

Abstract
Bromodomains are epigenetic reader domains that have recently become popular targets. In contrast to BET bromodomains, which have proven druggable, bromodomains from other regions of the phylogenetic tree have shallower pockets. We describe successful targeting of the challenging BAZ2B bromodomain using biophysical fragment screening and structure-based optimization of high ligand-efficiency fragments into a novel series of low-micromolar inhibitors. Our results provide attractive leads for development of BAZ2B chemical probes and indicate the whole family may be tractable.
Introduction
Bromodomains are epigenetic reader domains within proteins that specifically recognize acetylated lysine (Kac) in histones and other substrate proteins. There are 61 different bromodomains spread across 46 proteins in the human genome, many of which are medically relevant targets for areas such as cancer, inflammation, and neurological disease.1,2 A subfamily of bromodomains of the bromo and extra terminal (BET) proteins have been shown to have tractable Kac binding pockets computationally3 and by the development of potent inhibitors such as JQ14 and iBET.5 These compounds have helped to unravel the biology and therapeutic potential of BET proteins, leading to mounting interest in developing chemical probes for other bromodomains in the human genome.
One bromodomain-containing protein whose biological role is still elusive is the bromodomain adjacent to zinc finger domain protein 2B (BAZ2B). A chemical probe would provide a useful tool to help determine its function. The BAZ2B bromodomain has an unusually small Kac-binding pocket compared to the other 41 bromodomains for which structural information is available (92–105 Å3 volume vs 131–221 Å3 in BRD4(BD1)) which lacks many of the features of BET bromodomains such as a ZA channel and a hydrophobic groove adjacent to the WPF motif (Supporting Information Figure 1).3
Strategies that have been successfully employed in BET bromodomain inhibitor optimization exploit the aforementioned structural features and are therefore not transferrable to BAZ2B. Analysis by Vidler et al. predicts the BAZ2B bromodomain to be one of the least druggable in the family.3 Consistent with this, reported inhibitors for other bromodomains show no cross-reactivity with BAZ2B, even at relatively early stages of the design process.6
As the Kac binding site in all bromodomains is of a suitable size and shape to bind to organic solvents and low MW molecules (fragments),7 we reasoned that a fragment-based approach would provide insights into the different functionalities and the strategies required for ligand optimization in this more challenging Kac-binding pocket.
Results and Discussion
As a first step, an unbiased library of 1300 rule of three8 compliant commercially available fragments were screened. Primary screening was performed using a competitive AlphaScreen assay, which measures displacement of a histone H3 peptide acetylated at K14 (H3Kac14).7 Initial hits were defined as those that showed >50% inhibition at 1 mM compound. For these fragments IC50 values were measured using AlphaScreen, resulting in the identification of 10 fragments. All of these showed direct binding and displacement when validated using orthogonal ligand-observed NMR techniques STD,9 CPMG,10 and WaterLOGSY11 (Table 1), a hit rate of 0.8%. The same fragment library was screened against the bromodomains of BRD2-BD1 and CREBBP (see Supporting Information Figure 2 for overlapping hits). The initial hit rates for these proteins were much higher, 1.8% and 6.1% respectively, consistent with their predicted higher ligandability.12
Table 1. Structures, IC50 Values Determined by AlphaScreen, and Ligand Efficiencies of Validated Fragment Hitsa.
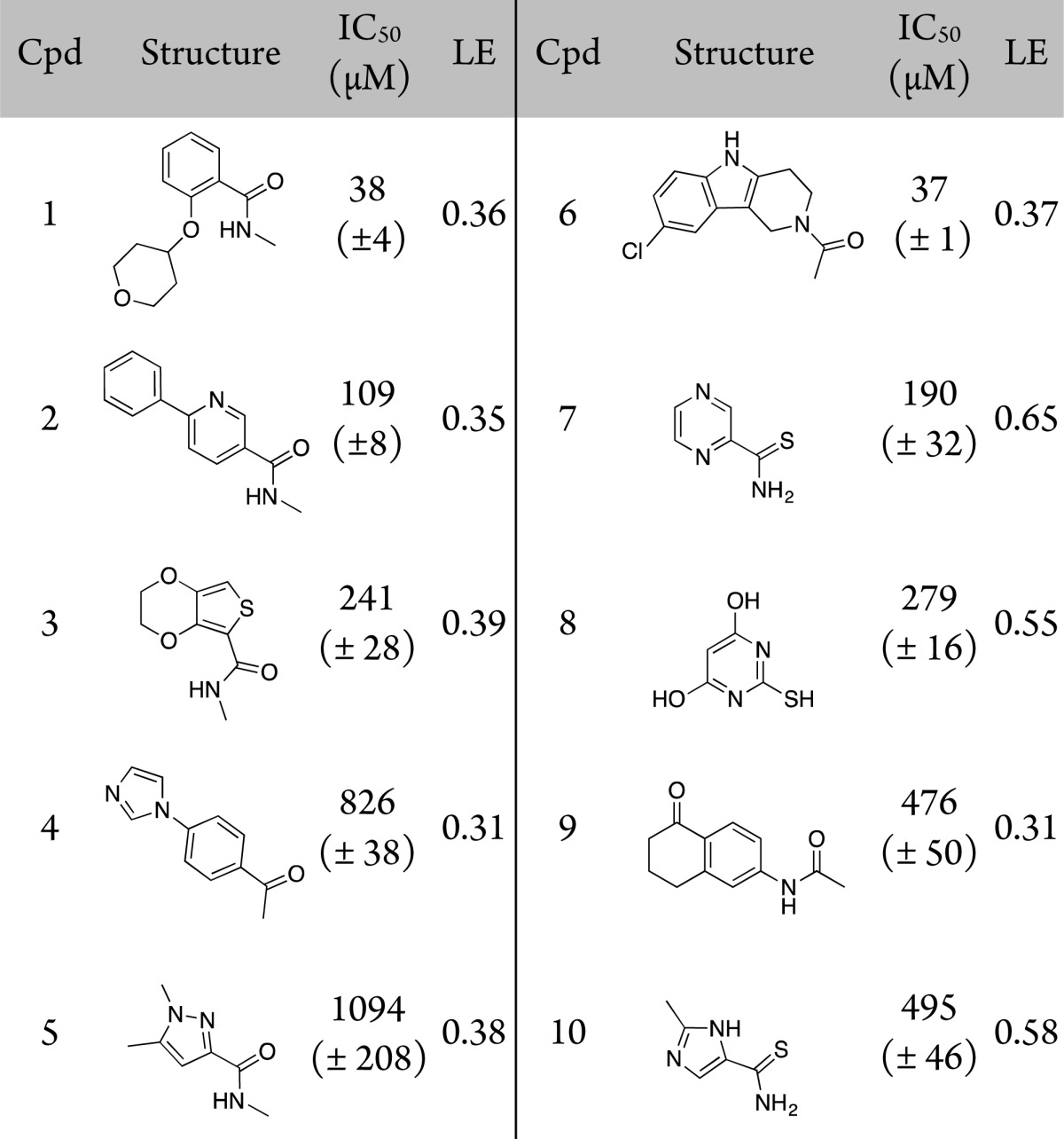
IC50 values are reported as the mean of three replicates (±standard error of the mean).
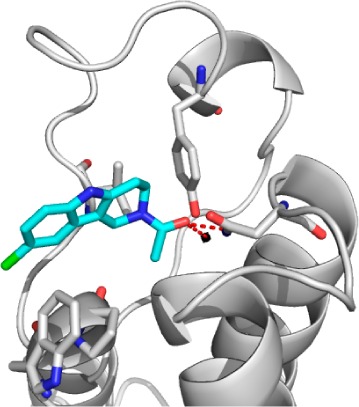
To elucidate the binding modes, the 10 fragment hits were soaked into apo-crystals of BAZ2B. It was possible to solve high-resolution crystal structures for fragments 1, 3, 6, and Kac (Figure 1).
Figure 1.
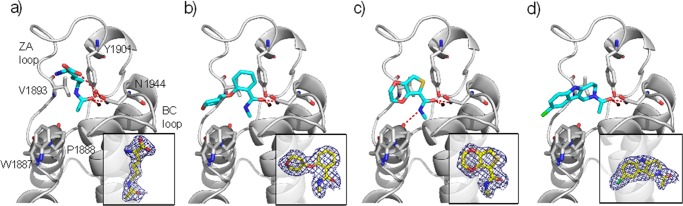
Crystal Structures of the BAZ2B bromodomain in complex with (a) acetyllysine PDB 4NR9, (b) 1 PDB 4NRB, (c) 3 PDB 4NRC, and (d) 6 PDB 4NRA. The bridging water molecule essential for the acetyllysine interaction is shown black. Other binding site waters are not shown for clarity. Hydrogen bonds are shown as red dashed lines. |2Fo| – |Fc| electron density maps contoured at 1σ for the bound ligands are shown in subpanels.
The ligand Kac makes hydrogen bonds to the side chain of Asn1944 and to Tyr1901 via a highly conserved water, as has been reported for other bromodomains.13 The alkyl chain has an unusual kink, which orients toward the BC loop. The carboxylic acid of Kac interacts with a water molecule that is also bound to the side chain of Asn1944.
Fragments 1 and 3 form hydrogen bonds to Asn1944 and the conserved water via their carbonyl group in a similar manner to Kac. The amide nitrogen forms a hydrogen bond to the backbone carbonyl of Pro1888. A similar interaction is observed by Magno et al. between the equivalent residue in TAF1 and ε-NH of Kac in MD binding simulations.14 The amide nitrogen of fragment 1 forms an intramolecular hydrogen bond to the adjacent ether group, which restricts rotational freedom and therefore may reduce the entropic penalty of binding. The tetrahydropyran ring of 1 lies orthogonal to its phenyl ring, providing an attractive trajectory for growing the ligand toward Val1893 (Figure 1b and Supplementary Figure 3).
The acetyl group of the most potent fragment, tetrahydro-γ-carboline (THγC) 6, bound to BAZ2B overlays with that in the Kac structure (Figure 1d and Supporting Information Figure 4). The unsaturated ring allows the fragment to twist such that the aromatic rings can make an edge-to-face π-stacking interaction with the side chain of Trp1887. The poor solubility of 6 meant direct ITC titration was not possible; therefore, a KD of 65 μM was determined using competitive ITC experiments with an H3Kac14 peptide (Table 2 and Supporting Information Table 4).
Table 2. IC50 Values Determined by AlphaLISA for Analogues of 6a.
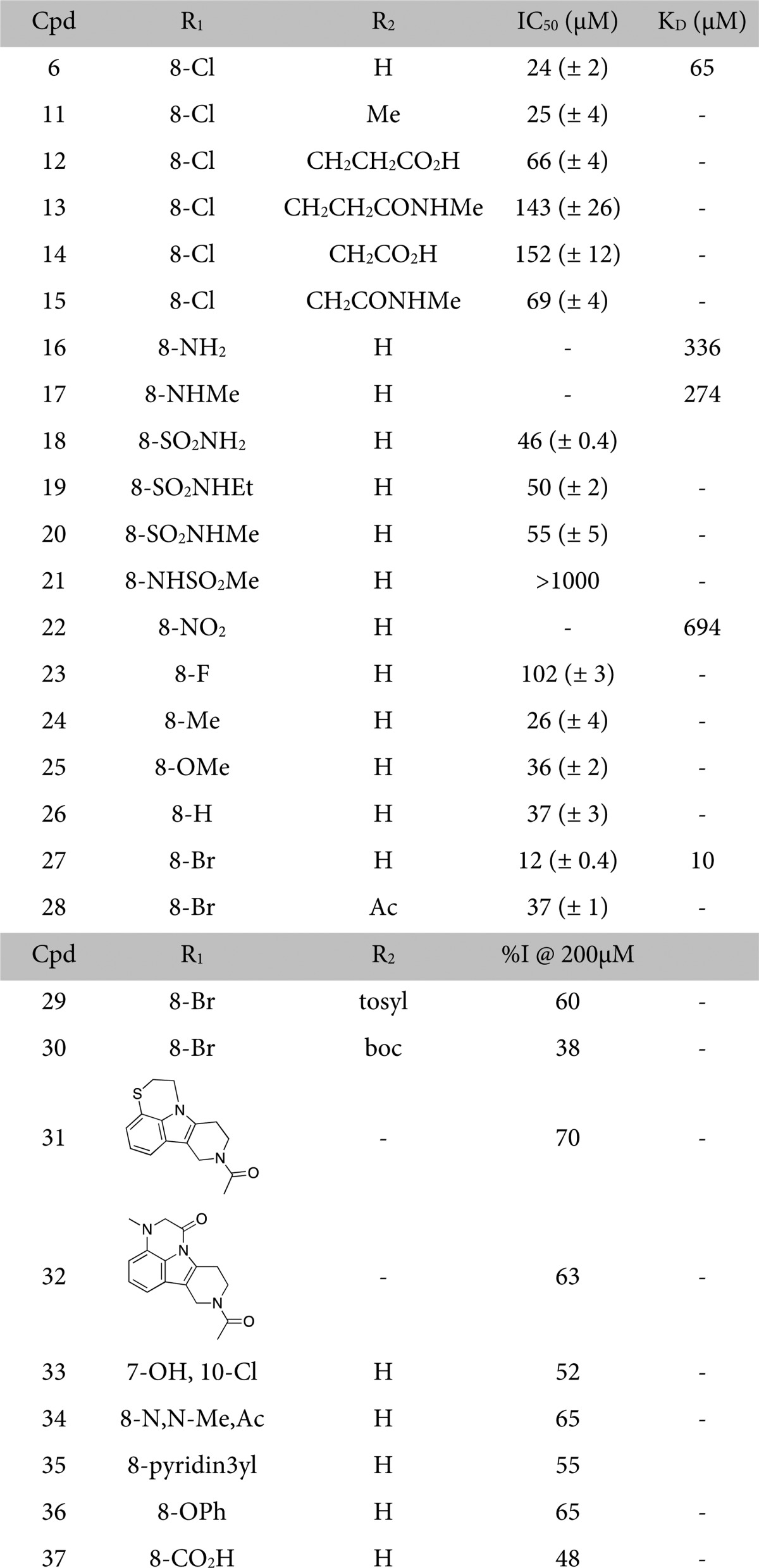
IC50 values are reported as the mean of three replicates (±standard error of the mean) and were not measured for molecules that resulted in an inhibition below 70% at 200 μM compound (%I).15KD values determined by ITC (Supporting Information Tables 4 and 5).
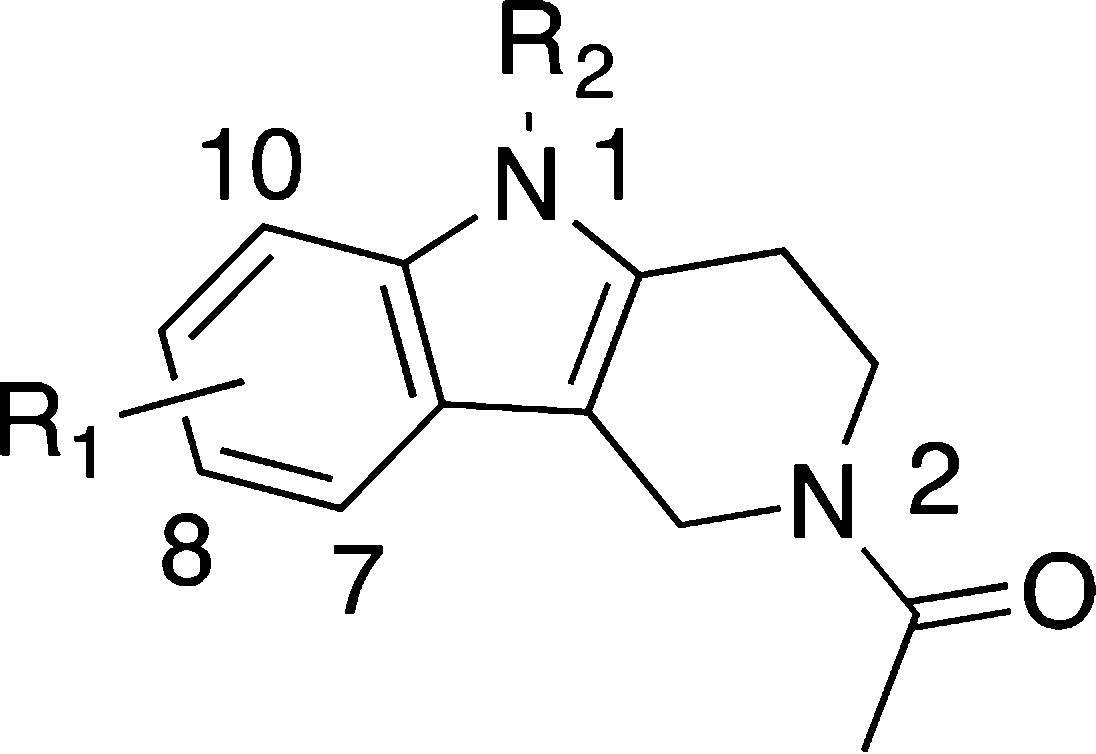
Encouraged by these initial results, we sought to optimize fragment 6 by varying the N1, N2, and aromatic substituents. The 1-position of the THγC scaffold presented an attractive vector for fragment growing to make interactions with the ZA loop. In the crystal structure the N1 proton is not interacting with the protein and the methylated compound (Table 2, 11) retained potency, suggesting further modifications could be exploited.
A set of compounds growing further from this position were synthesized; however, all modifications resulted in a reduction in potency (12–15, 28–32).
The distance between the backbone carbonyl oxygen of Trp1887 and the chlorine of 6 is 3.5 Å (combined van der Waals radii, 3.3 Å) and the angle between them is 109°, indicating no halogen bonding is occurring.16 On the basis of this observation, analogues were synthesized (16–21, 33–36) with alternative aryl substituents that preliminary docking studies17 suggested could hydrogen-bond to the protein and also provide a vector for growing the molecule out toward the ledge formed by residues 1893–1889, predicted to be a hotspot by FTMap solvent mapping software (Supporting Information Figure 5).18 Although some of these compounds showed a desirable improvement in solubility, all lost some potency, the best having an IC50 of 50 μM (19).
Another strategy implemented to optimize the aromatic substituent of 6 was using it to tune the charge distribution of the ring to strengthen the edge-to-face π–π interaction (22–27, 37). As expected, molecules containing strongly electron-withdrawing groups showed reduced potency, implying a weakened π–π interaction (18, 22, 23, 37). However, those with electron-donating groups (24, 25) did not result in the expected improvement in affinity.
Compound 27 proved the most potent of the tested analogues, showing a 6-fold improvement in KD over 6. This is possibly due to the weaker inductive electron-withdrawing properties of bromine vs chlorine aromatic substituents and increased hydrophobic contacts with the Trp1887 side chain.
Observation that the most ligand efficient fragments found in the screen (7, 8, and 10) contain thioamides, conceivably a Kac mimetic, led us to measure the KD of 7 using ITC, KD = 524 μM (Supporting Information Table 5). This encouraged us to synthesize thioamide 38 and thiourea 39. Neither of these bound to the protein. This indicates that the thioamide functionality in fragment hits 7, 8, and 10 adopts an orientation that cannot be achieved when attached to the larger THγC ring and that the BAZ2B bromodomain does not recognize the thiocarbonyl group.
Overlaying of the crystal structures of fragments 3 and 6 bound to BAZ2B (Figure 2) revealed that an additional H-bond could potentially be gained by merging the Kac mimetic of 3 with the scaffold of 6 to generate compound 40. Urea-containing 40 was synthesized and gratifyingly showed improved solubility, an 8-fold reduction in KD (determined by direct ITC titration), and a corresponding gain in ligand efficiency (Table 3). The 8-bromo analogue 41 showed slightly weakened binding compared to 27. The results show that optimization of the Kac bioisostere can play an important role in bromodomain inhibitor design.
Figure 2.
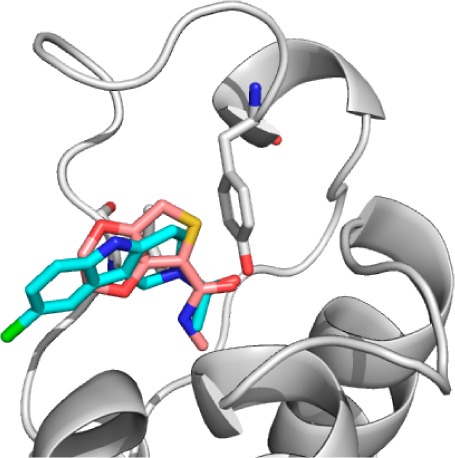
Overlay of the crystal structures of fragments 3 (shown pink) and 6 (offset, shown cyan) illustrates the rationale for fragment merging.
Table 3. IC50 Values Determined by AlphaLISA for Analogues of 6 in Which the Acetyllysine Mimetic Group Is Varieda.
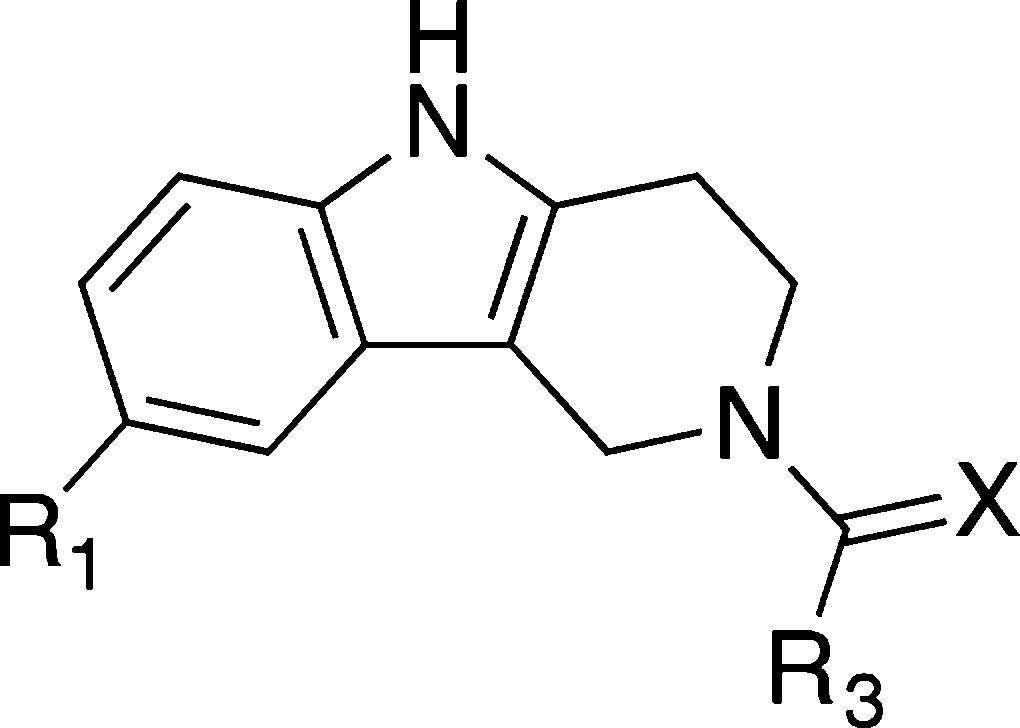
| Cpd | R1 | R3 | X | IC50 (μM) | KD (μM) |
|---|---|---|---|---|---|
| 38 | Cl | Me | S | >100 | |
| 39 | Cl | NH2 | S | >2000 | |
| 40 | Cl | NHMe | O | 9 (±2) | 8 |
| 41 | Br | NHMe | O | 14 (±0.1) | 17 |
IC50 values are reported as the mean of three replicates (±standard error of the mean). KD determined by ITC (Supporting Information Table 5).
Fragments containing an N-methylamide, e.g., 1–3 and 5, along with the THγC 6 were also hits against BRD2(1) and CREBBP in our fragment screen, which led us to profile 27 and 40 against these proteins using differential scanning fluorimetry (DSF), as previously described by Hewings et al.19 to allow for comparison (Table 4).
Table 4. ΔTm in °C Measured by DSF against CREBBP and BRD2 Bromodomainsa.
| bromodomain |
|||
|---|---|---|---|
| Cpd | CREBBP | BRD2 (BD1) | BRD2 (BD2) |
| 27 | 8.1 | 7.3 | 3.5 |
| 40 | 2.1 | –0.6 | –0.2 |
ΔTm values are reported as the mean of three replicates (standard error of the mean all <0.1 °C). Compound concentration, 100 μM; protein concentration, 2 μM.
Interestingly 27 causes a significant increase in thermal stability against CREBBP and BRD2 (BD1), indicating that this series may also provide useful leads for targeting these proteins. Hewings et al. describe a ΔTm of 4 °C in CREBBP corresponding to an IC50 of 28.1 μM;19 therefore, 27 is likely to be a low micromolar lead for CREBBP. Compound 40 shows markedly lower thermal stabilization for the three other bromodomains, implying that this compound may be more selective.
Conclusions
In summary, we describe biophysical screening, validation, and structural characterization for a number of chemically diverse fragment hits, which represent attractive starting points for inhibitor design against the BAZ2B bromodomain. A novel chemical series of BAZ2B bromodomain ligands based on a THγC scaffold are disclosed and SARs discussed. Efforts focused on optimization of interactions in and close to the Kac binding pocket proved the most productive, compared to those exploring its wider peripheries. These compounds make key interactions with the WPF motif, which complements another reported inhibitor, GSK2801,20 which makes key interactions with residues on the opposite side of the pocket. We also show that replacement of an acyl group with an N-methylamide as the KAc bioisostere can lead to a gain in binding affinity.
Our successful targeting of one of the least druggable bromodomains with small molecules highlights the possibility that the family as a whole may be tractable to small molecule modulation.
Experimental Section
Chemistry
General directions are in the Supporting Information. The syntheses of selected compounds are described below as representative. A full description of the synthetic protocol and spectroscopic analysis for each compound can be found in the Supporting Information. The purity of all tested compounds was analyzed by HPLC–MS (ESI) and is >95% unless otherwise stated.
General Procedure for Synthesis of THγCs 6, 18, 24–27
Substituted phenylhydrazine hydrochloride (1 equiv) and N-acyl-4-piperidone hydrochloride monohydrate (1 equiv) were refluxed in absolute EtOH for 3 h. The mixture was cooled to room temperature and the solvent concentrated in vacuo. The residue was purified by automated flash chromatography on silica or recrystallized from EtOH/H2O.
Acknowledgments
This work was supported by the UK Biotechnology and Biological Sciences Research Council (BBSRC, Grants BB/J001201/1 and BB/G023123/1 to A.C.). F.M.F. is funded by a BBSRC Ph.D studentship. The SGC is a registered charity (No. 1097737) that receives funds from the Canadian Institutes for Health Research, the Canada Foundation for Innovation, Genome Canada, GlaxoSmithKline, Pfizer, Eli Lilly, the Novartis Research Foundation, Takeda, the Ontario Ministry of Research and Innovation, and the Wellcome Trust. We thank Robert Pasteris and DuPont for the donation of 31–33 from their collection, Paul Brennan for useful discussions, and Charlotte Sutherell for synthesis of 24–26, 35.
Glossary
Abbreviations Used
- Alpha
amplified luminescent proximity homogeneous assay
- BAZ2B
bromodomain adjacent to zinc-finger domain 2B
- BD
bromodomain, BET, bromodomain and extra terminal
- BRD2
bromodomain containing protein 2
- CPMG
Carr–Purcell–Meiboom–Gill
- CREB
cyclic adenosine monophosphate responsive element binding protein
- CREBBP
CREB binding protein
- DSF
differential scanning fluorimetry, H3Kac14, histone H3 acetylated at lysine 14
- ITC
isothermal titration calorimetry
- Kac
Nε-acetyllysine
- SAR
structure–activity relationship
- STD
saturation transfer difference
- THγC
tetrahydro-γ-carboline
- WaterLOGSY
water ligand observed via gradient spectroscopy
- %I
percent inhibition of signal relative to solvent-only control
Supporting Information Available
Supplementary figures, supplementary tables, synthetic schemes, experimental procedures, synthesis and characterization of organic molecules, biophysical assays, ITC data, crystallographic refinement data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ A.C.: College of Life Sciences, University of Dundee, Dow Street, Dundee, DD1 5EH, U.K.
Author Present Address
⊥ J.R.C.M.: Institute of Physics of São Carlos, University of São Paulo, Avenida Trabalhador Sãocarlense 400, São Carlos, SP 13560-970, Brazil.
Author Present Address
# I.F.: Genome Centre, University of Sussex, Sussex House, Falmer, Brighton, BN1 9RH, U.K.
Author Present Address
∞ T.H.: Astex Therapeutics, 436 Cambridge Science Park, Cambridge, CB4 0QA, U.K.
The authors declare no competing financial interest.
Accession Codes
PDB accession codes of BAZ2B in complex with acetyllysine, 6, 1, and 3 are 4NR9, 4NRA, 4NRB, and 4NRC, respectively.
Supplementary Material
References
- Muller S.; Filippakopoulos P.; Knapp S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri I.; Cannizzaro E.; Dawson M. A. Bromodomains as therapeutic targets in cancer. Brief Funct. Genomics 2013, 123219–230. [DOI] [PubMed] [Google Scholar]
- Vidler L. R.; Brown N.; Knapp S.; Hoelder S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J. Med. Chem. 2012, 55177346–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 46873271067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C.-w.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 46873271119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish P. V.; Filippakopoulos P.; Bish G.; Brennan P. E.; Bunnage M. E.; Cook A. S.; Federov O.; Gerstenberger B. S.; Jones H.; Knapp S.; Marsden B.; Nocka K.; Owen D. R.; Philpott M.; Picaud S.; Primiano M. J.; Ralph M. J.; Sciammetta N.; Trzupek J. D. Identification of a chemical probe for bromo and extra C-terminal bromodomain inhibition through optimization of a fragment-derived hit. J. Med. Chem. 2012, 55229831–9837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott M.; Yang J.; Tumber T.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Keates T.; Felletar I.; Ciulli A.; Knapp S.; Heightman T. D. Bromodomain-peptide displacement assays for interactome mapping and inhibitor discovery. Mol. BioSyst. 2011, 7102899–2908. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Carr R.; Murray C.; Jhoti H. A “rule of three” for fragment-based lead discovery?. Drug Discovery Today 2003, 819876–877. [DOI] [PubMed] [Google Scholar]
- Mayer M.; Meyer B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem., Int. Ed. 1999, 38121784–1788. [DOI] [PubMed] [Google Scholar]
- Carr H. Y.; Purcell E. M. Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys. Rev. 1954, 943630–638. [Google Scholar]
- Dalvit C.; Pevarello P.; Tato’ M.; Veronesi M.; Vulpetti A.; Sundstrom M. Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water. J. Biol. NMR 2000, 18165–68. [DOI] [PubMed] [Google Scholar]
- Care must be taken when interpreting this result, as the peptides used in each assay have different affinities for their partnered bromodomains (IC50: CREBBP-H3K56ac, 7.8 μM; BRD2-H4K(5,8,12,16)ac, 3.0 μM; BAZ2B-H3Kac, 14 2.1 μM).
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J.-P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Müller S.; Pawson T.; Gingras A.-C.; Arrowsmith C. H.; Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 1491214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magno A.; Steiner S.; Caflisch A. Mechanism and kinetics of acetyl-lysine binding to bromodomains. J. Chem. Theory Comput. 2013, 994225–4232. [DOI] [PubMed] [Google Scholar]
- The protein immobilization technique of the assay was changed from Ni-chelate acceptor beads (AlphaScreen) to antipentahis antibody coated acceptor beads (AlphaLISA) in order to enable analysis of molecules containing functionalities known to chelate to Ni, such as sulfonamides. The IC50 of 6 was remeasured using this assay to provide a reference point.
- Wilcken R.; Zimmermann M. O.; Lange A.; Joerger A. C.; Boeckler F. M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2012, 5641363–1388. [DOI] [PubMed] [Google Scholar]
- Glide, version 5.9; Schrödinger, LLC: New York, NY, 2013. [Google Scholar]
- Brenke R.; Kozakov D.; Chuang G.-Y.; Beglov D.; Hall D.; Landon M. R.; Mattos C.; Vajda S. Fragment-based identification of druggable “hot spots” of proteins using Fourier domain correlation techniques. Bioinformatics 2009, 255621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewings D. S.; Wang M.; Philpott M.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Vuppusetty C.; Marsden B.; Knapp S.; Conway S. J.; Heightman T. D. 3,5-Dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J. Med. Chem. 2011, 54196761–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GSK2801: A Selective Chemical Probe for BAZ2B/A Bromodomains. http://www.thesgc.org/scientists/chemical_probes/GSK2801 (accessed August 5, 2013).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.