

Conspectus
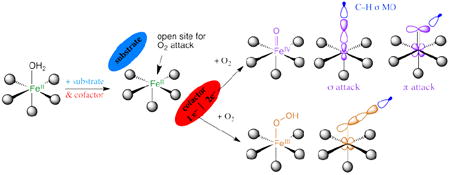
Mononuclear non-heme Fe (NHFe) enzymes play key roles in antibiotic biosynthesis, hypoxic response, DNA repair, anticancer therapy and many other biological processes. On a molecular level these enzymes catalyze a diverse range of oxidation reactions, including hydroxylation, halogenation, ring closure, desaturation and electrophilic aromatic substitution (EAS). Most of these enzymes use an FeII site to activate dioxygen. These ferrous active sites had been inaccessible to traditional spectroscopic methods. A methodology has been developed that provides detailed geometric and electronic structure insight for these NHFeII active sites. This has defined a general mechanistic strategy utilized by a wide range of these enzymes to control O2 activation by FeII coordination unsaturation only in the presence of cosubstrates to limit autooxidation and self-hydroxylation. Depending on the type of enzyme, O2 activation either involves a 2e− reduced FeIII–OOH intermediate or a 4e− reduced FeIV=O intermediate. The nature of these intermediates has been defined in terms of geometric structure using nuclear resonance vibrational spectroscopy (NRVS) and electronic structure using magnetic circular dichroism (MCD) to define the frontier molecular orbitals (FMOs) that control reactivity.
For FeIII–OOH intermediates the anticancer drug Activated Bleomycin is shown to be the non-heme Fe analog of compound 0 in heme (e.g. P450) chemistry but undergoes different reactivity where the low-spin (LS) FeIII–OOH can directly abstract an H atom from DNA. It is also shown that the transition states of LS and high-spin (HS) FeIII–OOH are fundamentally different in that the former goes through a hydroxyl radical while the latter is activated for EAS without O-O cleavage, which is important in one class of NHFe enzymes that utilizes a HS FeIII–OOH intermediate in dioxygenation.
For FeIV=O intermediates the LS form is shown to have a π-type FMO activated for attack perpendicular to the Fe–O bond while the HS form (present in the NHFe enzymes) has both π and σ FMOs that are activated for attack both perpendicular to and along the Fe–O bond, respectively. For the NHFe enzymes these π vs σ FMOs direct reactivity for EAS vs H-atom abstraction, and for the latter halogenation vs hydroxylation.
This study emphasizes that experimental spectroscopy is critical in evaluating the results of electronic structure calculations and thus key to bridging structure and reactivity with mechanism.
Introduction
Mononuclear non-heme Fe (NHFe) enzymes play key roles in metabolic pathways, hypoxic response, DNA repair, natural product and antibiotic synthesis, and bioremediation.1,2 Mutations in a number of these enzymes lead to disease states such as phenylketonuria in the case of phenylalanine hydroxylase. The NHFe antibiotic Bleomycin (BLM) is a key drug used in treating head, neck, and testicular cancers. At a molecular level these NHFe sites catalyze a variety of oxidation reactions that include H-atom abstraction for hydroxylation, halogenation, desaturation or ring closure of a substrate and electrophilic aromatic substitution (EAS) for mono- or dioxygenation. As presented in Table 1, these can be broadly divided into two classes. The lipoxygenases and intradiol dioxygenases use a high-spin (HS) NHFeIII center to activate singlet organic substrates for the spin-forbidden reaction with 3O2. However, most NHFe enzymes use a HS NHFeII center to activate O2. The latter group divides into six subclasses (Table 1A). For the pterin- and α-ketoglutarate (αKG)-dependent enzymes the cofactor and FeII each supply two electrons to reduce O2 to form an S = 2 FeIV=O intermediate.3 For the pterin family FeIV=O catalyzes EAS while in the αKG family this ferryl intermediate reacts with substrate via H-atom abstraction. For the extradiol dioxygenases two electrons are supplied by the catecholate substrate to reduce O2 and form a peroxy-quinone-bridged FeII intermediate.4 In the Rieske dioxygenases (RDOs), the Fe2S2 Rieske center is bridged through an H-bond to the FeII catalytic site and together these transfer two electrons to activate O2 via a high-spin FeIII–OOH for cis-dioxygenation of an organic substrate.4 There is also a group of NHFeII enzymes (represented by isopenicillin-N-synthase, IPNS, in Table 1A) that bind non-redox-active substrates and use O2 for either the two-electron or four-electron oxidation/oxygenation of the substrate. In IPNS this reaction appears to involve the initial, thermodynamically-difficult one-electron reduction of O2 to form a HS FeIII—O2•− intermediate.5
TABLE 1.
NHFe enzymes.
| A. Oxygen-Activating (FeII)
| |
| Extradiol Dioxygenases |
|
| Pterin-Dependent Hydroxylases |
|
| α-KG-Dependent Dioxygenases |
|
| Rieske Dioxygenases |
|
| Non-Redox Substrate Enzymes |
|
| Bleomycin |
|
|
| |
| B. Substrate-Activating (FeIII)
| |
| Lipoxygenases |
|
| Intradiol Dioxygenases |
|
Almost all the NHFeII enzymes utilize a 2-His-1-carboxylate facial triad ligand set of protein residues to bind the FeII, with additional coordination positions (occupied by waters in the resting enzyme) that are potentially available for O2, substrate and cofactor binding. The drug BLM also has a NH ligand set (vide infra). BLM uses FeII to bind O2 and with one exogenous electron forms activated BLM (ABLM), a low-spin (LS) FeIII–OOH site that H-atom abstracts from DNA for strand cleavage in its anti-cancer activity. ABLM is the NHFe analog of compound 0 in P450 heme chemistry (also a LS FeIII–OOH). The latter goes on to protonate and cleave heterolytically to generate compound I (a (Por•−)FeIV=O species6) that H-atom abstracts from strong C-H bonds of substrates.
In this Account we focus on the NHFeII enzymes and BLM (see ref. 7 for a perspective on NHFeIII substrate-activating enzymes). We first consider the geometric and electronic structures of the resting FeII centers and their interactions with substrates that turn on O2 activation. The NHFeII active site has been challenging to study as the π-π* transitions of heme Fe are absent, and these d6 ions have an S = 2 ground state, which is an integer-spin, non-Kramers system where all the spin degeneracy is eliminated by the low-symmetry protein environment and thus produces no EPR signal at X-band. The ferrous active sites in these enzymes have been made accessible through the development of a near-IR (NIR) variable-temperature, variable-field magnetic circular dichroism (VTVH MCD) spectroscopic methodology. This has led to the determination of a general mechanistic strategy used by the NHFeII enzymes in Table 1A. This methodology is surveyed in section 1.1 and used to develop structure/function correlations in section 1.2. For the RDOs and BLM, the O2 is activated at the hydroperoxo level. In section 2 we consider how the reactivity of the LS FeIII—OOH of the NHFe site in ABLM relates to that of compound 0 in P450, and the activation of HS FeIII—OOH for EAS as in the RDOs. The emphasis here is on the frontier molecular orbitals (FMOs) and how they change with spin. For the αKG- and pterin-dependent NHFe enzymes, the oxygen intermediate key to reactivity is the HS FeIV=O species. In section 3.1 we use spectroscopy on structurally-defined LS and HS FeIV=O model complexes to experimentally determine the FMOs that are important for reactivity, and in section 3.2 we consider how these FMOs direct H-atom abstraction vs. EAS and halogenation vs. hydroxylation.
1. Ferrous active sites
1.1 NIR VTVH MCD Methodology
From ligand field theory (LFT), an Oh FeII d6 HS site has a 5T2g ground state and at an energy 10Dq above this is the spin-allowed 5Eg excited state, where for the N/O facial triad and H2O ligation 10Dq ≈ 10,000 cm-1.1 These states are orbitally-degenerate and will split in energy in the low-symmetry protein environment. For a six-coordinate (6C) FeII site the 5Eg can split by up to 2000 cm-1. Loss of one ligand to form a five-coordinate (5C) square pyramidal FeII center leads to a large 5E splitting, producing LF transitions in the >10,000 cm-1 and ≈5000 cm-1 regions. In a 5C trigonal bipyramidal structure the LF changes and the transitions shift to <10,000 cm-1 and <5000 cm-1, and in a 4C distorted tetrahedral site, since 10Dq (Td) = −4/9 10Dq (Oh), there are only LF transitions in the ≈5000–7000 cm-1 region. Thus from Figure 1A the energy splittings of the d orbitals and associated d→d transitions are very sensitive to the LF of the FeII site.
FIGURE 1.
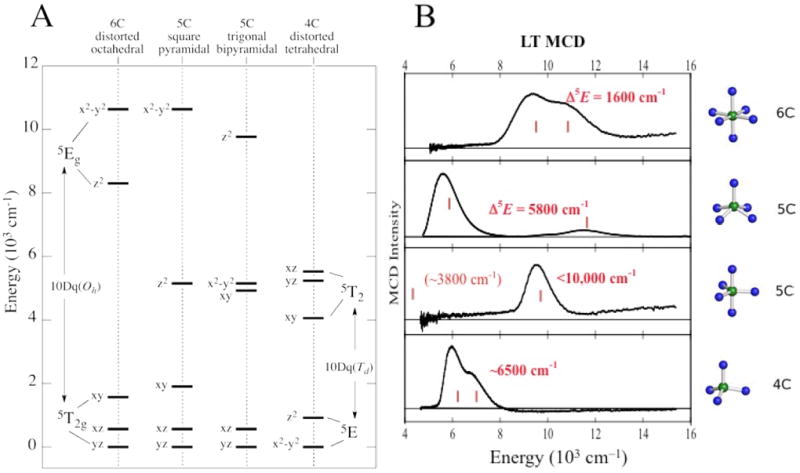
NIR MCD transitions for NHFeII sites. (A) Theoretical d orbital splittings for various geometries. (B) Experimentally-observed transitions.
Since d→d transitions are parity-forbidden, they are weak in absorption (abs.) and since these are in the NIR spectral region where intense vibrations from the protein and buffer contribute, these cannot be studied by abs. spectroscopy. However, since the ground state is S = 2, the sites can be directly studied by NIR MCD at low temperature (LT) where the “C-term” MCD signals of paramagnetic centers are orders of magnitude larger than those of the diamagnetic background. The LT MCD spectra of >20 structurally-defined FeII complexes with different coordination environments have been studied and found to be as predicted by LFT as shown in Figure 1B.1
From Figure 2A the threefold-degenerate 5T2 ground state also splits in energy in the low- symmetry protein environment, reflecting differences in dπ interactions with the ligands. This can be studied directly by VTVH MCD.1 A HS FeII center will produce a set of nested saturation magnetization curves as shown in Figure 2B. This nesting is due to the nonlinear energy splitting and mixing of the MS sublevels of the S = 2 ground state, and fitting these data gives the ground-state splitting (δ, g∥) that in turn is determined by the axial (Δ) and rhombic (V) splittings of the dπ orbitals. Thus for the NHFeII sites we are able to experimentally determine the energies of the five d orbitals (Figure 1A), which reflect the active site geometric and electronic structure.
FIGURE 2.
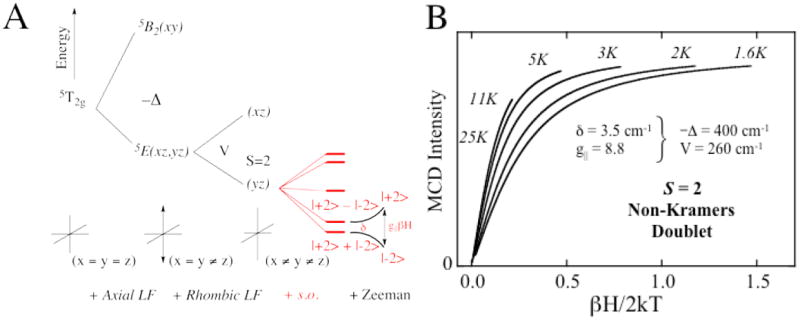
VTVH MCD for FeII sites. (A) Splitting of the 5T2g ground state upon axial and rhombic perturbations. S.O. Refers to the effect of spin-orbit coupling. (B) Nested MCD isotherms for an S = 2 non-Kramers doublet.
1.2 General Mechanistic Strategy
The above methodology has been applied to define structure/function correlations over a number of NHFeII enzymes in each of the subclasses in Table 1A. The data presented in Figure 3 on the pterin-dependent enzyme phenylalanine hydroxylase (PAH) (with Caradonna et al.8) are representative of the behavior we observe for all three pterin-dependent enzymes and in general for all the subclasses of NHFeII enzymes. Resting PAH exhibits the LT MCD spectrum in black, showing two LF transitions in the ~10,000 cm-1 region split by ~2,000 cm-1. Thus the resting FeII site is 6C. Upon addition of phenylalanine substrate the blue spectrum is obtained, showing a slightly perturbed 6C FeII site. Addition of pterin to the resting FeII enzyme produces the green LT MCD spectrum showing that the pterin cofactor does not bind to the FeII. Importantly when both pterin and substrate are added to resting PAH, the LT MCD spectrum in red is obtained with one LF transition at ≈10,000 cm-1 and one in the ≈5,000 cm-1 region. Also, the VTVH MCD saturation magnetization curves change dramatically upon substrate-plus-cofactor binding to resting FeII PAH (Figure 3B). These data give the experimental LF splittings of the five d orbitals in Figure 3C. In the resting, substrate-bound and only-cofactor-bound FeII forms, 10Dq is large and there is only a limited splitting of E and T2 orbital degeneracies, reflecting 6C sites. Alternatively when both substrate and cofactor are bound to the enzyme, 10Dq greatly decreases and there is a large splitting of E and T2 degeneracies, all showing that the FeII active site becomes 5C upon binding both cosubstrates.
FIGURE 3.
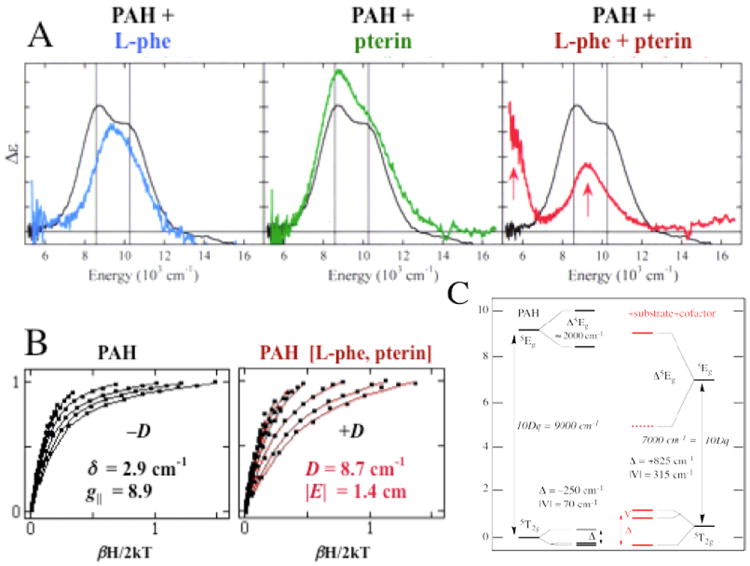
MCD studies of PAH. (A) MCD spectra of resting PAH (black) with substrate-bound (blue), pterin-bound (green) and substrate/pterin-bound (red). All spectra were collected at 5 K. (B) VTVH MCD data for resting (left) and substrate/pterin-bound (right) PAH. (C) Derived d-orbital energy level diagrams.
Parallel NIR VTVH MCD studies have been performed over a range of other NHFeII enzymes which have led to the general mechanistic strategy used by this class of enzymes shown in Figure 4. The resting FeII state and sites with either substrate or cofactor bound are 6C and relatively stable to reaction with O2, which at this stage would lead to uncoupled turnover and potentially self-hydroxylation. However, when both the substrate and the cofactor are bound to the enzyme the FeII site becomes 5C, with an open coordination position for O2 activation.
FIGURE 4.
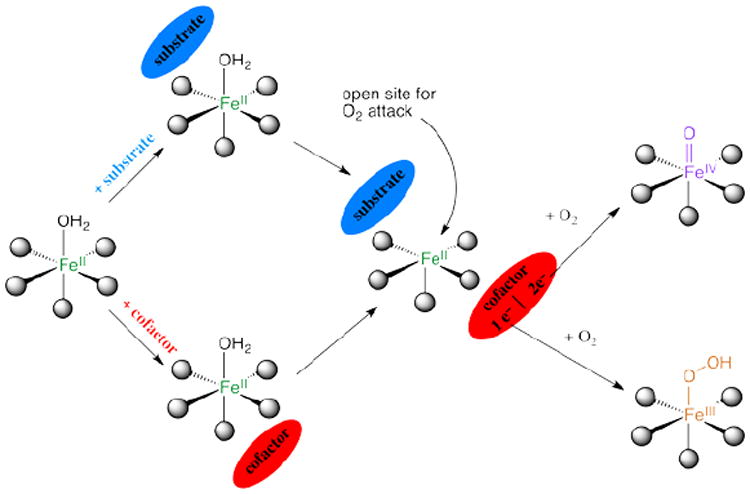
General mechanistic strategy for O2-activation by NHFeII enzymes.
This 6C→5C conversion involves the loss of an H2O ligand. The molecular basis for the substrate-induced H2O loss has been defined (in collaboration with Knapp et al.9) in FIH (involved in hypoxic signaling) and has been found to involve both steric clashes between H2O and substrate in the 6C form and stabilization of the 5C form through H-bonding to the facial triad carboxylate (Figure 5A).
FIGURE 5.
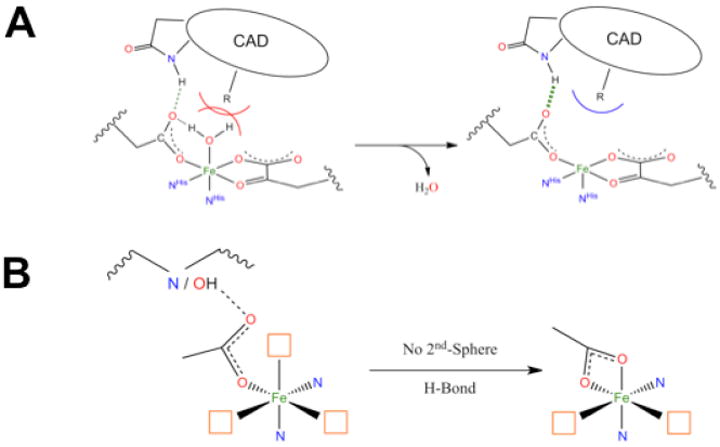
(A) Steric and H-bonding contributions to loss of coordinated H2O. CAD stands for C-terminal activation domain of the substrate HIF-1α. (B) Effect of a second-sphere H-bonding partner on the facial triad carboxylate.
With respect to the facial-triad carboxylate, there are interesting differences among the different subclasses that reflect second-sphere interactions that are important for O2 reactivity. For the αKG-dependent and extradiol enzymes the cofactor/cosubstrate binds bidentate to FeII. As O2 must also bind to FeII for catalysis, three coordination sites are needed for reactivity (Figure 5B, left). However, for a 5C FeII site the facial-triad carboxylate will switch to bidentate coordination (Figure 5B, right), which would prevent catalysis.9 Extradiol dioxygenases and αKG-dependent enzymes avoid this by having a second-sphere residue available to H-bond to the facial-triad carboxylate. For the pterin-dependent and RDO enzymes, neither substrate nor cofactor binds to the FeII. Members of these subclasses do not have a second-sphere residue H-bond to the facial triad carboxylate; thus it coordinates to the FeII in a bidentate mode, allowing O2 to bind in a single orientation for proper reactivity (Figure 5B, right). For pterin-dependent enzymes, O2 must bind in an end-on fashion for bridging between FeII and the pterin cofactor, cleave, and rearrange for reaction with substrate. For Rieske dioxygenases, a side-on bidentate binding mode for O2 is required for formation of the reactive intermediate.
2. FeIII–hydroperoxy intermediates
FeIII–OOH intermediates are important in ABLM which is LS (S = 1/2) and in the RDOs which are HS (S = 5/2). ABLM is the NHFe intermediate closest to that in heme chemistry and in section 2.1 we contrast their reactivity. In section 2.2 we evaluate how spin state influences the FMOs and affects the transition state (TS), and its relation to function in the RDOs.
2.1 Low-Spin Non-Heme vs. Heme
(BLM)FeII binds O2 and takes up an exogenous e− to form ABLM, a LS ferric-hydroperoxide (Figure 6).10 ABLM is the intermediate before H-atom abstraction from the C4’ position of DNA.11 In the heme analog, compound 0 of P450, this LS FeIII–OOH protonates and heterolytically cleaves to form compound I, an FeIV=O porphyrin radical, that H-atom abstracts from strong H–C bonds. There has been significant interest in whether ABLM parallels heme chemistry to generate an unobserved compound I analog that does the DNA cleavage. Thus, we used calculations supported by the spectroscopic data to elucidate the relative energies of the protonation/heterolytic cleavage reactions in ABLM and compound 0 in P450. From Table 2, this reaction is calculated to be 71 kcal/mol less favorable in ABLM relative to P450 and endergonic by 13 kcal/mol.12 This reflects the fact that delocalization of the hole over the π system of the porphyrin and its −2 charge facilitate ligand oxidation in heme relative to a non-heme ligand set, as evidenced by the much-higher-energy of the lignd-to-metal charge transfer (LMCT) transitions of (BLM)FeIII as compared to heme (Figure 7).
FIGURE 6.
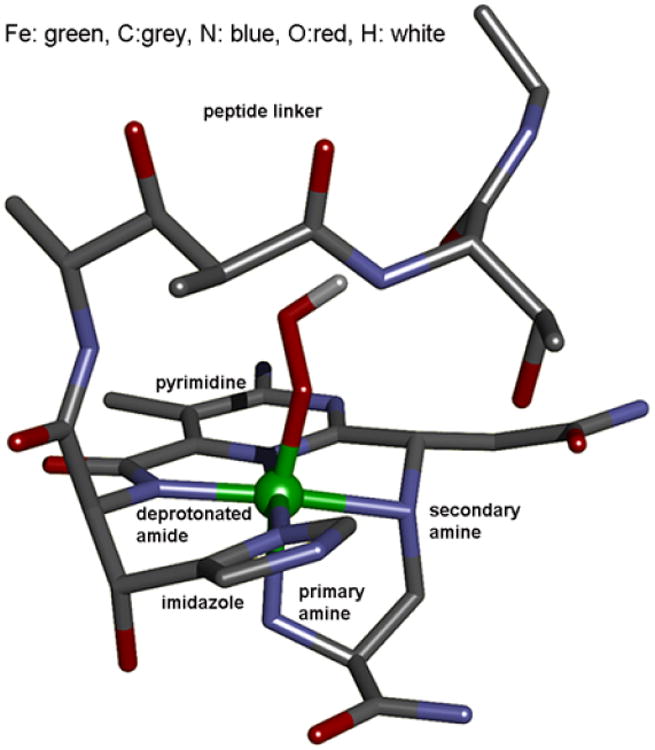
Optimized structure of ABLM.
TABLE 2.
Thermodynamics of heterolytic cleavage of O–O bonds in P450 and ABLM and direct H-atom abstraction by ABLM.
| Reactant | Thermodynamics (kcal/mol) | |||
|---|---|---|---|---|
| ΔE | ΔE(solv) | −TΔS | ΔG | |
| FeIII(Por)(SCH3)–OOH Heterolytic | −75 | −52 | −6 | −58 |
| FeIII(BLM)–OOH Heterolytic | +99 | +20 | −7 | +13 |
| FeIII(BLM)–OOH Direct H-atom | +13 | +9 | −16 | −7 |
FIGURE 7.
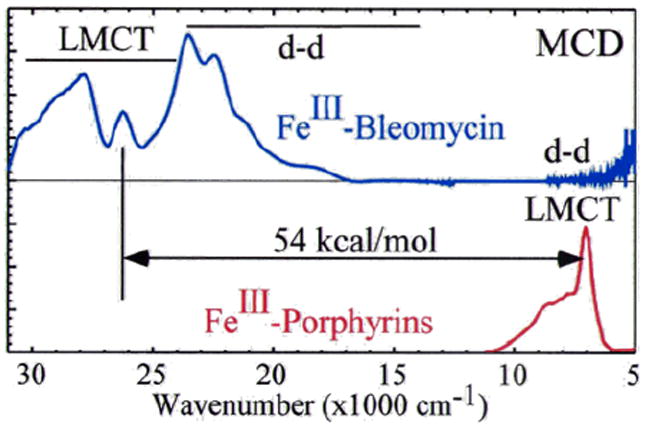
MCD spectral comparison of LMCT energies of (BLM)FeIII (deprotonated amide to FeIII) and a prototypical FeIII-heme (porphyrin to FeIII).
These considerations lead to the alternative hypothesis of direct H-atom abstraction from the substrate by ABLM.12 From the calculations listed in Table 2, this reaction is 20 kcal/mol more favorable than the heterolytic cleavage reaction and exergonic by 7 kcal/mol.
However, a computational study considering the peroxo-shunt reaction argued for a (BLM)FeIII—H2O → (BLM)FeIII—H2O2 conversion, and the presence of the additional proton would make the heterolytic cleavage more favorable by 15 kcal/mol, comparable to direct H-atom abstraction by (BLM)FeIII–OOH.13
Thus it was necessary to experimentally determine the nature of the axial ligands in (BLM)FeIII and ABLM. Since ABLM is unstable in a laser, we turned to nuclear resonance vibrational spectroscopy (NRVS). From Figure 8, the spectrum for (BLM)FeIII shows an H2 18O isotope-sensitive vibration at 567 cm−1 which, combined with an EXAFS distance of 1.86 Å and computational modeling (Figure 84, bottom), assigns the axial ligand as hydroxide.10 It also exhibits a dominant feature at 407 cm−1, which reflects the degenerate pair of trans-axial bends (Figure 8C) that has dominant Fe motion. In going to the NRVS spectrum of ABLM, this pair splits into two peaks at 438 and 398 cm−1 and an additional feature is present at 328 cm−1. This trans-axial splitting is only reproduced by the FeIII–OOH structure of ABLM where the 328 cm−1 peak is the Fe–O–O bend and this mixes with the in-plane trans-axial bend leading to the peak splitting.
FIGURE 8.
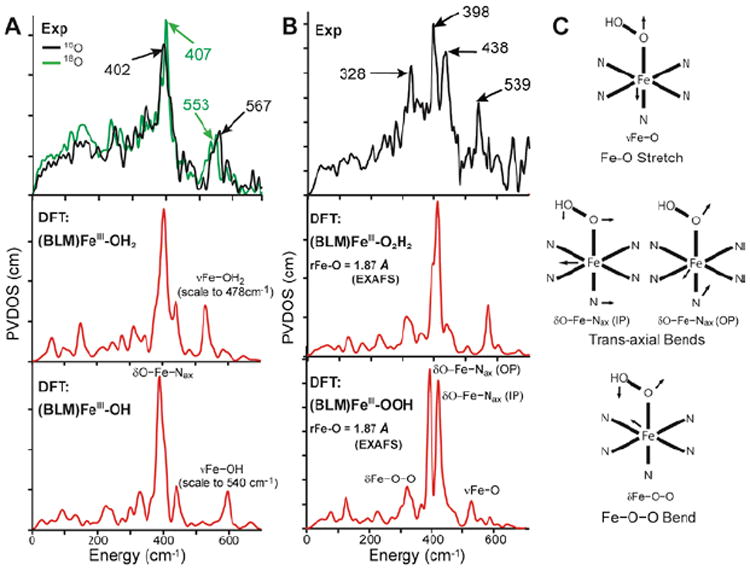
(A) NRVS spectra (PVDOS) of 16O and 18O (BLM)FeIII (top) and DFT-simulated PVDOS spectra of two models with H2O (middle) and OH− (bottom) as axial ligands. (B) NRVS spectrum of ABLM (top) and DFT-simulated spectra with H2O2 (middle) and OOH− (bottom) as axial ligands. (C) Major NRVS-active vibrational modes of ABLM.
ABLM is confirmed to be a LS FeIII–OOH species; thus its protonation and heterolytic cleavage to produce a heme-type compound I is unfavorable by 20 kcal/mol relative to its direct H-atom abstraction from DNA, Table 2. The direct reaction of ABLM requires that DNA participate in the TS. DNA does in fact accelerate this reaction by ~2.5 times and changes ABLM’s decay kinetics from having a relatively-small primary KIE (3.6 ± 0.9, from self-decay) to a large secondary KIE with DNA (1.7 ± 0.2, non-exchangeable H-C4’ group).14
These experimental data were modeled with a 2D potential energy surface (PES) for the H-atom abstraction from the H–C bond of a substrate by ABLM (Figure 9A).15 A TS was found that reproduces experiment with an Ea of 13 kcal/mol, about half that calculated for heterolytic cleavage and H-atom abstraction. The TS for direct FeIII–OOH attack on DNA also reproduces the small primary and large secondary KIEs that reflect a TS that is late in O–O cleavage but early in H-atom abstraction (Figure 9B).
FIGURE 9.
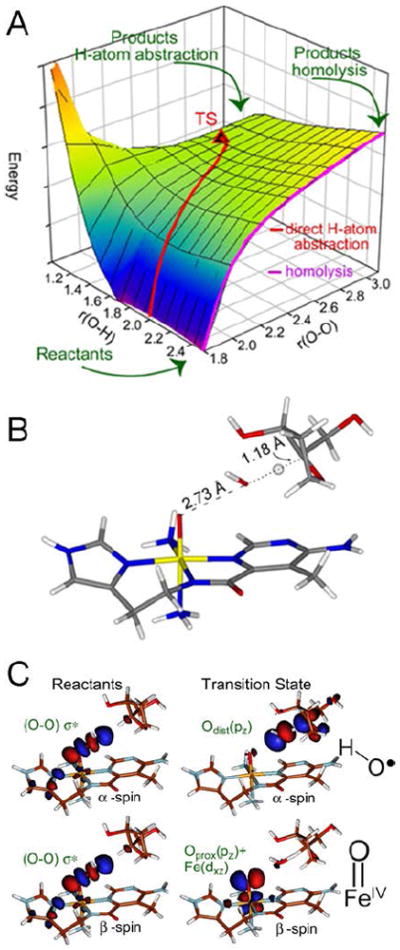
(A) PES for H-atom abstraction by ABLM. (B) TS of direct H-atom abstraction. (C) FMOs of ABLM before (left) and at the TS (right).
This relates to the FMO of the LS FeIII–OOH, which is the lowest-unoccupied hydroperoxide σ* orbital. From Figure 9C, elongation of the O–O to the TS greatly weakens the peroxo σ/σ* interaction and allows its α- and β-spin holes to polarize: one on the distal O making this an OH• radical that is efficient in H-atom abstraction, and the other localizes on the proximal O generating an FeIV=O species. The latter is effective in performing a second H-atom abstraction from DNA, leading to double-strand cleavage important for BLM’s anticancer activity.
2.2 Low-Spin to High-Spin FeIII-OOH FMOs
In going to the RDOs the FeIII–OOH intermediate is HS.4 Spectroscopic studies were performed (with Nam and co-workers16) on S = 1/2 (N4Py)FeIII–OOH and the S = 5/2 (TMC)FeIII–OOH models and correlated to electronic-structure calculations to understand differences in their reactivities. For LS FeIII–OOH the TS for H-atom abstraction is again late in O–O cleavage, generating an OH• for H-atom abstraction.
In contrast, in the HS16 FeIII–OOH complex the TS is early in O–O cleavage and later in H-atom abstraction, Figure 10A. The origin of this difference in TSs is the spin-state dependence of O–O bond homolysis (Figure 10B), where the HS FeIII–OOH has an additional 12 kcal/mol barrier. This derives from the different interactions between redox active orbitals in LS and HS FeIII–OOH (Figure 10 C vs. D).17
FIGURE 10.
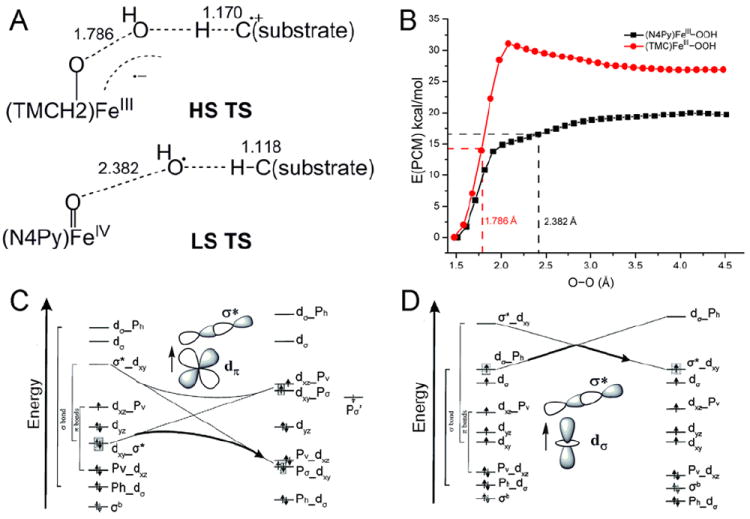
(A) TSs of HS (TMC)FeIII–OOH and LS (N4Py)FeIII–OOH in H-atom abstraction. (B) PESs of HS (TMC)FeIII–OOH and LS (N4Py)FeIII–OOH O–O homolysis. Calculations were performed using the Polarizable Continuum Model (PCM) with a dielectric constant of acetone. (C) and (D) Forbidden and allowed orbital crossings for O–O bond homolysis of LS and HS FeIII–alkylperoxo complexes. (See Ref 17 for details)
The relatively-low barrier for O–O homolysis in the LS system allows generation of an OH• for H-atom abstraction. Alternatively for the HS FeIII–OOH complex, the larger barrier for homolysis leads to a relatively-short O–O bond (1.79 Å) at the TS for H-atom abstraction (Figure 10A). At this TS significant electron density is transferred from the substrate into the FeIII–OOH LUMO due to the higher reduction potential of the HS complex. This leads to an important spin-state dependence of the reaction barrier on the substrate. LS FeIII–OOH (eg. ABLM) is favored for H-atom abstraction from strong H–C bonds while HS FeIII–OOH is activated for electrophilic attack on substrates without O–O bond cleavage. The latter is important for the RDOs where both oxygen atoms of the hydroperoxide are inserted into cis positions of aromatic substrates.
3. FeIV=O intermediates
For the αKG and pterin-dependent enzymes the cofactor and FeII each donate 2e− to dioxygen to generate S = 2 FeIV=O intermediates. The pterin enzymes perform electrophilic aromatic substitution (EAS); the αKG-dependent enzymes abstract an H-atom, followed by rebound hydroxylation for the facial-triad enzymes, or halogenation in the halogenases where the facial-triad carboxylate is replaced by a halide. In section 3.1 we consider the FMOs available for these reactivities in structurally-defined S = 1 and S = 2 FeIV=O model complexes.18,19 In section 3.2 we extend these FMO concepts to understand the factors that control EAS vs. H-atom abstraction, and for the latter the factors that control hydroxylation vs. halogenation.
3.1 FMOs
The bonding description of the first structurally-defined S = 1 FeIV=O model complex (TMC)FeIV=O18 is shown in Figure 11: at high energy is the dz2 orbital which is strongly σ antibonding with the oxo pz, and at lower energy is the Fe π*-dxz,yz – oxo px,y combination, forming the dπ* β FMOs available for reactivity. These FMOs were studied directly (with Que et al.20) by using abs. and MCD spectroscopies to probe the dxy(nb)→dxz,yz(π*) excitation. The weak, relatively-uninformative abs. band at ~12,000 cm−1 (Figure 12A) results from three overlapping transitions as revealed by VT MCD (Figure 12B). These three transitions have different temperature-dependent MCD behaviors that reflect the different polarizations of the electronic transitions. Importantly, overlapping band I is the negative, sharp band II, the lowest-energy (x,y)-polarized dxy→dxz,yz LF transition. Band II directly experimentally probes the dπ* FMO of this FeIV=O complex and shows (from the associated vibrational progression, Figure 12C) a strong π-antibonding interaction which shifts significant O pπ character into the dπ* orbitals, activating them for electrophilic attack perpendicular to the Fe—O bond.20
FIGURE 11.
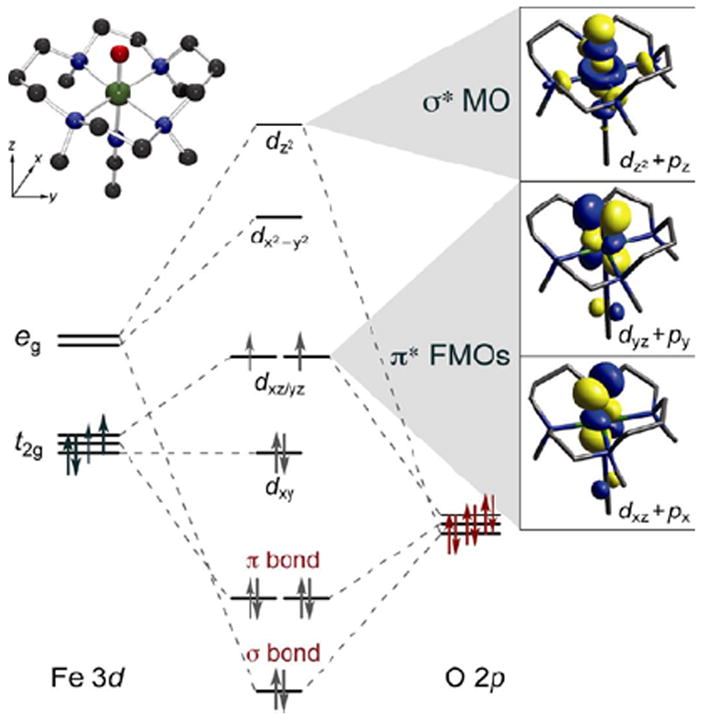
MO diagram for S = 1 (TMC)FeIV=O (inset, top left) producing π* FMOs and σ* MO (right).
FIGURE 12.
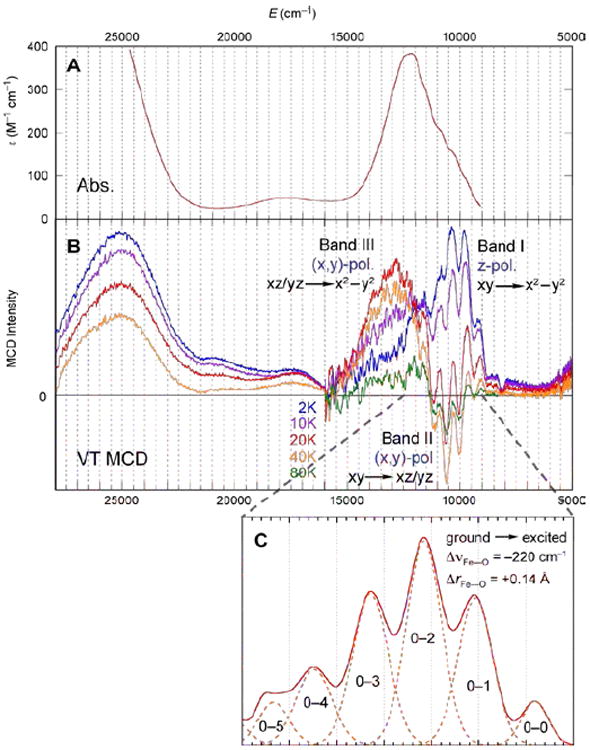
(A) 233 K Abs. and (B) VT MCD spectra of (TMC)FeIV=O. (C) Vibronic progression of band II (40 K; plotted positive).
In going from an S = 1 to an S = 2 ground state of FeIV=O, a β e− in dxy is transferred into the dx2−y2 orbital. This does not affect the FeIV=O bond; however, there is a significant effect on the low-lying unoccupied FMOs. From the spin-unrestricted MO diagram in Figure 13, excitation of a β e− from the dxy into the α-dx2−y2 orbital leads to stabilization of the α-spin manifold due to the spin polarization associated with differences in exchange. This brings the α-dz2 σ* orbital down to an energy comparable to the dπ* FMO. However, the dz2 σ* has oxo pz character oriented along the Fe—O bond (Figure 13C). Thus, for the S = 2 NHFeIV=O intermediates there are two low-lying unoccupied FMOs available for H-atom abstraction and EAS: the dπ* FMO with oxo px,y character perpendicular to the Fe—O bond, and the dσ* FMO with oxo pz character for attack along the Fe—O bond.
FIGURE 13.
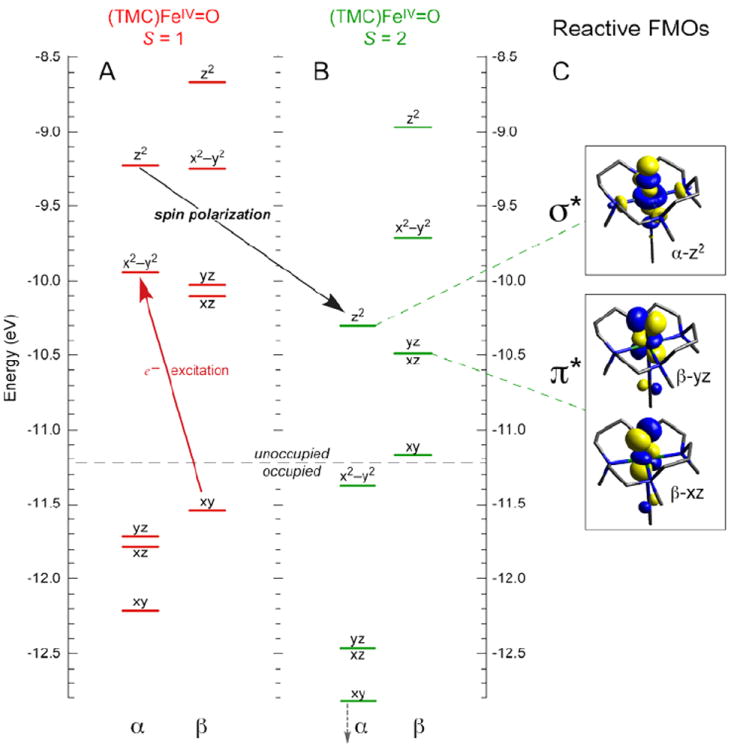
MO diagrams of (TMC)FeIV=O in (A) S = 1 (ground) and (B) S = 2 states, showing that excitation of β-dxy e− into α-dx2–y2 orbital leads to spin-polarization of the α-manifold and a low-energy α-dz2 FMO for reactivity. (C) Isosurface plots of S = 2 FMOs.
Studies on the first structurally-defined S = 2 FeIV=O complex (TMG3tren)FeIV=O19 (with Que et al.21) revealed relatively-uninformative low-energy weak and high-energy intense abs. bands, but high-information-content LT MCD spectra (Figure 14). The band at ~12,000 cm−1 is assigned as the lowest-energy 5A → 5E (dxz,yz → dz2) LF transition, based on its pseudo-A (derivative) shape and temperature-dependent behavior. Interestingly, the sharp positive peak overlapping this band corresponds to a dip in abs. (dashed line), indicating a Fano antiresonance of this allowed 5E state with a spin-forbidden 3A. Furthermore, the two different vibronic progressions associated with this 5E indicate strong spin-orbit coupling (SOC) with another nearby triplet (3E) which distorts the 5E PES. Also, from the MCD spectra the low-energy shoulder in the abs. spectrum at ~20,000cm−1 corresponds to a pronounced, negatively-signed z-polarized (i.e. Fe—O) vibronic progression with a νFe—O of 490 cm−1, greatly reduced from that of the ground state (~830 cm-1). These excited-state properties led to the assignment of this band as the oxo π → dxz,yz CT transition. Thus, spectroscopy on (TMG3tren)FeIV=O reveals three FMOs: the dz2 σ* ground state, the low-lying dπ* and a higher-energy oxo π FMO.21
FIGURE 14.
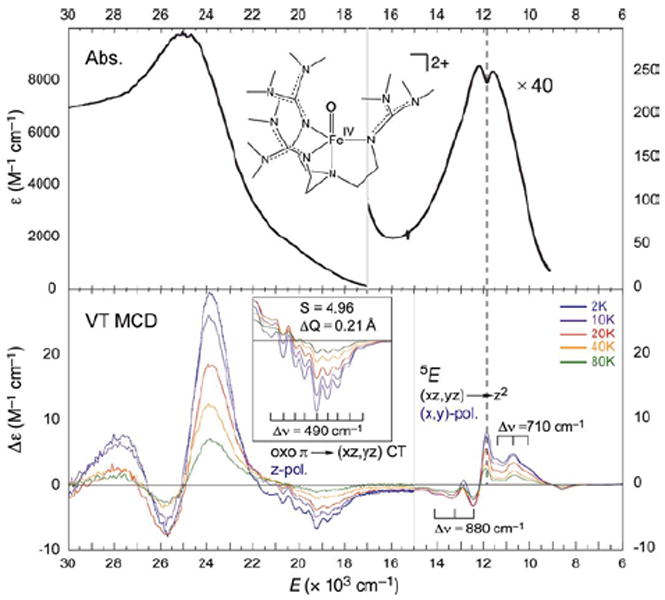
233 K Abs. (top) and VT MCD (7T, bottom) spectra of (TMG3tren)FeIV=O showing structured features corresponding to FMOs.
CASPT2 calculations reproducing these spectral features (Figure 15) were extrapolated to the Fe—O bond length associated with the TS for H-atom abstraction.21 These low-lying states generate (Figure 15, right) three comparable (in energy and coefficients on the oxo) FMOs – one σ and two π’s – with the former oriented for attack along the Fe—O bond and the latter two oriented for perpendicular attack on substrates. Importantly, all three FMOs polarize at the TS, generating FeIII—oxyl species with high oxygen p hole character for effective electrophilic reactivity.
FIGURE 15.
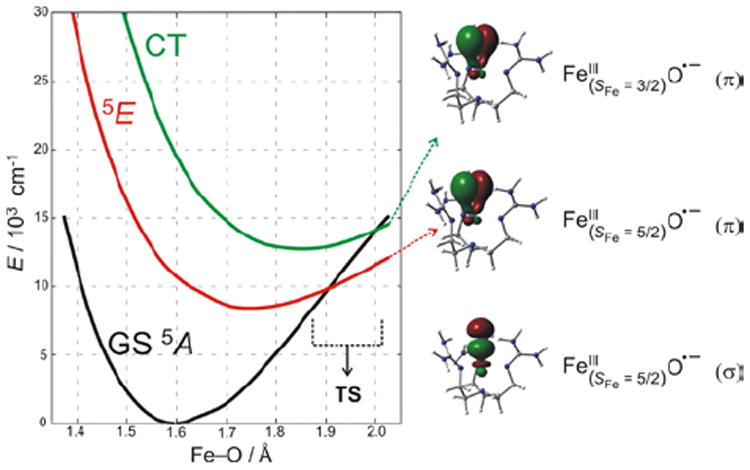
(left) PES of S = 2 states along the Fe—O coordinate for (TMG3tren)FeIV=O, which lead to one σ and two π FMOs of FeIII—oxyl character (right) at the TS.
Thus, spectroscopy correlated to electronic-structure calculations has revealed that in contrast to FeIV=O S = 1 species where only a π-FMO is available for electrophilic reactivity perpendicular to the FeIV=O bond, FeIV=O S = 2 intermediates have both σ and π-FMOs available for reactivity, respectively along and perpendicular to the FeIV=O bond.
3.2 Structure/Function Correlations
3.2.1 H-atom abstraction vs. Electrophilic aromatic substitution
(4-hydroxyphenyl)pyruvate dioxygenase (HPPD) and (4-hydroxy)mandelate synthase (HmaS) both react with the same substrate, (4-hydroxyphenyl)pyruvate (HPP); HPPD performs EAS on the ring, while HmaS performs H-atom abstraction (Figure 16). HPP has an α-keto acid group that binds in a bidentate mode that directly tethers this substrate to the FeII. From the CD data (with Moran and Spencer et al.22) in Figure 17, HPP is bound to the two FeII enzymes with different conformations of the ring, consistent with their different reaction products (Figure 16). The O2 reaction22 produces the two FeIV=O intermediates (Figure 18). For HPPD, the Fe–O bond is oriented for σ electrophilic attack on the aromatic ring, while in HmaS the Fe–O bond is oriented perpendicular to the H–C bond of the substrate and activated for π H-atom abstraction. For both reaction coordinates the Fe–O bond elongates and polarizes at the TS to become FeIII–O•−. This provides low-lying unoccupied oxo pσ (for HPPD) and pπ (for HmaS) FMOs, leading to comparable low-energy barriers for EAS (16 kcal/mol, HPPD) and H-atom abstraction (14 kcal/mol, HmaS).22
FIGURE 16.
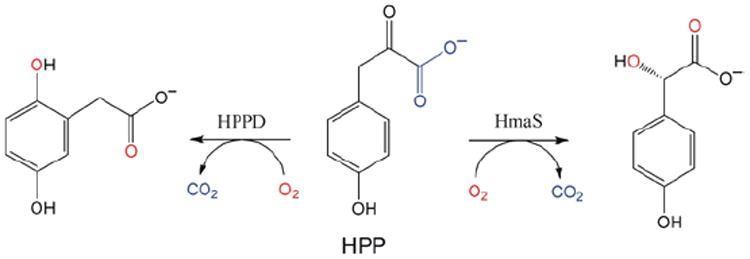
Reactions of HPP with HPPD (left, EAS) and HmaS (right, H-atom abstraction) to produce homogentisate and (4-hydroxy)mandelate respectively.
FIGURE 17.
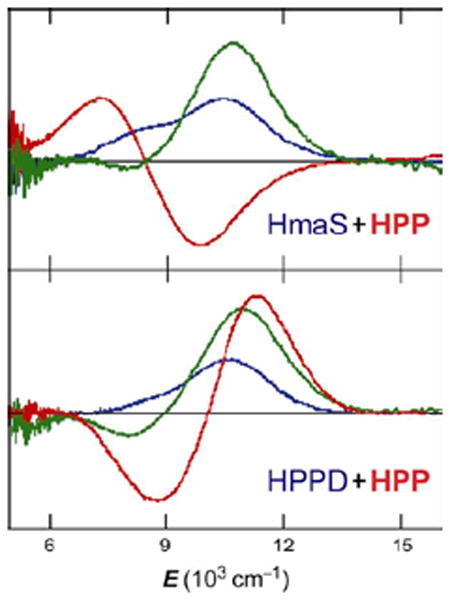
CD spectra of HPP binding (red lines) to HmaS (top) and HPPD (bottom) FeII sites. Resting FeII d-d transitions (blue), and pyruvate-bound-FeII (green) as controls.
FIGURE 18.
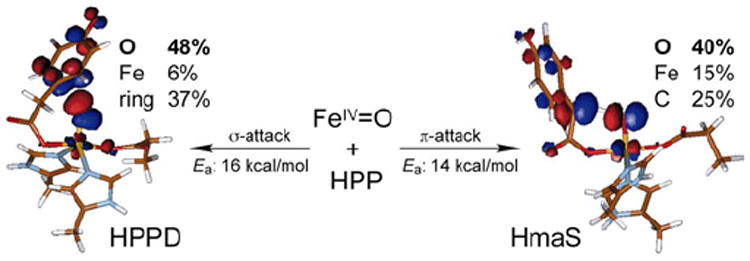
Reactivities of FeIV=O intermediates in HPPD (left) and HmaS (right), with different substrate (HPP) conformations leading to a σ TS for EAS in HPPD and a π TS for H-atom abstraction in HmaS. Formation of FeIII—O•− species at TS provides FMOs primed for reactivity.
3.2.2 Halogenation vs. Hydroxylation
For the halogenases that include SyrB2, which catalyzes the chlorination of L-Thr in syringomycin E biosynthesis, the carboxylate ligand of the FeII facial triad is replaced by a halide (Cl−/Br−).3 These halogenases bind αKG and react with O2 to generate an FeIV=O intermediate3 that H-atom abstracts, and in the case of SyrB2 with its native substrate rebounds to form the halogenated product 4-Cl-l-Thr (Figure 19, top). With the alternative substrate l-Nva (Figure 19, bottom), the FeIV=O intermediate H-atom abstracts but rebounds to produce a hydroxylated product.23 Thus, in complement to the studies described in section 3.2.1 where two NHFe enzymes perform different reactions with the same substrate (HPP), for the halogenases one NHFe enzyme perfoms different reactions depending on substrate.
FIGURE 19.
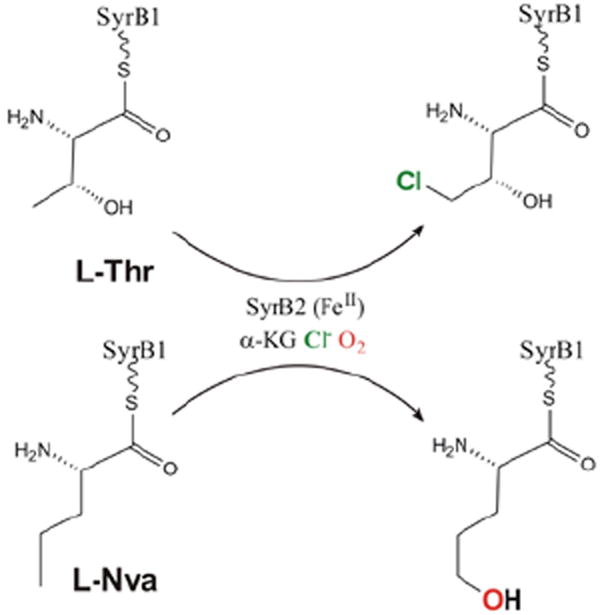
SyrB2 catalyzes both chlorination of native substrate l-Thr (top) and hydroxylation of alternative substrate l-Nva (bottom).
In collaboration with Bollinger and Krebs et al.24 the first NRVS spectra of any oxygen intermediate were obtained. This FeIV=O intermediate can be generated with either Cl− or Br− bound to NHFe active site, providing an important mass perturbation on the NRVS data. The data in Figure 20 show that the FeIV=O intermediate has three dominant peaks in its NRVS spectrum at around 370, 300 and 250 cm−1, and the intensity shifts from the higher-energy peaks in the Cl cognate into the lowest-energy peak in the Br cognate. These NRVS spectra and their Cl→Br perturbation are only reproduced with a 5C TBP structure with the FeIV=O bond defining the trigonal axis (Figure 20, bottom). This is the first direct structural insight into any NHFeIV=O intermediate.24
FIGURE 20.
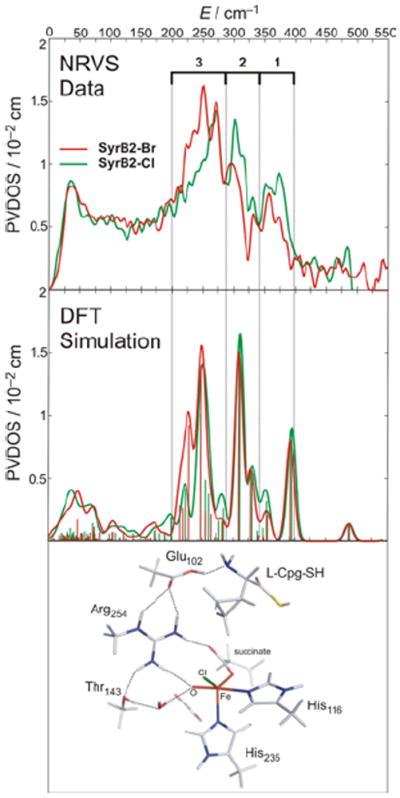
Experimental (top) and simulated (middle) NRVS spectra of FeIV=O intermediate of SyrB2, ligated at FeIV=O active site by either Cl− (green) or Br− (red). Simulated spectra based on TBP structure with inert substrate l-Cpg (bottom).
For correlation to these data, a reaction coordinate was evaluated for SyrB2 using a computational approach calibrated by spectroscopic studies25 on the stable {FeNO}7 analog of the unobserved {FeO2}8 intermediate in HPP-bound HPPD (vide supra). (Figure 21, top right) This is a 5C TBP structure consistent with the NRVS data (Figure 20), with its Fe—O axis oriented perpendicular to the H–C bond of the native substrate. This structure was used to evaluate the H-atom abstraction reaction. The perpendicular orientation leads to a π-attack on the substrate and produces the H-atom abstracted first product with the halide well-oriented for efficient rebound and the hydroxide ligand oriented away from the radical and stabilized by several hydrogen bonds (Figure 22, green).
FIGURE 21.
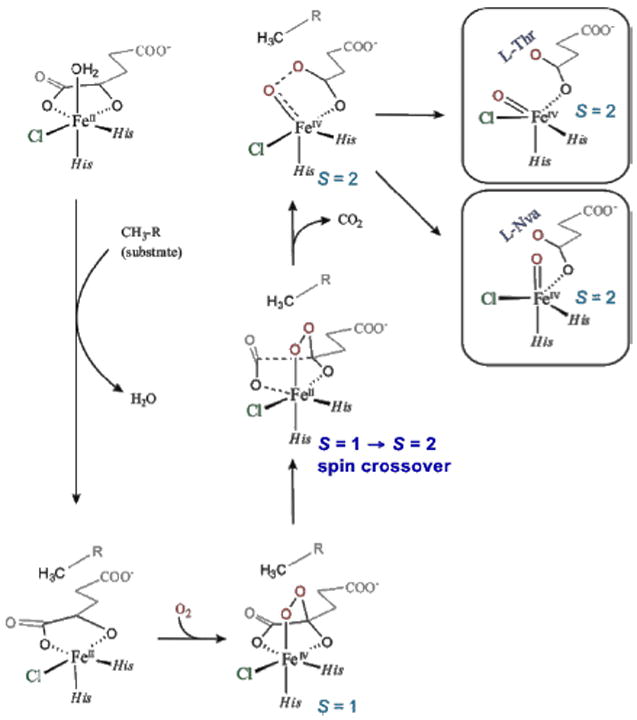
O2 activation in SyrB2 leading to two FeIV=O intermediate orientations relative to substrate (right): perpendicular (native l-Thr) and parallel (non-native l-Nva).
FIGURE 22.
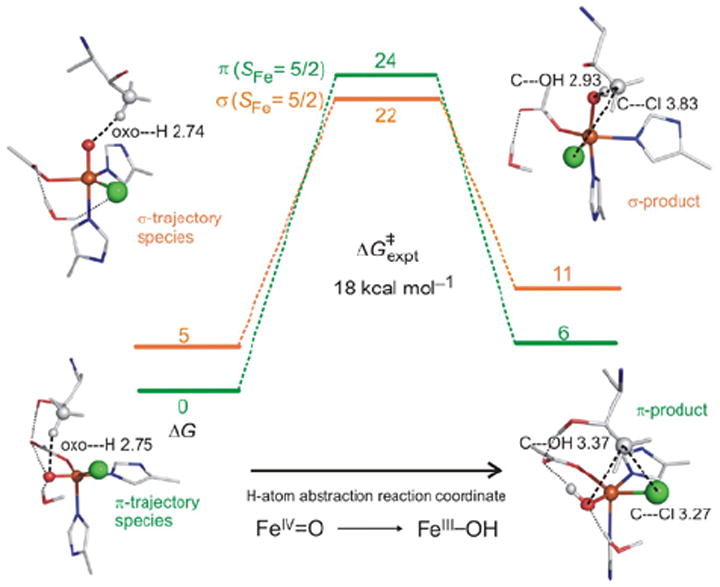
SyrB2 H-atom abstraction reaction coordinates for π-trajectory (green) and σ-trajectory (orange).
In parallel, the O2 reaction coordinate in Figure 21 was evaluated with the alternative substrate l-Nva, where the difference in substrate hydrogen bonding to the peroxy species generated along the reaction coordinate can lead to an FeIV=O intermediate with its Fe—O axis oriented toward the H–C group of the substrate.24 The reaction coordinate with this orientation (Figure 22, orange) involves a σ attack on the H–C bond of the substrate with a comparable barrier to the native substrate. This produces a first product with the halide oriented away from the C radical but with the coordinated hydroxide group well-oriented for efficient rebound to form the hydroxylated product as observed (Figure 22).
As shown by our studies on the HPP-dependent enzymes and the halogenases, the S = 2 FeIV=O intermediate provides both π and σ FMOs. These have different orientation dependencies, providing an active site flexibility in using this orientation to control reactivity.
4. Concluding Comments
Crystallography, combined with enzymology, synthesis and kinetics have revealed important structural types of NHFe enzymes and their reactivities. These are often used as the bases for electronic structure calculations to elucidate mechanism. Here we have emphasized that experimental spectroscopy is key to bridging structure and reactivity with mechanism.
Acknowledgments
This research is funded by the National Institutes of Health (GM 40392).
Biographies
Ed Solomon received his Ph.D. from Princeton (with D.S. McClure) and postdoc-ed at the H.C. Ørsted Institute (with C.J. Ballhausen) and at Caltech (with H.B. Gray). He started his career at MIT in 1975 and moved to Stanford in 1982 where he is the Monroe E. Spaght Professor of Humanities and Sciences.
Kenneth Light received a B.S. and M.S. in chemistry from the Colorado School of Mines and is a PhD student at Stanford University. His research focus has included FeII enzyme control of active site coordination and its contribution to reactivity.
Lei Liu received his B.S. in chemistry from Beijing Normal University and M.S. in chemistry from the University of British Columbia. Lei recently received his Ph.D. from Stanford Univirsity where he focused on peroxo-level intermediates in iron enzymes and models.
Martin Srnec received his Ph.D. from Charles University in Prague and is a postdoctoral fellow at Stanford University. His major interest is in metalloenzymatic reaction mechanisms.
Shaun Wong received his B.S. in chemistry from UC Berkeley and his Ph.D. from Stanford University. His work centered on elucidating the electronic structures and reaction mechanisms of low-and high-spin Fe(IV)–oxo species in enzymes and model complexes.
References
- 1.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 2.Que L, Jr, Ho RYN. Dioxygen Activation by Enzymes with Mononuclear Non-Heme Iron Active Sites. Chem Rev. 1996;96:2607–2624. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- 3.Krebs C, Galonić Fujimori D, Walsh CT, Bollinger JM. Non-heme Fe(IV)-oxo intermediates. Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kovaleva EG, Niebergall MB, Chakrabarty S, Lipscomb JD. Finding Intermediates in the O2 Activation Pathways of Non-Heme Iron Oxygenases. Acc Chem Res. 2007;40:475–483. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown CD, Neidig ML, Lipscomb JD, Solomon EI. VTVH-MCD and DFT Studies of Thiolate Bonding to {FeNO}7/{FeO2}8 Complexes of Isopenicillin N Synthase: Substrate Determination of Oxidase versus Oxygenase Activity in Nonheme Fe Enzymes. J Am Chem Soc. 2007;129:7427–7438. doi: 10.1021/ja071364v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-Containing Oxygenases. Chem Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 7.Pau MYM, Lipscomb JD, Solomon EI. Substrate activation for O2 reactions by oxidized metal centers in biology. Proc Natl Acad Sci USA. 2007;104:18355–18362. doi: 10.1073/pnas.0704191104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kemsley JN, Mitić N, Zaleski KL, Caradonna JP, Solomon EI. Circular Dichroism and Magnetic Circular Dichroism Spectroscopy of the Catalytically Competent Ferrous Active Site of Phenylalanine Hydroxylase and Its Interaction with Pterin Cofactor. J Am Chem Soc. 1999;121:1528–1536. [Google Scholar]
- 9.Light KM, Hangasky JA, Knapp MJ, Solomon EI. Spectroscopic Studies of the Mononuclear Nonheme FeII Enzyme FIH: Second–Sphere Contributions to Reactivity. J Am Chem Soc. 2013;135:9665–9674. doi: 10.1021/ja312571m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu LV, Bell CB, Wong SD, Wilson SA, Kwak Y, Chow MS, Zhao J, Hodgson KO, Hedman B, Solomon EI. Definition of the intermediates and mechanism of the anticancer drug bleomycin using nuclear resonance vibrational spectroscopy and related methods. Proc Natl Acad Sci USA. 2010;107:22419–22424. doi: 10.1073/pnas.1016323107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burger RM, Peisach J, Horwitz SB. Activated Bleomycin - a Transient Complex of Drug, Iron, and Oxygen That Degrades DNA. J Biol Chem. 1981;256:1636–1644. [PubMed] [Google Scholar]
- 12.Neese F, Zaleski JM, Zaleski KL, Solomon EI. Electronic structure of activated bleomycin: Oxygen intermediates in heme versus non-heme iron. J Am Chem Soc. 2000;122:11703–11724. [Google Scholar]
- 13.Kumar D, Hirao H, Shaik S, Kozlowski PM. Proton-Shuffle Mechanism of O–O Activation for Formation of a High-Valent Oxo–Iron Species of Bleomycin. J Am Chem Soc. 2006;128:16148–16158. doi: 10.1021/ja064611o. [DOI] [PubMed] [Google Scholar]
- 14.Chow MS, Liu LV, Solomon EI. Further insights into the mechanism of the reaction of activated bleomycin with DNA. Proc Natl Acad Sci USA. 2008;105:13241–13245. doi: 10.1073/pnas.0806378105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Decker A, Chow MS, Kemsley JN, Lehnert N, Solomon EI. Direct hydrogen-atom abstraction by activated bleomycin: An experimental and computational study. J Am Chem Soc. 2006;128:4719–4733. doi: 10.1021/ja057378n. [DOI] [PubMed] [Google Scholar]
- 16.Liu LV, Hong S, Cho J, Nam W, Solomon EI. Comparison of High-Spin and Low-Spin Nonheme FeIII–OOH Complexes in O–O Bond Homolysis and H-Atom Abstraction Reactivities. J Am Chem Soc. 2013;135:3286–3299. doi: 10.1021/ja400183g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehnert N, Ho RYN, Que L, Jr, Solomon EI. Electronic structure of high-spin iron(III)-alkylperoxo complexes and its relation to low-spin analogues: Reaction coordinate of O-O bond homolysis. J Am Chem Soc. 2001;123:12802–12816. doi: 10.1021/ja011450+. [DOI] [PubMed] [Google Scholar]
- 18.Rohde J-U, In J-H, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L., Jr Crystallographic and Spectroscopic Characterization of a Nonheme Fe(IV)=O Complex. Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 19.England J, Guo Y, Farquhar ER, Young VG, Jr, Münck E, Que L., Jr The Crystal Structure of a High-Spin Oxoiron(IV) Complex and Characterization of Its Self-Decay Pathway. J Am Chem Soc. 2010;132:8635–8644. doi: 10.1021/ja100366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Decker A, Rohde J-U, Klinker EJ, Wong SD, Que L, Jr, Solomon EI. Spectroscopic and Quantum Chemical Studies on Low-Spin FeIV=O Complexes: Fe—O Bonding and Its Contributions to Reactivity. J Am Chem Soc. 2007;129:15983–15996. doi: 10.1021/ja074900s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Srnec M, Wong SD, England J, Que L, Jr, Solomon EI. π-Frontier molecular orbitals in S=2 ferryl species and elucidation of their contributions to reactivity. Proc Natl Acad Sci USA. 2012;109:14326–14331. doi: 10.1073/pnas.1212693109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neidig ML, Decker A, Choroba OW, Huang F, Kavana M, Moran GR, Spencer JB, Solomon EI. Spectroscopic and electronic structure studies of aromatic electrophilic attack and hydrogen-atom abstraction by non-heme iron enzymes. Proc Natl Acad Sci USA. 2006;103:12966–12973. doi: 10.1073/pnas.0605067103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matthews ML, Neumann CS, Miles LA, Grove TL, Booker SJ, Krebs C, Walsh CT, Bollinger JM. Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc Natl Acad Sci USA. 2009;106:17723–17728. doi: 10.1073/pnas.0909649106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong SD, Srnec M, Matthews ML, Liu LV, Kwak Y, Park K, Bell CB, Alp EE, Zhao J, Yoda Y, Kitao S, Seto M, Krebs C, Bollinger JM, Jr, Solomon EI. Elucidation of the iron(IV) –oxo intermediate in the non-haem iron halogenase SyrB2. Nature. 2013 doi: 10.1038/nature12304. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diebold AR, Brown-Marshall CD, Neidig ML, Brownlee JM, Moran GR, Solomon EI. Activation of α-Keto Acid-Dependent Dioxygenases: Application of an {FeNO}7/{FeO2}8 Methodology for Characterizing the Initial Steps of O2 Activation. J Am Chem Soc. 2011;133:18148–18160. doi: 10.1021/ja202549q. [DOI] [PMC free article] [PubMed] [Google Scholar]