

Abstract
Objective
Pallido-ponto-nigral degeneration (PPND), caused by an N279K mutation of the MAPT gene, is 1 of a family of disorders collectively referred to as frontotemporal dementia and parkinsonism linked to chromosome 17. This study aims to characterize the nature of the sleep disturbance in PPND and compare these findings to those in other progressive neurological illnesses. Pathological findings are also provided.
Methods
Ten subjects were recruited from the PPND kindred; 5 affected and 5 unaffected. The subjects underwent clinical assessment, polysomnography, and wrist actigraphy. Available sleep-relevant areas (pedunculopontine/laterodorsal tegmentum, nucleus basalis of Meynert, thalamus, and locus ceruleus) of affected subjects were analyzed postmortem.
Results
The affected group's total sleep time was an average of 130.8 minutes compared to 403.6 minutes in the control group (p < 0.01). Initial sleep latency was significantly longer in affected subjects (range, 58–260 minutes vs 3–34 minutes). Affected subjects also had an increase in stage I sleep (8.5% vs 1%), and less stage III/IV sleep (8.5% vs 17%). At the time of autopsy, all cases had severe neuronal tau pathology in wake-promoting nuclei, as well as decreases in thalamic cholinergic innervations. There was no difference in orexinergic fiber density in nucleus basalis of Meynert or locus ceruleus compared to controls.
Interpretation
The PPND kindred showed severe sleep disturbance. Sleep abnormalities are common in neurodegenerative illnesses, but this is the first study of sleep disorders in PPND. Unlike most neurodegenerative conditions, PPND is characterized by decreased total sleep time, increased sleep latency, and decreased sleep efficiency, without daytime hypersomnolence.
Sleep disturbances are frequently seen in neurodegenerative conditions. They are common in tauopathies,1–3 synucleinopathies,4–8 and prion disorders.9,10 The sleep disorders in Alzheimer disease,3 Parkinson disease (PD),5,6,8,11 and fatal familial insomnia (FFI)9,10 have been well characterized. Less is known about the sleep dysfunction in progressive supranuclear palsy,12,13 Pick disease, corticobasal degeneration,14,15 and frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17).16
Pallido-ponto-nigral degeneration (PPND) is a familial neurodegenerative disorder pathologically characterized by the presence of 4 repeat tau deposits in neurons and glia due to an N279K mutation in the gene for the microtubule associated protein tau (MAPT).17,18 PPND belongs to a cluster of disorders collectively labeled as FTDP-17. Clinical, pathological, and neuropsychometric findings in PPND were previously published.16,18,19
Despite recent progress in the understanding of motor and cognitive symptoms, pathology, and genetics of FTDP-17, little is known about sleep disturbances in this disorder. A previous study determined that the PPND kindred demonstrated progressive, generalized slowing on electroencephalography (EEG) as the clinical severity of the disorder increased.16 Daytime recordings captured no sleep, although patients were encouraged to fall asleep. The sleep difficulties observed during the baseline EEG study prompted further polysomnographic investigation.20 Initial polysomnographic data revealed that members of this kindred did not have clinical or electrophysiological features of rapid eye movement (REM) sleep behavior disorder.21
The prevailing theory of sleep generation is a flip-flop switch that governs sleep versus arousal.22 The ascending arousal system is composed of multiple anatomically distinct groups of neurons, including the tuberomammillary nucleus (TMN), the locus ceruleus (LC), the nucleus basalis of Meynert (nbM), and the pedunculopontine tegmentum/laterodorsal tegmentum (PPN/LDT). Meanwhile, the preoptic area of the hypothalamus has been demonstrated to be responsible for generating and maintaining non-REM (NREM) sleep via efferent connections from the median preoptic nucleus (MnPN) and the ventrolateral preoptic nucleus (VLPO), respectively.23,24 These γ-aminobutyric acidergic neurons are inhibitory for the PPN/LDT and LC, thus promoting sleep. Orexin nuclei in the hypothalamus are understood to stabilize the switch via efferent connections to the LC and nbM.25,26 Pathology in the anteroventral and dorso-medial thalamus has also been implicated in sleep disorders, specifically FFI.9 Positron emission tomography (PET) studies of the PPND kindred showed decreased acetylcholinesterase activity in the thalamus,27 which may parallel underlying behavioral similarities between PPND and FFI.
The goal of this study was to examine NREM and REM sleep in both affected family members and those genealogically at risk for the disease. We sought to specifically characterize the nature of the sleep disturbance in PPND and compare these findings to those in other progressive neurological illnesses. Based on the inability to sleep noted in the initial study,16 we hypothesized that the PPND patients would have impaired sleep initiation and reduced sleep efficiency. Polysomnographic findings of 6 affected and 5 at-risk individuals from the PPND family are reported. The polysomnographic results prompted further investigation by retrospective, postmortem pathological analysis of available sleep-related areas of the brain.
Subjects and Methods
Consent
The study protocol was approved by the Mayo Foundation Institutional Review Board. All subjects or their legal guardians signed an informed consent document to participate in this study. Consent was also obtained for brain autopsy.
Subjects
Participants for this study (n = 10) were recruited from the PPND kindred,28 and grouped according to clinical condition: affected versus unaffected. The unaffected group was composed of 5 subjects out of the 74 living members of the kindred; these subjects were genealogically at risk for the MAPT mutation, but their genotypes were not known at the time of enrollment.
After completion of the initial study, an additional member of the kindred presented for clinical evaluation. She underwent polysomnography on two occasions, separated by 9 months. This permitted observation of the progression of her sleep dysfunction in concert with her deteriorating clinical condition. These data are included for illustrative purposes, but not included in statistical analyses, as she did not undergo the same clinical and polysomnographic protocols as the specifically recruited subjects.
Clinical Scales
The initial 10 subjects were evaluated with a structured history and physical examination by 2 subspecialty movement disorder neurologists (Z.K.W. and R.J.U.). Each subject was categorized into 1 of 5 clinical stages, with stage 0 representing no evidence for PPND, stage 1 early disease, stages 2 and 3 worsening symptoms, and stage 4 terminal disease (Table 1).16 The Beck Depression Inventory (BDI) was also administered.
TABLE 1.
Clinical Staging of PPND
| Stage | Name | Disease Duration, yr | Clinical Characteristics |
|---|---|---|---|
| 0 | At risk | 0 | Asymptomatic |
| 1 | Early | 0–2 | Mild to moderate bradykinesia and rigidity, personality changes, occasionally violent behavior and aggressiveness, mild cognitive impairment |
| 2 | Mid | 2–4 | Moderate bradykinesia, rigidity, postural instability, at times mild tremulous activity, dysarthria, pyramidal signs, eye movement abnormalities, further progression of cognitive impairment |
| 3 | Late | 4–6 | Weight loss, prominent parkinsonism (no tremor), perseverative vocalizations, dysphagia, severe cognitive impairment |
| 4 | Terminal | 6–8 | Severe cachexia, eyelid opening and closing apraxia, dystonia, fixed joint contractures, mutism, urinary and fecal incontinence |
Modified with permission from Wszolek and pfeiffer.38
Genetic Testing
Prior to study enrollment, subjects provided blood samples to test for the N279K mutation of the MAPT gene. Testing was performed as previously described using standardized techniques.29 Following the study, genetic testing results were made available to the at-risk subjects upon their request, with appropriate genetic counseling.
Electrophysiological Monitoring
Each subject was monitored with wrist actigraphy to observe baseline sleep/wake activity for 6 days prior to polysomnography. Polysomnography was performed on 2 consecutive nights, with the first night used to allow subjects to acclimate to the equipment, and the second night used to record data for analysis. Following the second night, the Multiple Sleep Latency Test (MSLT) was performed. No adjustments were made to subjects' home medication regimens. A sleep specialist (S.L., P.F., or J.K.) interpreted all polysomnograms as described in the supplemental material. Polysomnography and wrist actigraphy data were calculated for total sleep time (TST), sleep efficiency (SE), initial sleep latency (ISL), initial REM latency, apnea/hypopnea index (AHI), and periodic limb movements. Data were analyzed using Kruskal-Wallis analysis of variance by rank.
Neuropathology
Autopsies were performed and brains were processed as previously described.21 All cases underwent a neuropathological assessment that included evaluation of gross and microscopic findings. A complete description of histopathologic methods can be found in the supplemental material.
Due to the retrospective nature of the pathologic evaluation and previous tissue sampling for routine diagnostic neuropathology, certain sleep-related nuclei (eg, MnPN, VLPO, TMN) were not available for analysis. Hence, data analyses were performed (by D.W.D. and B.N.D.) with specific emphasis on available regions of the brain known to be involved in sleep, including the anteroventral thalamus at the level of the mammillothalamic tract, PPN/LDT at the level of the inferior colliculus, nbM at the level of the infundibulum, and LC at the level of the isthmus. Orexinergic innervation of the nbM and LC in the PPND cohort and choline acetyl transferase staining in the anteroventral thalamus was compared to 7 age- and sex-matched normal controls. All areas were delineated in reference to human brain atlases.30,31
To further characterize the neuronal phenotype in which tau inclusions were identified, double immunohistochemistry was performed for phospho-tau and neurotransmitter markers, specific to cholinergic (ie, choline acetyl transferase) and catecholaminergic (ie, tyrosine hydroxylase) neuronal types. Further descriptions of the methods are described in the supplemental materials.
Results
Subjects
Of the 10 subjects, 5 were clinically affected, and 5 showed no clinical signs, but were genealogically at risk. The affected group was composed of 3 men and 2 women. The unaffected group consisted of 2 men and 3 women. The additional subject was a 41-year-old, clinically affected female.
Genetic Testing
All 5 subjects in the affected group, the additional affected subject, and 2 of the unaffected subjects (#7 and #9 in Table 2) were confirmed to be carriers of the N279K MAPT gene mutation. One asymptomatic carrier remains asymptomatic (#7); the other progressed to symptomatic PPND and has since died (#9).This subject constitutes the seventh member of the kindred included in the autopsy data. The other 3 members of the control group were not carriers. To ensure that the mutation carriers included in the control group did not influence the results, statistical analyses were performed comparing the carriers to the wild-type subjects within the control group. No significant differences between members of these subgroups were identified on any of the reported measures.
TABLE 2.
Polysomnography Data in Pallido-Ponto-Nigral Degeneration and Controls
| Subject | Age | Sex | TST | SE% | ISL | WASO | I % | II % | III/IV % | REM % | SE % by Actigraphy | AHI | MSLT, min | Epworth |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Affected | ||||||||||||||
| 1 | 48 | M | 0 | 0 | NA | NA | NA | NA | NA | NA | 49.2 | NA | 15.6 | 23 |
| 2 | 59 | M | 43 | 10 | 59 | 339 | 13 | 76 | 0 | 11 | 35.2 | 1 | 19.0 | 8 |
| 3 | 43 | M | 234 | 57 | 72 | 108 | 13 | 56 | 13 | 18 | 65.4 | 2 | 20.0 | 0 |
| 4 | 44 | F | 159 | 36 | 260 | 25 | 4 | 80 | 6 | 10 | 81.2 | 2 | 16.5 | |
| 5 | 45 | F | 218 | 51 | 58 | 153 | 4 | 75 | 11 | 10 | 64.3 | 1 | 14.3 | 0 |
| Median | 45a | 159b | 36b | 65a | 130.5 | 8.5a | 75.5 | 8.5a | 10.5 | 64.3 | 1.5 | 16.5 | 4 | |
| 11a | 41 | F | 370 | 87 | 21 | 35 | 5 | 20 | 51 | 24 | 2 | 0 | ||
| 11b | 42 | F | 4 | 1 | 280 | 135 | 100 | 0 | 0 | 0 | 0 | |||
| At risk | ||||||||||||||
| 6 | 33 | F | 350 | 85 | 15 | 49 | 2 | 59 | 19 | 12 | 93.3 | 8 | 11.8 | 4 |
| 7 | 37 | F | 421 | 97 | 5 | 10 | 1 | 58 | 15 | 26 | 96.9 | 2 | 14.8 | 5 |
| 8 | 24 | M | 488 | 95 | 3 | 21 | 1 | 52 | 23 | 24 | 91.8 | 1 | 9.6 | 4 |
| 9 | 45 | F | 314 | 79 | 34 | 52 | 9 | 69 | 15 | 7 | 90.7 | 2 | 19.0 | 1 |
| 10 | 23 | M | 445 | 95 | 9 | 15 | 0 | 58 | 17 | 25 | 80.7 | 1 | 6.4 | 7 |
| Median | 32a | 421b | 95b | 9a | 21 | 1.0a | 58 | 17a | 24 | 91.8b | 2 | 11.8 | 4 | |
| Normal adults | 370.2 | 90.2 | 8.4 | 4.3 | 54.6 | 10.9 | 20.4 | <5 | >10 | <10 |
Subject 11 underwent two studies nine months apart that are denoted as 11a and 11b, respectively.
Subject 11 is the additional subject who underwent polysomnography on two occasions. Normal sleep values for healthy adults aged 40 to 49 years can be found in Natale et al33 and Starkstein et al.34
p < 0.05,
p < 0.01,
Kruskal-Wallis analysis of variance by rank.
TST = total sleep time; SE = sleep efficiency; ISL = initial sleep latency; WASO = wake after sleep onset; I % = % of stage I; II % = % stage II; III/IV % = % stage III/IV; REM % = % rapid eye movement sleep; AHI = apnea/hypopnea index; MSLT = mean sleep latency test; Epworth = Epworth Sleepiness Scale score; NA = not available.
Clinical Scales
Of the 5 affected subjects, 1 was clinical stage 1, 2 were stage 2, and 2 were stage 3 using the staging scheme described in Table 1. No end-stage patients were included. Both mutation carriers in the unaffected group were, by definition, stage 0. The 11th subject's symptoms were first detected 18 months prior to her first polysomnogram. She was in stage 1 at the time of her first polysomnogram and stage 2 during her second study 9 months later.
Epworth Sleepiness Scale scores were obtained for 9 of the 10 patients, with no significant difference between the 2 groups. Only 1 patient had a high score (23), but this subject's scale was completed by the subject's wife, as he was too cognitively impaired to complete it for himself. BDI scores were obtained for 8 subjects, 4 from each group. One patient from the affected group scored 48, indicating severe depression; however, none of the other subjects from either group scored higher than 10.
Electrophysiological Data
Three subjects showed severe sleep impairment. These subjects were the most clinically affected by PPND, including a late stage 2 subject and the 2 stage 3 subjects (see Table 2). One of the stage 3 subjects achieved no sustained sleep, but microsleep intrusions were observed; TST was 0. Less clinically affected subjects also showed sleep restriction. The at-risk subject with the poorest sleep efficiency entered clinical stage 1 within 6 months of undergoing polysomnography. The control group was within established normal limits on all sleep parameters.32,33
Overall TST in the affected group averaged 130.8 minutes compared to a mean of 403.6 minutes in the control group (p < 0.01). Sleep efficiency was also significantly reduced in the affected group (30.8% vs 90.2%, p < 0.01). Excluding the subject who achieved no sleep, initial sleep latency was significantly longer in affected subjects (range, 58–260 minutes vs 3–34 minutes). There was also an increase in stage I sleep (8.5% vs 1%), and less stage III/IV sleep (8.5% vs 17%) in the affected as compared to the unaffected subjects. No statistical differences were detected between the at-risk subjects who subsequently proved to be mutation carriers and those who were not carriers. When the affected group was compared directly to the wild-type subgroup of the controls, total sleep time (130.8 minutes vs 427.7 minutes, p = 0.005) and sleep efficiency (30.8% vs 91.7%, p = 0.007) were significantly different. Comparing affected to wild-type subjects in initial sleep latency showed a trend toward significance (112.3 minutes vs 9 minutes, p = 0.14).
During the additional patient's first polysomnogram, her TST was 370 minutes. Nine months later, this dropped to just 4 minutes. Likewise, her SE fell from 87% to 1%, and her ISL increased from 21 minutes to 280 minutes. She achieved no stage II, III, IV, or REM sleep. Subjectively, she had no complaints of sleepiness. This lack of perceived sleepiness was common among affected subjects.
Wrist actigraphy demonstrated significantly more activity at night in the affected group (p 0.016). Figure 1A displays a normal wrist actigraphy study from 1 of the unaffected subjects. An abnormal study from a subject in clinical stage 3 demonstrating severely decreased motionless time is presented in Figure 1B.
FIGURE 1.

Wrist actigraphy from (A) a normal subject and (B) a severely affected pallido-ponto-nigral degeneration subject. In the normal subject, there is minimal movement detected during the night, presumably due to the subject sleeping. Far fewer and shorter periods of stillness were detected in the affected subject, thought to be due to reduced sleep. Note: for 8 hours at the beginning of the sixth day, the affected subject removed the recording device, creating an artifactual flatline during this period, and the control subject removed the device at 6 AM on the final morning.
As previously reported, electromyographic atonia was normal during REM sleep; no dream enactment behavior was exhibited, nor were there any other REM or NREM parasomnias. Mild disordered breathing was observed in 1 of the at-risk subjects (AHI 8) and none of the affected subjects (p = 0.624). Periodic leg movement was elevated in 3 of the affected and 1 of the at-risk subjects. None of the affected and only 1 of the at-risk subjects exhibited excessive daytime sleepiness as measured by MSLT. The 1 abnormal MSLT was from a subject who is not a mutation carrier. There was no significant difference between the groups on MSLT (p = 0.095). No sleep onset REM periods were observed.
Neuropathology
Seven subjects involved in prospective sleep studies came to autopsy. Six of these subjects were symptomatic at the time of their first polysomnograms, and 1 was classified in the unaffected group, but later developed genetically confirmed PPND. Some of the cases were included in previously published pathologic studies of PPND.18,28 All cases had neuropathologic findings, including widespread neuronal and glial tau pathology, consistent with the usual findings in PPND. The mean calculated fixed whole brain weight was 1,063g (range, 1,000–1,100g; normal, 1,300–1,400g). On gross examination, all cases had atrophy of the hippocampus and the amygdala and a decrease in neuromelanin pigmentation in the substantia nigra and LC.
Given the relatively young age, the brains had minimal to no amyloid plaques and sparse Alzheimer type neurofibrillary tangles that could be readily visualized with thioflavin S fluorescent microscopy. This was consistent with previous results.28 There were also no Lewy bodies with α-synuclein immunohistochemistry. All cases had severe neuronal loss and gliosis in the globus pallidus, mesencephalic tegmentum, and substantia nigra. The brains also exhibited frontal and medial temporal lobe degeneration with extensive pretangle tau pathology in the form of fine punctate and granular cytoplasmic tau immunoreactivity in neurons that was negative with thioflavin S fluorescent microscopy. Immunohistochemistry with tau antibodies revealed extensive pretangles and tau-positive processes (tau threads and grains) in both the gray and white matter of other brain areas.
Analysis of the LC, nbM, PPN/LDT, and anteroventral thalamus revealed similar pathologies in all cases, including tau-positive neuronal, astrocytic, and oligodendroglial inclusions, as well as many tau-positive processes (ie, threads and grains) (Fig 2). In the regions of analysis, tau pathology in sleep-related nuclei was uniform across the rostral caudal extent of the examined nuclei. There were sparse astrocytic and oligodendroglial inclusions in the nbM, PPN/LDT, and anteroventral thalamus; glial tau pathology was absent in the LC. Using double-labeling immunohistochemistry, abnormal tau immunoreactivity was present in many choline acetyltransferase (ChAT)-positive neurons of the nbM (33% average; range, 16–59%), PPN/LDT (17% average; range, 0–41%), and tyrosine hydroxylase-positive neurons of the LC (49% average; range, 38–62%) (Fig 3). Antibody labeling to fluorescein isothiocyanate and rhodamine fluorochrome channels did not identify any nonspecific associative artifacts. The PPN/LDT, due to its diffuse nature, demonstrated the highest variability of tau in ChAT-positive cells, with most cases containing very few double-labeled cells. Of the numerous tau positive neurites, none was noted to be ChAT positive. Most wake-promoting nuclei had many tau-positive neurites, with the LC having significantly lower density compared to the other regions (Fig 4). Orexin immunoreactive neuritic staining revealed no statistically significant differences between PPND and controls in the LC (Fig 5) or the nbM (Fig 6). However, there was decreased ChAT immunoreactivity in the anteroventral thalamus compared to controls (Fig 7).
FIGURE 2.
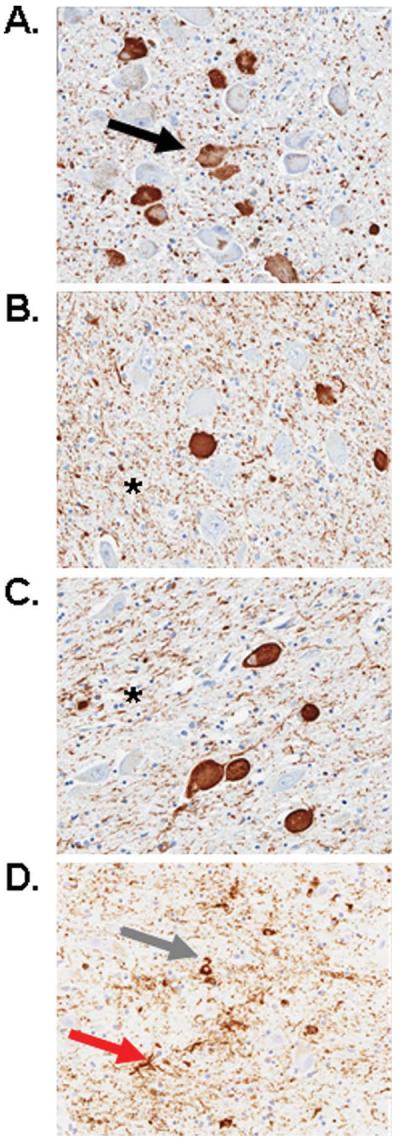
Photomicrographs of tau staining in the locus ceruleus (A), pedunculopontine tegmentum/laterodorsal tegmentum (PPN/LDT) (B), nucleus basalis of Meynert (C), and anteroventral thalamus (D) of a pallido-ponto-nigral degeneration case (original magnification, ×20). Numerous neurofibrillary tangles (black arrow in A) and tau grains and threads (asterisk in B, C) were located in all regions examined. Coiled bodies (gray arrow in D) and tau astrocytes (red arrow in D) were only sparsely observed in the PPN/LDT and anteroventral thalamus. [Color figure can be viewed in the online issue, which is available at annalsofneurology.org.]
FIGURE 3.

Double-labeling immunohistochemistry of neurotransmitter markers (red), tyrosine hydroxylase in the locus ceruleus (LC), and choline acetyltransferase in the pedunculopontine tegmentum/laterodorsal tegmentum (PPN/LDT) and nucleus basalis of Meynert (nbM), with tau (CP13, green). White arrows demonstrate tau inclusions in neurotransmitter marker-positive neurons. Original magnification, ×20. Bar 5 200μm. [Color figure can be viewed in the online issue, which is available at annalsofneurology.org.]
FIGURE 4.

Neuritic tau staining in the locus ceruleus (LC), pedunculopontine tegmentum/laterodorsal tegmentum (PPN/LDT), nucleus basalis of Meynert (nbM), and anteroventral thalamus (thalamus). (A) Photomicrographs taken of each area (original magnification, ×20). (B) Quantification of the percentage of neuritic tau staining (% area) in the LC, PPN/LDT, nbM, and thalamus. Three 5μm sections from each of the cases were stained with CP13 and quantified using a positive pixel density algorithm specific for neuritic morphology. *LC contained significantly less neuritic tau staining when compared to the other areas analyzed (p < 0.05). [Color figure can be viewed in the online issue, which is available at annalsofneurology.org.]
FIGURE 5.

No differences in orexinergic innervations to the locus ceruleus (LC) were identified in pallido-ponto-nigral degeneration (PPND) cases when compared to age-matched controls. (A) Photomicrographs of orexin staining in the LC of a PPND case (left) and a normal subject (right) (original magnification, ×20). Black arrows indicate positive staining. (B) Quantitation of orexin staining. There were no significant differences between PPND subjects and normal controls (p 5 0.11). [Color figure can be viewed in the online issue, which is available at annalsofneurology.org.]
FIGURE 6.

No differences in orexinergic innervations to the nucleus basalis of Meynert (nbM) were identified in pallido-ponto-nigral degeneration (PPND) cases when compared to age-matched controls. (A) Photomicrographs of orexin staining in the nbM of a PPND case (left) and a normal subject (right) (original magnification, ×20). Black arrows indicate positive staining. (B) Quantitation of orexin staining in the nbM. There were no significant differences between PPND subjects and normal controls (p = 0.61). [Color figure can be viewed in the online issue, which is available at annalsofneurology.org.]
FIGURE 7.

Choline acetyltransferase (ChAT) staining of neurites in the anterolateral thalamus is decreased in pallido-ponto-nigral degeneration (PPND) cases when compared to age-matched controls. (A) Photomicrographs of ChAT staining in the anteroventral thalamus of a PPND case (left) and a normal subject (right) (original magnification, ×20). Black arrows indicate positive staining. (B) Quantitation of ChAT staining in the anteroventral thalamus. *PPND cases contained less ChAT staining when compared to normal controls (p < 0.05). [Color figure can be viewed in the online issue, which is available at annalsofneurology.org.]
Discussion
In this study, we report severe sleep dysfunction, particularly sleep initiation, in a large and well-characterized FTDP-17 kindred due to N279K mutation in MAPT, the PPND kindred. Significant reductions in total sleep time, sleep efficiency, and in stage I, III, and IV sleep along with prolonged initial sleep latency were observed. There was also a trend toward a lower REM sleep percentage. There was no compensatory daytime sleep demonstrated on MSLT. Results of the wrist actigraphy study showed frequent nocturnal activity, suggesting that the lack of sleep seen on polysomnography was not unique to the testing environment. Additionally, Epworth Sleepiness Scores argue against pathological daytime sleepiness. Although 1 subject with poor nocturnal sleep was determined by his wife to have a high Epworth score, we do not know if the behavior she observed was truly sleep (as would be determined by EEG) or simply appeared to be sleep. A similar phenomenon occurs with wrist actigraphy, which overestimates the true amount of sleep time by measuring motionlessness. For those patients able to complete the forms themselves, no excessive daytime sleepiness was elicited.
Unlike PD patients,34 the BDI scores and clinical evaluations of the PPND family show no evidence for the contribution of either depression or pain to their reduced sleep. There was also no evidence that tremor was an impairment to sleep initiation.
The severe loss of sleep seen in PPND has been identified in only 2 other neurodegenerative illnesses: a prion disease (FFI) and a synucleinopathy (E46K mutation in the SNCA gene, seen in a Spanish kindred).35 FFI is a prion disorder specifically characterized by the inability to sleep. FFI patients have demonstrated disordered sleep initiation, poor sleep maintenance, prolonged sleep onset latency, reduced total sleep time, decreased sleep spindles and K complexes, and the absence of slow wave sleep.9,10 The SNCA mutation has a phenotype of parkinsonism and dementia with Lewy bodies. These patients were found to have severe sleep disturbances with loss of both REM and NREM sleep. Two subjects did not achieve any stage II, III, or IV sleep.35 Daytime hypersomnolence was not seen in any of these 3 conditions, which makes them unique among neurodegenerative disorders. Further comparisons of these disorders are displayed in Table 3.
TABLE 3.
Similar Sleep Dysfunctions Can Be Seen in Otherwise Dissimilar Neurodegenerative Illnesses9,10,16,35
| Disease | Pathology | Affected Gene | Age of Onset, yr | Clinical Syndrome | Sleep Dysfunction |
|---|---|---|---|---|---|
| PPND | Tau | MAPT | 30s–40s | Atypical parkinsonism, supranuclear gaze palsy, dementia | Decreased TST and SE. Increased ISL. No daytime hypersomnolence. |
| FFI | Prion | PRNP | 40s–60s | Autonomic hyperactivity, myoclonus, pyramidal signs | Decreased TST and SE. Increased ISL. No daytime hypersomnolence. Oneiric stupor.a |
| E46K | α-Synuclein | SNCA | 50s–60s | Dopamine-responsive parkinsonism and dementia | Decreased TST and SE. Increased ISL. No daytime hypersomnolence. |
Oneiric stupor consists of complex dream enactment behavior that can be recalled upon arousal. Later in the course of FFI, oneiric stupor periods become more frequent and recall for the events diminishes.
PPND = pallido-ponto-nigral degeneration; TST = total sleep time; SE = sleep efficiency; ISL = initial sleep latency; FFI = fatal familial insomnia.
A weakness of the present study is the retrospective nature of the neuropathologic analysis, which therefore placed constraints on availability of tissue for all sleep-related nuclei of interest (eg, MnPN and VLPO). All cases that came to autopsy had advanced end-stage disease, with long intervals between sleep study and autopsy, which precluded meaningful correlation with pathologic findings and results of sleep studies. All cases, including those affected and unaffected at the time of sleep evaluation, had substantial tau burden in the LC, nbM, PPN/LDT, and anteroventral thalamus.
Although the pathological data revealed significant tau burden in wake-promoting areas, the clinical phenotype was characterized by decreased sleep, consistent with pathology in sleep-promoting regions as well. We speculate that the loss of sleep, despite extensive pathology in the wake-promoting nuclei, is caused by the simultaneous loss of MnPN function, leading to a perpetual state of wakefulness and essentially locking the flip-flop switch in this direction. The lack of pathology in the orexinergic system would also help promote perpetual wakefulness if the sleep-promoters are damaged. Future research should specifically examine the MnPN in PPND and other neurodegenerative disorders characterized by pathological loss of sleep. Likewise, recent research has identified a role for the basal ganglia in the sleep–wake cycle; specifically, globus pallidus lesions increased wakefulness in rats.36 What effect pallidal pathology has on sleep in PPND is another avenue for further investigation.
Clinical data on the sleep dysfunction associated with FTDP-17 are lacking. One strength of this study is that it fills this void. The 6 affected subjects represent a substantial percentage of available PPND patients. Recruitment of additional affected subjects was limited due to logistical complications and health concerns. Another strength of this study is the identification of a third neurodegenerative disorder in which sleep disappears as the disease progresses. Our hope is that this finding opens the door to further neuropathological analysis in search of the biological underpinnings of sleep.
Future research could investigate the role of polysomnography in the early diagnosis and prognosis of neurodegenerative diseases. One recent study found imaging differences between presymptomatic gene carriers from the same PPND kindred and normal controls using magnetic resonance imaging and PET.37 Similarly, polysomnography accurately predicted which asymptomatic subject would soon develop symptoms. Potentially, polysomnography could help predict longevity for affected patients.
Subjects in this study, particularly those with advanced clinical symptoms, had severe abnormalities in generating and maintaining NREM sleep. Pathologic analyses demonstrated substantial tau burden in each of the sleep-relevant regions examined. Intriguingly, there was no compensatory daytime hypersomnolence, indicating that these subjects lost either the ability or the need to sleep. This pattern of sleep disturbance is seen infrequently across the spectrum of neurodegenerative conditions, which could aid in shedding light on specific anatomical regions necessary for sleep initiation.
Supplementary Material
Acknowledgments
This study was partially supported by a CR20 grant Mayo Clinic Florida Research Committee (Z.K.W., R.J.U., A.J.S., S.-C.L.), National Institute of Neurological Disorders and Stroke grant P50 #NS072187 to the Morris K. Udall Center of Excellence in Parkinson's Disease Research (Z.K.W., R.J.U., D.W.D., A.J.S.), and a gift from Carl Edward Bolch, Jr. and Susan Bolch.
We thank the members of this family for their continued support and willingness to cooperate in this study; Dr O. I. Iweala, Dr J. Dominik, and K. Viola for contributing to the editing of the manuscript; and Dr J. Lucas for assisting with statistical analysis.
Footnotes
Potential Conflicts of Interest Nothing to report.
Additional Supporting Information can be found in the online version of this article.
References
- 1.Loewenstein RJ, Weingartner H, Gillin JC, et al. Disturbances of sleep and cognitive functioning in patients with dementia. Neurobiol Aging. 1982;3:371–377. doi: 10.1016/0197-4580(82)90025-2. [DOI] [PubMed] [Google Scholar]
- 2.Martin PR, Loewenstein RJ, Kaye WH, et al. Sleep EEG in Korsakoff's psychosis and Alzheimer's disease. Neurology. 1986;36:411–414. doi: 10.1212/wnl.36.3.411. [DOI] [PubMed] [Google Scholar]
- 3.Vitiello MV, Prinz PN, Williams DE, et al. Sleep disturbances in patients with mild-stage Alzheimer's disease. J Gerontol. 1990;45:M131–M138. doi: 10.1093/geronj/45.4.m131. [DOI] [PubMed] [Google Scholar]
- 4.Ferman TJ, Smith GE, Boeve BF, et al. DLB fluctuations: specific features that reliably differentiate DLB from AD and normal aging. Neurology. 2004;62:181–187. doi: 10.1212/wnl.62.2.181. [DOI] [PubMed] [Google Scholar]
- 5.Happe S, Klosch G, Lorenzo J, et al. Perception of sleep: subjective versus objective sleep parameters in patients with Parkinson's disease in comparison with healthy elderly controls. Sleep perception in Parkinson's disease and controls. J Neurol. 2005;252:936–943. doi: 10.1007/s00415-005-0785-0. [DOI] [PubMed] [Google Scholar]
- 6.Ondo WG, Dat Vuong K, Khan H, et al. Daytime sleepiness and other sleep disorders in Parkinson's disease. Neurology. 2001;57:1392–1396. doi: 10.1212/wnl.57.8.1392. [DOI] [PubMed] [Google Scholar]
- 7.Stevens S, Cormella CL, Stepanski EJ. Daytime sleepiness and alertness in patients with Parkinson disease. Sleep. 2004;27:967–972. doi: 10.1093/sleep/27.5.967. [DOI] [PubMed] [Google Scholar]
- 8.Tandberg E, Larsen JP, Karlsen K. Excessive daytime sleepiness and sleep benefit in Parkinson's disease: a community-based study. Mov Disord. 1999;14:922–927. doi: 10.1002/1531-8257(199911)14:6<922::aid-mds1003>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 9.Sforza E, Montagna P, Tinuper P, et al. Sleep-wake cycle abnormalities in fatal familial insomnia. Evidence of the role of the thalamus in sleep regulation. Electroencephalogr Clin Neurophysiol. 1995;94:398–405. doi: 10.1016/0013-4694(94)00318-f. [DOI] [PubMed] [Google Scholar]
- 10.Tinuper P, Montagna P, Medori R, et al. The thalamus participates in the regulation of the sleep-waking cycle. A clinico-pathological study in fatal familial thalamic degeneration. Electroencephalogr Clin Neurophysiol. 1989;73:117–123. doi: 10.1016/0013-4694(89)90190-9. [DOI] [PubMed] [Google Scholar]
- 11.Arnulf I, Leu S, Oudiette D. Abnormal sleep and sleepiness in Parkinson's disease. Curr Opin Neurol. 2008;21:472–477. doi: 10.1097/WCO.0b013e328305044d. [DOI] [PubMed] [Google Scholar]
- 12.Aldrich MS, Foster NL, White RF, et al. Sleep abnormalities in progressive supranuclear palsy. Ann Neurol. 1989;25:577–581. doi: 10.1002/ana.410250609. [DOI] [PubMed] [Google Scholar]
- 13.Pareja JA, Caminero AB, Masa JF, Dobato JL. A first case of progressive supranuclear palsy and pre-clinical REM sleep behavior disorder presenting as inhibition of speech during wakefulness and somniloquy with phasic muscle twitching during REM sleep. Neurologia. 1996;11:304–306. [PubMed] [Google Scholar]
- 14.Kimura K, Tachibana N, Aso T, et al. Subclinical REM sleep behavior disorder in a patient with corticobasal degeneration. Sleep. 1997;20:891–894. doi: 10.1093/sleep/20.10.891. [DOI] [PubMed] [Google Scholar]
- 15.Roche S, Jacquesson JM, Destee A, et al. Sleep and vigilance in corticobasal degeneration: a descriptive study. Neurophysiol Clin. 2007;37:261–264. doi: 10.1016/j.neucli.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Wszolek ZK, Lagerlund TD, Steg RE, McManis PG. Clinical neurophysiologic findings in patients with rapidly progressive familial parkinsonism and dementia with pallido-ponto-nigral degeneration. Electroencephalogr Clin Neurophysiol. 1998;107:213–222. doi: 10.1016/s0013-4694(98)00064-9. [DOI] [PubMed] [Google Scholar]
- 17.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 18.Reed LA, Schmidt ML, Wszolek ZK, et al. The neuropathology of a chromosome 17-linked autosomal dominant parkinsonism and dementia (“pallido-ponto-nigral degeneration”) J Neuropathol Exp Neurol. 1998;57:588–601. doi: 10.1097/00005072-199806000-00006. [DOI] [PubMed] [Google Scholar]
- 19.Ferman TJ, McRae CA, Arvanitakis Z, et al. Early and pre-symptomatic neuropsychological dysfunction in the PPND family with the N279K tau mutation. Parkinsonism Relat Disord. 2003;9:265–270. doi: 10.1016/s1353-8020(02)00098-6. [DOI] [PubMed] [Google Scholar]
- 20.Arvanitakis Z, Witte RJ, Dickson DW, et al. Clinical-pathologic study of biomarkers in FTDP-17 (PPND family with N279K tau mutation) Parkinsonism Relat Disord. 2007;13:230–239. doi: 10.1016/j.parkreldis.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Boeve BF, Lin S-C, Strongosky A, et al. Absence of rapid eye movement sleep behavior disorder in 11 members of the pallidopontonigral degeneration kindred. Arch Neurol. 2006;63:268–272. doi: 10.1001/archneur.63.2.268. [DOI] [PubMed] [Google Scholar]
- 22.Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. comment, 1207. [DOI] [PubMed] [Google Scholar]
- 23.Szymusiak R, McGinty D. Hypothalamic regulation of sleep and arousal. Ann N Y Acad Sci. 2008;1129:275–286. doi: 10.1196/annals.1417.027. [DOI] [PubMed] [Google Scholar]
- 24.Sherin JE, Elmquist JK, Torrealba F, Saper CB. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci. 1998;18:4705–4721. doi: 10.1523/JNEUROSCI.18-12-04705.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horvath TL, Peyron C, Diano S, et al. Hypocretin (orexin) activation and synaptic innervation of the locus coeruleus noradrenergic system. J Comp Neurol. 1999;415:145–159. [PubMed] [Google Scholar]
- 26.Moore RY, Abrahamson EA, Van Den Pol A. The hypocretin neuron system: an arousal system in the human brain. Arch Ital Biol. 2001;139:195–205. [PubMed] [Google Scholar]
- 27.Hirano S, Shinotoh H, Kobayashi T, et al. Brain acetylcholinesterase activity in FTDP-17 studied by PET. Neurology. 2006;66:1276–1277. doi: 10.1212/01.wnl.0000208515.50924.94. [DOI] [PubMed] [Google Scholar]
- 28.Wszolek ZK, Pfeiffer RF, Bhatt MH, et al. Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-pontonigral degeneration. Ann Neurol. 1992;32:312–320. doi: 10.1002/ana.410320303. [DOI] [PubMed] [Google Scholar]
- 29.Clark LN, Poorkaj P, Wszolek Z, et al. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci U S A. 1998;95:13103–13107. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mai JK, Assheuer J, Paxinos G. Atlas of the human brain. Academic Press; San Diego, CA: 1997. [Google Scholar]
- 31.Paxinos G, Huang XF. Atlas of the human brainstem. Academic Press; San Diego, CA: 1995. [Google Scholar]
- 32.Hirshkowitz M, Moore CA, Hamilton CR, III, et al. Polysomnography of adults and elderly: sleep architecture, respiration, and leg movement. J Clin Neurophysiol. 1992;9:56–62. [PubMed] [Google Scholar]
- 33.Natale V, Plazzi G, Martoni M. Actigraphy in the assessment of insomnia: a quantitative approach. Sleep. 2009;32:767–771. doi: 10.1093/sleep/32.6.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Starkstein SE, Preziosi TJ, Robinson RG. Sleep disorders, pain, and depression in Parkinson's disease. Eur Neurol. 1991;31:352–355. doi: 10.1159/000116692. [DOI] [PubMed] [Google Scholar]
- 35.Zarranz JJ, Fernandez-Bedoya A, Lambarri I, et al. Abnormal sleep architecture is an early feature in the E46K familial synucleinopathy. Mov Disord. 2005;20:1310–1315. doi: 10.1002/mds.20581. [DOI] [PubMed] [Google Scholar]
- 36.Qiu MH, Vetrivelan R, Fuller PM, Lu J. Basal ganglia control of sleep-wake behavior and cortical activation. Eur J Neurosci. 2010;31:499–507. doi: 10.1111/j.1460-9568.2009.07062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyoshi M, Shinotoh H, Wszolek ZK, et al. In vivo detection of neuropathologic changes in presymptomatic MAPT mutation carriers: a PET and MRI study. Parkinsonism Relat Disord. 2010;16:404–408. doi: 10.1016/j.parkreldis.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 38.Wszolek ZK, Pfeiffer RF. Rapidly progressive autosomal dominant Parkinsonism and dementia with pallido-ponto-nigral degeneration. In: Stern MB, Koller WC, editors. Parkinsonian Syndromes. Marcell Dekker, Inc.; Newyork, NY: 1993. pp. 297–312. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.