monophasic combined oral contraceptive containing nomegestrol acetate and 17β-oestradiol compared with one containing levonorgestrel and ethinylestradiol on haemostasis, lipids and carbohydrate metabolism. Eur J Contracept Reprod Health Care. 2011;16:444–457. doi: 10.3109/13625187.2011.604450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Polakof S, Skiba-Cassy S, Choubert G, Panserat S. Insulin-induced hypoglycaemia is co-ordinately regulated by liver and muscle during acute and chronic insulin stimulation in rainbow trout (Oncorhynchus mykiss) J Exp Biol. 2010;213:1443–1452. doi: 10.1242/jeb.037689. [DOI] [PubMed] [Google Scholar]
- 248.Sesti G. Pathophysiology of insulin resistance. Best Pract Res Clin Endocrinol Metab. 2006;20:665–679. doi: 10.1016/j.beem.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 249.Leclercq IA, Da Silva Morais A, Schroyen B, Van Hul N, Geerts A. Insulin resistance in hepatocytes and sinusoidal liver cells: mechanisms and consequences. J Hepatol. 2007;47:142–156. doi: 10.1016/j.jhep.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 250.Bernsmeier C, Duong FH, Christen V, Pugnale P, Negro F, Terracciano L, Heim MH. Virus-induced over-expression of protein phosphatase 2A inhibits insulin signalling in chronic hepatitis C. J Hepatol. 2008;49:429–440. doi: 10.1016/j.jhep.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 251.Kawaguchi T, Yoshida T, Harada M, Hisamoto T, Nagao Y, Ide T, Taniguchi E, Kumemura H, Hanada S, Maeyama M, et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am J Pathol. 2004;165:1499–1508. doi: 10.1016/S0002-9440(10)63408-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Pazienza V, Clément S, Pugnale P, Conzelman S, Foti M, Mangia A, Negro F. The hepatitis C virus core protein of genotypes 3a and 1b downregulates insulin receptor substrate 1 through genotype-specific mechanisms. Hepatology. 2007;45:1164–1171. doi: 10.1002/hep.21634. [DOI] [PubMed] [Google Scholar]