

Abstract
Weak protein interactions between ubiquitin and the ubiquitin-proteasome system (UPS) enzymes that mediate its covalent attachment to substrates serve to position ubiquitin for optimal catalytic transfer. We show that a small molecule inhibitor of the E2 ubiquitin conjugating enzyme Cdc34A, called CC0651, acts by trapping a weak interaction between ubiquitin and the E2 donor ubiquitin binding site. A structure of the ternary CC0651-Cdc34A-ubiquitin complex reveals that the inhibitor engages a composite binding pocket formed from Cdc34A and ubiquitin. CC0651 also suppresses the spontaneous hydrolysis rate of the Cdc34A-ubiquitin thioester, without overtly affecting the interaction between Cdc34A and the RING domain subunit of the E3 enzyme. Stabilization of the numerous other weak interactions between ubiquitin and UPS enzymes by small molecules may be a feasible strategy to selectively inhibit different UPS activities.
Introduction
The ubiquitin-proteasome system (UPS) regulates all cellular processes through precise spatial and temporal control of protein stability, activity and/or localization 1, and is frequently dysregulated in cancer and other diseases 2,3. The conserved E1-E2-E3 enzyme cascade activates and transfers ubiquitin through step-wise thioester linkages that culminate in covalent conjugation of ubiquitin to free amino groups on substrate proteins. The resultant mono- or polyubiquitination of the substrate typically leads to altered protein interactions or destruction by the 26S proteasome, respectively 1,4,5. The E2 enzymes lie at a crucial nexus in the UPS hierarchy as they exhibit specific interactions with E1 enzymes, E3 enzymes, deubiquitinating enzymes and substrates. E2 enzymes contain an essential catalytic cysteine that forms the ubiquitin thioester and an adjacent invariant asparagine residue that stabilizes the oxyanion transition state 6,7. Weak protein interactions between the E2 and ubiquitin are important for catalysis. In particular, the donor site tethers the thioesterified ubiquitin to prevent steric occlusion of the reaction centre and allow efficient attack of the thioester by the incoming substrate nucleophile, whereas the acceptor site orients the incoming ubiquitin to guide formation of the appropriate ubiquitin chain linkage 8-10. The detailed structural understanding of the ubiquitin transferase reaction has been hampered by the transient and structurally complex nature of these non-covalent catalytic intermediates.
The cullin-RING ligases (CRLs) form the largest family of E3 enzymes and are built on a core cullin-based architecture that recruits many hundreds of substrates through cohorts of different adaptor proteins 11-13. The Rbx1 RING domain subunit provides the docking site for Cdc34A (Ube2R1) and Cdc34B (Ube2R2), which are the principal E2s for the CRL family. Weak electrostatic interactions between the acidic C-terminus of Cdc34A and a basic cleft on the cullin subunit facilitate rapid cycles of E2 loading/unloading in the complex 14 and stabilize the E2-cullin interaction 15. CRL enzyme activity depends on the reversible modification of the cullin subunit by the ubiquitin-like modifier Nedd8, which triggers a conformational release of the Rbx1 subunit and the docked E2 enzyme to enable the E2 to access the bound substrate 16. Global CRL activity has been validated as a cancer target through development of a Nedd8 activating enzyme (NAE1) inhibitor called MLN4924 that traps NAE1 in a stable intermediate with Nedd8 and drives all CRLs into inactive non-neddylated forms 17,18. MLN4924 potently inhibits cancer cell proliferation in pre-clinical models, primarily through perturbation of cell cycle, DNA replication and DNA damage/repair functions 3.
As a parallel strategy to inhibit CRL activity, we recently identified a small molecule called CC0651 as a specific inhibitor of the human E2 enzyme Cdc34A 19. Like MLN4924, CC0651 stabilizes the CDK inhibitor p27 in cultured cells and inhibits the proliferation of human cancer cell lines. A previous structure of the CC0651-Cdc34A complex showed that CC0651 binds a cryptic pocket on the Cdc34A surface that is far removed from the active site cysteine but did not explain the mechanism of inhibition 19. Here, we show that CC0651 unexpectedly traps the weak interaction between ubiquitin and the donor site of Cdc34A and thereby impedes catalysis.
Results
Interactions between CC0651, Cdc34A and free ubiquitin
A partial overlap between the CC0651 binding site and a predicted donor ubiquitin binding surface on Cdc34A 19 lead us to investigate the interactions between CC0651, Cdc34A and free ubiquitin. We developed a synthetic route for CC0651 in order to produce sufficient quantities for structural and biophysical studies, and showed that the preparations used were of virtually identical purity and properties as previous material (see Online Methods). We used nuclear magnetic resonance spectroscopy (NMR) to assess the interaction of Cdc34A with 15N-ubiquitin by chemical shift perturbation (CSP) and peak intensity analysis of the heteronuclear single quantum coherence (HSQC) spectra. In striking contrast to expectations that CC0651 might disrupt the donor ubiquitin interaction 10,19, CC0651 caused a pronounced interaction to occur between ubiquitin and the core catalytic domain of Cdc34A (Cdc34ACAT), which lacks the acidic C-terminal tail (Fig. 1a,b). Peak shifts and peak broadening of ubiquitin resonances occurred at residues Lys6, Thr7, Leu8, Gln40, Gln41, Arg42, Leu43, Ile44, Phe45, Gly47, Lys48, Gln49, Leu50, Leu67, His68, Val70, Leu71, Arg72 and Leu73. As none of these shifts were evident in the absence of CC0651 (Fig. 1c; Supplementary Results, Supplementary Fig. 1a) or with ubiquitin alone in the presence of CC0651 (Fig. 1d, Supplementary Fig. 1b), we concluded that CC0651 specifically stabilizes the normally low affinity interaction between ubiquitin and the catalytic domain of Cdc34A. Addition of unlabeled full length Cdc34A (Cdc34AFL), including the acidic C-terminal extension, caused a similar pattern of resonance shifts as for the CC0651-Cdc34A-ubiquitin complex (Supplementary Fig. 1c,e), consistent with a previous observation that the acidic tail directly contacts ubiquitin 20,21. The combination of Cdc34AFL and CC0651 caused further peak shifts and broadening (Supplementary Fig. 1d,f), suggesting that CC0651 and the acidic tail additively potentiate the interaction of free ubiquitin with Cdc34A. The affected residues formed a contiguous surface on ubiquitin that closely matched the previously inferred contact surface for the catalytic domain of E2 enzymes (Fig. 1e) 8,9,22-25. Comparison of the interaction surfaces on ubiquitin for Cdc34ACAT and Cdc34AFL in the presence of CC0651 with a previously determined disulfide tethered Cdc34AFL-ubiquitin complex demonstrated that the CC0651-induced complex likely reflects a native-like interaction between Cdc34A and the covalently linked donor ubiquitin (Fig. 1e,f; Supplementary Fig. 1g). Collectively, these results demonstrate that CC0651 acts to stabilize the low affinity interaction between Cdc34A and donor ubiquitin.
Figure 1.

CC0651 potentiates the interaction between Cdc34A and 15N-Ub. (a) Superposition of the 1H, 15N-HSQC spectra of 15N-Ub:Cdc34ACAT (black) and 15N-Ub:Cdc34ACAT:CC0651 (red) at the indicated molar ratios. (b-d) Peak intensity change versus residue number for HSQC spectra of (b) 15N-Ub:Cdc34ACAT versus 15N-Ub:Cdc34ACAT:CC0651, (c) 15N-Ub versus 15N-Ub:Cdc34ACAT and (d) 15N-Ub versus 15N-Ub:CC0651. Superimposed 1H, 15N-HSQC spectra corresponding to panel c and d are provided in Supplementary Fig. 1a,b. (e-f) Interaction surfaces on ubiquitin for (e) Cdc34ACAT induced by CC0651 and (f) disulfide tethered UBC1 derived from PDB 1FXT 22. Interaction surface in (e) was obtained using the peak intensity change cut-off indicated by red dashed line in panel b.
A series of experiments with 15N-Cdc34ACAT showed that CC0651 caused resonance shifts on Cdc34ACAT in absence of ubiquitin and further shifts in the presence of a four-fold excess of free ubiquitin (Fig. 2a,b), whereas unlabeled ubiquitin alone did not cause any detectable shifts on Cdc34ACAT even in large excess (Supplementary Fig. 2a). We used chemical shift changes for three representative peaks at different concentrations of CC0651 to estimate the affinity of CC0651 for Cdc34ACAT in the presence and absence of free ubiquitin. Concordant with a model where CC0651 and ubiquitin bind cooperatively to Cdc34A, quantitation of chemical shifts in the NMR titrations showed that CC0651 bound to Cdc34ACAT alone with an EC50 of 267 μM (Fig. 2c) but that in the presence of ubiquitin this affinity was increased to an EC50 of 19 μM (Fig. 2d). To confirm this observation, we used a time-resolved Förster resonance energy transfer (TR-FRET) assay at dilute concentrations of fluorescently labeled Cdc34A and ubiquitin. In this assay, CC0651 potentiated binding of Cdc34AFL to ubiquitin from undetectable levels to an EC50 of 14 ± 2 μM, whereas the Cdc34B (Ube2R2) isoform, which is insensitive to CC0651 19, did not respond to CC0651 (Supplementary Fig. 2b). The induced binding effect in the TR-FRET assay was dependent on the acidic tail of Cdc34A, consistent with the additive effect of the tail observed by NMR. A quantitative SCF ubiquitination assay with a β-Catenin substrate peptide yielded a value of IC50 of 18 ± 1 μM for CC0651 inhibition (Supplementary Fig. 2c), similar to the effective concentrations observed in the NMR and TR-FRET assays.
Figure 2.

CC0651 potentiates the interaction between 15N-Cdc34A and Ub. (a-c) Superposition of 1H, 15N-HSQC spectra at the indicated molar ratios for (a) 15N-Cdc34ACAT (black) versus 15NCdc34ACAT:CC0651 (red), (b) 15N-Cdc34ACAT:Ub (black) versus 15N-Cdc34ACAT:Ub:CC0651 (red). (c-d) Chemical shift perturbation for three representative resonances of (c) 15N-Cdc34ACAT or (d) 15N-Cdc34ACAT:Ub as a function of titrated CC0651. Calculated EC50 value is the mean of the three displayed curves (upper panel) for which one representative resonance peak is shown (lower panel).
Structure of a CC0651-Cdc34A-ubiquitin complex
To investigate the precise mode of binding between CC0651, Cdc34A and ubiquitin, we crystallized an equimolar mixture of constituents and solved the X-ray crystal structure at 2.6 Å resolution by molecular replacement (see Supplementary Table 1 for X-ray data collection and refinement statistics and Supplementary Fig. 3 for representative electron density maps). The structure revealed that CC0651 engages a composite binding pocket nestled at the periphery of the Cdc34A-ubiquitin interface composed of residues from both Cdc34A and ubiquitin (Fig. 3a-d, Supplementary Fig. 4a-c). The ternary complex was formed without any overt structural perturbation to either Cdc34A or to ubiquitin (RMSD of ubiquitin = 0.67Å; RMSD of Cdc34A-CC0651 complex = 0.75Å). Notably, regions of CC0651 that were highly solvent exposed in the CC0651-Cdc34A binary complex 19 were shielded by ubiquitin in the ternary complex. Of a total accessible surface area of 658 Å2 on CC0651, 528 Å2 (81%) and 66 Å2 (10%) were buried by contact of CC0651 with Cdc34A and ubiquitin respectively, with only 61 Å2 (9%) of the compound surface exposed to solvent. In addition, the CC0651 contacts with Cdc34A described previously 19 were unchanged by the presence of ubiquitin. The biphenyl ring system of CC0651 made a number of direct contacts with ubiquitin (Fig. 3c, Supplementary Fig. 4a), including van der Waals contacts with the backbone of Gly47Ub, Lys48Ub, and Gln49Ub and hydrophobic contact with the apolar portion of the Lys48Ub side chain. These structural features suggest that CC0651 acts as a molecular bridge between Cdc34A and ubiquitin.
Figure 3.

Crystal structure of a CC0651-Cdc34A-ubiquitin complex. (a) Ribbons representation with CC0651 in yellow, Cdc34A in cyan, and ubiquitin in pink. (b) Slabbed surface representation of the CC0651 pocket from Cdc34A and ubiquitin. (c) Zoom-in of the Cdc34A-ubiquitin interface with main chains colored as in panel a. Contact residues are shown as sticks with carbon atoms colored according to main chain, and oxygen in red, nitrogen in blue, and chlorine in green. The Lys48 side chain is omitted for clarity. See Supplementary Fig. 3 for more detailed views. (d) Reciprocal interaction surfaces on ubiquitin (left) and Cdc34A (right). Complementary interacting triad of hydrophobic residues on ubiquitin and pentad of residues on Cdc34A are demarcated by black boxes. Residues that form an inter-molecular salt bridge are indicated by red circles. (e) Orientation of ubiquitin and Cdc34A induced by CC0651 is similar to previously characterized E2-ubiquitin interactions. Overlays of the CC0651-Cdc34A-ubiquitin complex with a Ubc1-Ub covalent complex (PDB 1FXT 22) and a UbcH5A/B-Ub complex (PDB 4AP4 23; PDB 4AUQ 24) are shown. Superpositions were performed using the E2 coordinates. Rotation angles relate the orientations of ubiquitin subunits.
The Cdc34A-donor ubiquitin interface
The direct interaction of ubiquitin with Cdc34A accounts for 1092 Å2 out of a total of 2485 Å2 buried surface area in the CC0651-Cdc34A-ubiquitin complex. The contact surface on Cdc34A was composed of helix α2, helix 310, and the linkers that join helices α2-α3, helices 310-α2, and helix α1 and strand β1. The reciprocal contact surface on ubiquitin was composed of strands β1, β3, and β4 and intervening linkers joining β1-β2 and β3-β4 (see Fig. 3c,d for all contact residues). Notable electrostatic and H-bond interactions included a network between the Asn132Cdc34A side chain and backbone NH of Gln49Ub, the Glu133Cdc34A side chain and side chains of Gln49Ub, Arg42Ub, and Arg72Ub, and lastly between the Ser129Cdc34A side chain and the side chains of Gln49Ub and Arg72Ub (Fig. 3c; Supplementary Fig. 4b). The main hydrophobic contacts occurred between a ridge formed by Leu8Ub, Ile44Ub, and Val70Ub, and a complementary groove formed by Thr122 Cdc34A, Leu125 Cdc34A, Ser126 Cdc34A, Ile 128 Cdc34A and Ser129 Cdc34A (Fig. 3c,d; Supplementary Fig. 4c).
The binding mode of ubiquitin to Cdc34A induced by CC0651 was reminiscent of donor ubiquitin-E2 interactions deduced previously for Ubc1 22, for Cdc34A and Ube2S 8,9, and for UbcH5A/B 23,24. This similarity extended to the interaction of the ubiquitin-like modifier SUMO with its cognate E2 Ubc9 26,27. The contact surfaces on both ubiquitin/SUMO and the E2s are similar but the structures are nevertheless differentiated by moderate rotations (23° to 43°) of ubiquitin/SUMO subunits on their respective E2 surfaces (Fig. 3e; Supplementary Fig. 5). This rotational variability at the donor site was evident even between different native ubiquitin/SUMO bound E2 structures, and was not specific to the inhibitor-stabilized Cdc34A-ubiquitin structure. Despite the fact that all E2s engage the same surface of ubiquitin, the contact residues on the E2s are poorly conserved (Supplementary Fig. 6). These features suggest that the donor binding site on the E2 is topologically conserved but that there is considerable plasticity in the recognition of the same ubiquitin surface by different E2s.
The CC0651-Cdc34A-ubiquitin structure validated a previously proposed interaction between donor ubiquitin and Cdc34A, as inferred from mutational analysis 8. In particular, perturbation of the hydrophobic environment surrounding Ser129Cdc34A or disruption of a presumptive salt bridge between Glu133Cdc34A and Arg42Ub compromise Cdc34A function 8. Both of these residues lie at the nexus of interactions between Cdc34A and ubiquitin in the ternary structure (Fig. 3c). Consistently, mutation of either Glu133Cdc34A or Ser129Cdc34A to Arg (but not the latter to Leu) abolished all CC0651-dependent resonance shifts and peak intensity changes in the 15N-Ub HSQC spectrum, thereby demonstrating that the X-ray structure represents a snapshot of the CC0651-Cdc34A-ubiquitin complex observed in solution (Fig. 4a,b; Supplementary Fig. 7a-f). In contrast to the Cdc34ASer129Arg mutant, which was completely compromised for function, the Cdc34AGlu133Arg mutant retained wild type activity when assayed against the substrate Sic1 and yet displayed insensitivity to CC0651 in vitro (Fig. 4c; Supplementary Fig. 7g). Notably however, sensitivity of the Cdc34AGlu133Arg mutant to CC0651 was restored in the presence of a compensatory charge reversal UbArg42Glu mutant (Fig. 4c). These results demonstrate that the ubiquitin-Cdc34A interface is integral to CC0651 action and that the inhibitor strengthens a native-like interaction of ubiquitin and Cdc34A. We also observed that CC0651 inhibited the normally rapid hydrolysis of the ubiquitin~Cdc34A thioester to water (Fig. 4d), suggesting that CC0651 can engage the covalent ubiquitin-Cdc34A thioester, in addition to the non-covalent complex between free ubiquitin and Cdc34A. We infer that the structural alterations imposed by CC0651 must impair the intrinsic reactivity of the labile Cdc34A-ubuiquitin thioester intermediate.
Figure 4.

Mutations in Cdc34A that disrupt interaction with ubiquitin impair sensitivity to CC0651. (a,b) Peak intensity change versus residue number for the 1H, 15N-HSQC spectra of 15N-ubiquitin:Cdc34ACAT versus 15N-ubiquitin:Cdc34ACAT:CC0651 for (a) wild type Cdc34A and (b) Cdc34AGlu133Arg. The corresponding superimposed 1H, 15N-HSQC spectra are shown in Supplementary Fig. 7a,b. (c) Sensitivity of the SCFCdc4 Sic1 ubiquitination reaction to CC0651 using wild type Cdc34A and ubiquitin or the indicated mutants. (d) Spontaneous hydrolysis of the ubiquitin~Cdc34AFL thioester in the presence and absence of CC0651. Quantification of signal intensity is presented as mean +/− S.E.M., n=2.
Effect of CC0651 on RING domain interactions
The RING domain has been shown to stimulate catalytic efficiency of Cdc34 and other E2 enzymes 10,23,24,28. CC0651 does not overly affect the interaction of full length Cdc34A with an Rbx1-Cul1 subcomplex 19, but it is possible that effects of CC0651 on the Rbx1 interaction may be mitigated by interaction of the acidic tail region of Cdc34A with the basic canyon region of Cul1 14. Additionally, the ubiquitin-charged form of Cdc34A has been reported to interact more strongly with the Rbx1 RING domain subunit than uncharged Cdc34A 25, although this differential affinity is not apparent in the context of the fully active Rbx1-Cul1Nedd8 complex 14,29. To determine if CC0651 might exert positive or negative effects on the interaction of Cdc34A with Rbx1, we performed NMR analysis on an 15N-labelled Rbx1 fragment that contains the RING domain (residues 12-108) (Fig. 5; Supplementary Fig. 8). Addition of ubiquitin and CC0651 in 2-fold molar excess to 15N-Rbx1 did not cause any discernable chemical shift perturbations (Supplementary Fig. 8a-c). In contrast, addition of Cdc34AFL in 4-fold molar excess caused pronounced chemical shift perturbations to the HSQC spectrum of 15N-Rbx1 (Fig. 5a; Supplementary Fig. 8d). The Cdc34A-Rbx1 interaction was abrogated by Thr117Glu and Tyr70Arg mutations on the RING domain-binding surface of Cdc34A (Fig. 5b,c; Supplementary Fig. 8e,f), which render Cdc34A incompetent for E3-dependent ubiquitin transfer to substrate but do not affect charging by E1 (Supplementary Fig. 9a,b). Addition of a six-fold molar excess of both ubiquitin and CC0651 caused a modest increase in Cdc34A-induced perturbations to the HSQC spectrum of 15N-Rbx1 (Fig. 5d,e; Supplementary Fig. 8g). Peak broadening at higher Cdc34A concentrations precluded more accurate quantification of binding affinities (data not shown). Projection of the subset of resonances most affected by CC0651 onto the folded region of the solution structure of Rbx1 mapped to the canonical E2 binding surface of the RING domain (Fig. 5f). We conclude that CCC0651 does not disrupt the interaction of Cdc34A with the RING domain of Rbx1, and may in fact moderately stabilize this interaction.
Figure 5.

Effect of CC0651 and ubiquitin on the interaction of 15N-Rbx1 with Cdc34A. Chemical shift perturbation analysis for the 1H, 15N-HSQC spectra of (a) 15N-Rbx1 versus 15N-Rbx1:Cdc34AFL, (b) 15N-Rbx1 versus 15N-Rbx1:Cdc34AFL-Thr117Glu, (c) 15N-Rbx1 versus 15NRbx1:Cdc34AFL-Tyr70Arg, and (d) 15N-Rbx1:CC0651:ubiquitin versus 15NRbx1:CC0651:Cdc34AFL:ubiquitin. Corresponding 1H, 15N-HSQC spectra from which CSPs were derived are shown in Supplementary Fig. 8d-g. (e) Chemical shift perturbations for selected Rbx1 residues affected by addition of CC0651, Cdc34A and ubiquitin at the indicated molar ratios. (f) Front and back surface representations of the Rbx1 RING domain (PDB 2LGV 25) with E2 enzyme contacts highlighted in red and purple. Apo-Cdc34A (top) and CC0651-Cdc34A-ubiquitin contacts on Rbx1 (middle) were derived from panels a and d, respectively. Purple indicates contacts unique to the CC0651-Cdc34A-ubiquitin interaction with Rbx1, and may represent a direct contact between Rbx1 and ubiquitin. Predicted Cdc34A contacts on Rbx1 (bottom) were obtained by superposition of Cdc34A coordinates onto a Cbl RING-UbcH7 structure (PDB 1FBV 40). Peak intensity change cutoffs highlighted by red line in panels a and d were used to assign interaction surfaces. (g) CC0651 titration analysis of Cdc34A binding to ubiquitin in the presence or absence of the indicated concentrations of Rbx1 or Rbx1-Cul1 complex by TR-FRET assay. Data presented as mean +/− S.E.M., n=2.
We predicted that in turn Rbx1 might stabilize the ternary interaction between CC0651, Cdc34A and ubiquitin, and/or that CC0651 might stabilize a potential interaction between Rbx1 and ubiquitin, as recently documented for other RING domain E3s 23,24. Using the TR-FRET assay, we found that 1 μM or 10 μM free Rbx1 caused a modest 2-fold increase in the affinity of Cdc34A for ubiquitin in the presence of CC0651 (Fig. 5g). Addition of 1 μM Rbx1-Cul1 complex to the CC0651 titration yielded a smaller enhancement effect (Fig. 5g), possibly because competition for the acidic tail of Cdc34A by the cullin subunit 14,20 may render the tail less available for interaction with free ubiquitin. These results suggested that the CC0651-Cdc34A-ubiquitin complex is at minimum fully competent for interaction with the E3, concordant with the previously observed dominant negative effect of the complex 19. Consistently, the dominant negative effect was accentuated at lower relative concentrations of ubiquitin and E3 compared to E2, but was relatively insensitive to the concentration of E1 (Supplementary Fig. 10). This dominant negative effect reveals another facet of the CC0651 inhibitory mechanism, namely out-competition of the E3 interaction with charged and catalytically competent E2 enzyme.
Structure-guided CC0651 derivatives
To probe features of the CC0651 pocket from the chemical ligand perspective, we undertook a chemical structure-activity relationship (SAR) analysis focused on the core dichloro-biphenyl moiety of CC0651, as guided by the CC0651-Cdc34-ubiquitin crystal structure (Table 1; Supplementary Fig. 11,12). We developed a modified synthetic scheme for CC0651 to allow comparisons across a set of six biphenyl derivatives and analyzed each analog for inhibition of SCFCdc4-Cdc34A mediated ubiquitination reactions in vitro and stabilization of the Cdc34A-ubiquitin interaction in a TR-FRET assay. We observed that both parameters were tightly correlated and that the relative bioactivities of the analogs could be rationalized by their fit the into the binding pocket. In particular, substitution of both chlorine atoms (atomic radius = 1.75 Å) with similarly sized methyl groups (van der Waals radius = 2.0 Å) was equally well tolerated for inhibitory activity and binding effects (analog 2) whereas substitution with smaller fluorine atoms (atomic radius = 1.47 Å) was detrimental (analog 3). Repositioning of one of the two meta methyl groups in analog 2 to the para position (analog 4) strongly impaired inhibitor activity, consistent with an absence of an accommodating pocket in the crystal structure. More dramatic substitutions at all three para/meta positions with O-methyl groups (analog 5) abolished all inhibitory and binding activity. Notably, deletion of one chlorine atom and substitution of the other with a bulky benzyoxy group at ring position 3 (analog 6) also abolished activity. While the original CC0651-Cdc34A binary structure suggested that a bulky group at this position should be tolerated by projection into solvent, in the CC0651-Cdc34A-ubiquitin ternary structure, the benzyoxy group would be predicted to sterically clash with ubiquitin. These results validate the utility of the ternary structure as a guide for SAR and further demonstrate that stabilization of the Cdc34A-ubiquitin interaction is an essential aspect of the CC0651 mechanism of action.
Table 1.
CC0651 Structure Activity Relationship.
Activities of the indicated derivatives were measured in a Sic1-SCFCdc4 ubiquitination assay and a Cdc34A-ubiquitin TR-FRET binding assay. See Supplementary Fig. 11 for corresponding activity and binding profiles. Bn indicates benzyl moiety. Data is represented as the mean +/− S.E.M., n=2.
| ID | Structure | Ubiquitination assay IC50 |
Binding assay EC50 |
|---|---|---|---|
|
1 (CC0651) |
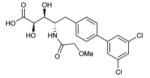
|
2.5±1 μM | 51±2 μM |
| 2 |
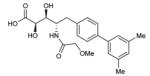
|
4.4±2 μM | 170±20 μM |
| 3 |
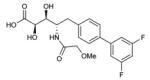
|
>100 μM | >300 μM |
| 4 |
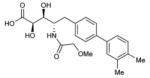
|
>100 μM | >300 μM |
| 5 |
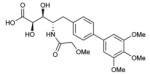
|
inactive | inactive |
| 6 |
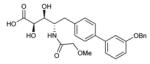
|
inactive | inactive |
Discussion
Our biochemical analysis and structure determination of the CC0651-Cdc34A-ubiquitin complex unexpectedly reveals that the small molecule inhibitor serves to stabilize the normally low affinity interaction between the E2 and ubiquitin at the donor site. Thus, in addition to the cryptic binding pocket on Cdc34A into which it docks, CC0651 exploits the weak interaction interface between Cdc34A and the donor ubiquitin to elaborate a more extensive binding pocket. This effect was unexpected because in isolation the surfaces on Cdc34A and ubiquitin to which CC0651 binds would otherwise appear flat and undruggable. The enhanced binding of free ubiquitin to Cdc34A induced by CC0651 is likely due to a combination of factors including direct bridging contacts between CC0651 and ubiquitin, distortion of the Cdc34A surface to improve complementarity of fit with ubiquitin, and/or reduction of entropic penalty by restriction of Cdc34A conformer space. The structure of the CC0651-Cdc34A-ubiquitin ternary complex appears to reflect the native-like docking pose of ubiquitin during the catalytic reaction, as suggested by the overall similarity to prior structures of ubiquitin and ubiquitin-like modifiers covalently linked to other E2 enzymes 8,9,22,23,24,26,27. However, until the structure of the labile Cdc34A-ubiquitin thioester complex is determined we cannot exclude the possibility that CC0651 locks the donor ubiquitin in a non-natural orientation.
We observed that CC0651 exerts multiple inhibitory effects on Cdc34A. Since CC0651 does not inhibit E2 charging, both ubiquitin-charged and -uncharged states of Cdc34A are accessible for CC0651 to interfere with the catalytic cycle. Stabilization of the donor ubiquitin interaction, either as a non-covalent complex or as the covalent thioesterified complex, would sterically hinder the catalytic transfer reaction. The intrinsic catalytic transfer function of the E2 is also severely impaired in the ternary complex. The E2~ubiquitin thioester can adopt open and closed (folded-back) conformations, the latter of which represents the catalytically poised conformer 10,23. While a folded back conformer is evidently locked into place by CC0651, the inhibitor-bound E2 may lack the necessary strain across the scissile thioester bond, which may be actuated upon E3 binding 23. Alternatively, subtle perturbations of E2 active site residues caused by CC0651 19 may decrease the reactivity of the thioester linkage. Because free thioester linkages are exceedingly stable under physiological conditions 30, and other E2 enzyme thioester linkages are much more stable than the Cdc34A-ubiquitin thioester 31, the orientation of Cdc34A catalytic residues may be highly optimized for rapid hydrolysis, consistent with the fast reaction kinetics exhibited by this enzyme 32. Indeed, this effect of CC0651 on hydrolysis rate recapitulates that of specific E2 catalytic site mutations 23. Finally, because the trapped CC0651-Cdc34A-ubiquitin complex engages the E3 with similar or slightly higher affinity than free Cdc34A, the complex has the capacity to competitively interfere with E2 enzymes that are resistant to the inhibitor. This inhibitory function can occur independent of the thioester formation but requires the acidic C-terminal tail of Cdc34A 19. The various inhibitory effects caused by CC0651 are all likely to be manifestations of the natural E2 enzyme catalytic cycle.
The specificity of CC0651 for Cdc34A is explained by the variability of the donor ubiquitin surface across the E2 family, which despite the conserved overall topology, exhibits considerable sequence variation 33. This variability, and the occurrence of likely partial binding pockets in other E2s 19, suggests that it will be possible to exploit stabilization of the donor ubiquitin-E2 interaction as a general means to isolate specific small molecule inhibitors of different E2 enzymes. This in principle positions the E2 enzyme family as a new drug target class in the UPS, despite the lack of an overt catalytic cleft in the E2 structure. The ternary structure reveals additional crevasses at the Cdc34A-ubiquitin interface (Fig. 3a) that explain our current SAR data and which may be exploited to further increase the affinity of CC0651.
The malleable nature of ubiquitin-E2 interactions at the donor site is revealed by the distinct orientations of different ubiquitin-E2 complexes, which are mediated by variable residues on the different E2s. These differential contacts appear to be augmented by additional ubiquitin interactions with the E3 subunit, which can vary from the minor interactions between ubiquitin and Rbx1 observed in this study, to the much more extensive contacts that occur between ubiquitin and the RING domain subunits of the dimeric RNF4 and BIRC7 enzymes 23,24. The multi-contact nature of the enzymatically active complexes is underscored by the fact that in no instance does free ubiquitin itself interact detectably with the RING domain.
Despite precedents of small molecules that freeze dynamic or low affinity interactions, there remain surprisingly few instances of protein complexes that are stabilized by inhibitors. Examples include the stabilization of microtubules by paclitaxel, the stabilization of the transient interaction of small GTPases with exchange factor Arf1 by Brefeldin A, the binding of FKBP12 to calcineurin and TOR complexes in the presence of FK506 and rapamycin, respectively 34. Typically, these protein interactions are of low affinity in the absence of inhibitor, much like the weak interaction between ubiquitin and the donor site on the E2. While most interaction stabilizers identified to date are structurally complex natural products, in principle the clefts and crevices often formed at protein interfaces should be amenable to smaller synthetic molecules, as shown here for CC0651. We note that the mechanism whereby MLN4924 inhibits the NAE1 enzyme also entails stabilization of the enzyme-substrate complex, although in this instance the stabilized complex is covalent adduct 18.
Weak non-covalent interactions of ubiquitin occur not only with many E2 enzymes 6,7, but also with E1 enzymes 35, E3 enzymes 36, deubiquitinating enzymes 37, and a host of ubiquitin binding domains 38. These weak interactions serve to modulate catalytic output, orient ubiquitin for efficient formation of different chain types, and enable recognition of different types of modification. In total, ubiquitin forms many hundreds of unique interfaces with the UPS enzyme hierarchy. These low affinity interactions can often be optimized by directed mutagenesis to yield variants that bind with high affinity and specificity to particular target surfaces 39. By analogy to CC0651, we suggest that it will be possible to identify unique small molecule inhibitors that selectively stabilize many other protein interactions with ubiquitin, or for that matter other ubiquitin-like modifiers, with any target of interest within the UPS or related ubiquitin-like modifier systems.
Online Methods
Protein expression and purification
Cdc34AFL (residues 1-236), Cdc34ACAT (residues 7-184), Cdc34AFL-Ser129Arg, Cdc34AFL-Thr117Glu, Cdc34AFL-Tyr70Arg, Cdc34ACAT-Ser129Arg, Cdc34AFL-Glu133Arg, Cdc34ACAT-Glu133Arg, Cdc34BFL (residues 1-238), Cdc34BCAT (residues 7-186), Rbx112-108-SSQS (residues 12 to 108, containing point mutations of Trp27Ser, Val30Ser, Leu32Gln and Trp33Ser to improve protein solubility 25), and Rbx136-108 (residues 36 to 108) were expressed from the pProEx-HTa plasmid in E.coli BL21(DE3) Codon+ cells (Agilent Technologies) with a Tobacco Etch Virus (TEV) cleavable N-terminal polyhistidine tag by induction with 0.25 mM isopropyl 1-thio-β-D-galactopyranoside (IPTG) for 14-18 hours at 18°C. Harvested cells were resuspended in 20 mM HEPES pH 7.5, 400 mM NaCl, 5 mM β-mercaptoethanol and lysed by passage through a cell homogenizer (Avestin Inc.). Following centrifugation at 30,000g supernatant was applied to a HiTrap nickel chelating HP column (GE Healthcare) equilibrated in lysis buffer with 5 mM Imidazole. Protein eluted with buffer containing 300 mM Imidazole was incubated overnight with TEV protease and 2 mM DTT. Cleaved protein was dialyzed in HiTrap loading buffer and flowed over a subtractive HiTrap nickel chelating column then concentrated for injection onto a 120 mL Superdex S75 or S200 column (GE Healthcare) equilibrated in 20 mM HEPES pH 7.5, 100 mM NaCl, 5 mM β-mercaptoethanol. Fractions (>95% pure) were concentrated to 10-35 mg/mL.
Cdc34AFL-Ser129Leu, Cdc34ACAT-Ser129Leu, Ub, UbCys0, UbArg42Glu, were expressed as TEV cleavable glutathione-S-transferase (GST) fusions using a modified vector pGEX-2T-TEV. Harvested cells were resuspended and lysed as per poly histidine tagged fusions. Lysates were incubated with glutathione sepharose resin (GE Healthcare) at 4°C, washed extensively and incubated overnight with TEV protease and 2 mM DTT. TEV protease, which was polyhistidine tagged, was removed by subtraction over a 1 mL HiTrap nickel chelating HP column and flow through was concentrated for injection onto a 120 mL Superdex S75 or S200 column equilibrated in 20 mM HEPES pH 7.5, 100 mM NaCl, 5 mM β-mercaptoethanol. Protein fractions at >95% purity were pooled and concentrated to 10-35 mg/mL.
Ubiquitin and ubiquitination reactions
UbCys0 bearing an additional cysteine residue preceding the initiator methionine was labeled with 5-iodoacetamidofluorescein (5-IAF, Molecular Probes) to enable quantitative measurement of fluorescence incorporation after in vitro ubiquitination. Incubation of 100 μM purified UbCys0 with 500 μM of 5-IAF and 2 mM DTT for 3 hours at 20°C was followed by size exclusion chromatography over an S-75 sepharose column equilibrated in 20 mM HEPES pH 7.5, 100 mM NaCl, 5 mM β-mercaptoethanol. Fluorescein-labelled UbCys0 was concentrated to 1.75 mM and stored at −80°C. E1, SCFCdc4 and Sic1 were expressed and purified as described 41.
For gel-based Sic1 ubiquitination assays (Fig. 4c; Table 1; Supplementary Figures 7g, 9b, 10, 11 and 12b), CC0651 was pre-incubated at the indicated concentrations with 0.5 μg E1, 1 μg Cdc34A, Cdc34B, or the indicated mutants, and 50 ng SCFCdc4 for 15 min at 20°C in 20μL reaction buffer (50 mM HEPES pH 7.5, 10 mM MgCl2, 2 mM ATP, 50 μM DTT). Reactions were initiated by addition of 1 μg ubiquitin or ubiquitinArg42Glu or fluorescein derivatized ubiquitinCys0 and 50 ng Sic1 phosphorylated by Cln2-Cdc28. Reactions were incubated at 30°C for 30 min and products visualized by immunoblot 41 or by direct fluorescence detection using a Typhoon 9500 imager (GE Healthcare). For β-catenin peptide gel-based ubiquitination assay in Supplementary Fig. 2b, 1 μM Cdc34A, 250 nM E1, 60 uM ubiquitin, and 100 nM neddylated SCFβ–TrCP were incubated for 2 min in reaction buffer containing 30 mM Tris pH 7.5, 150 mM NaCl, 5 mM MgCl2, 2 mM ATP and 2 mM DTT. Indicated concentrations of CC0651 were added to each reaction for 1 additional min followed by reaction initiation with 5 μM 32P-labeled β-catenin peptide substrate 29. Reactions were incubated for 20 min, quenched and separated by SDS-PAGE. Images were detected on a Typhoon Phosphor Imager and quantified using ImageQuant. Hydrolysis of a pre-formed ubiquitin-Cdc34A thioester was monitored by incubating 10 μM Cdc34A in the presence of 1 μM E1 and 5 μM 32P-labeled ubiquitinK48R for 5 min. Additional treatments of reactions with either 100 μM CC0651 and apyrase (Sigma) or apyrase alone were performed for 1 min. Aliquots were quenched at the indicated time points and resolved by SDS-PAGE. Data were fit to a single exponential decay function using GraphPad Prism.
E2 charging assays were performed with 25 μM Cdc34A, 200 μM ubiquitin (1:5 ratio of fluorescein labeled ubiquitinCys0: unlabeled ubiquitin) and 1 μM E1 were preincubated for 10 minutes at 23°C in reaction buffer (20 mM HEPES pH 7.5, 100 mM NaCl). The reaction was initiated with 4 mM ATP at 30°C, terminated at indicated time points by gel loading buffer with or without 5 mM DTT, resolved by SDS-PAGE and detected on a Typhoon Phosphor Imager.
TR-FRET assay
Binding reactions contained 1 μM His-Cdc34ACAT or His-Cdc34AFL, or His-Cdc34BFL (purified with omission of the TEV cleavage step), 25 μM fluorescein labeled UbCys0 in the absence or presence of Rbx1 or Rbx1:Cul1 complex with 0-100 μM CC0651. 2 nM of anti-His6-Tb3+ antibody (Invitrogen) was then added to the reaction and TR-FRET signal was measured using an excitation filter at 340 nm and emission filters for Tb3+ at 490 and Alexa488 at 520 nm, respectively, on a BMG PHERAstar instrument (BMG Labtech). EC50 values were computed with GraphPad Prism software.
NMR Spectroscopy
15N-ubiquitin, 15N-Rbx1 and 15N-Cdc34ACAT were expressed in E.coli using M9 medium supplemented with 15NH4Cl. Purification was performed as described for unlabelled proteins. NMR data were acquired at 25°C on 800MHz or 600 MHz Bruker AVANCE III spectrometers. The 800 MHz spectrometer was equipped with a 5 mm TCI CryoProbe and the 600 MHz with a 1.7 mm TCI CryoProbe. In NMR titrations, 0.1-0.3 mM 15N-ubiquitin, 15N-Rbx1 or 15N-Cdc34ACAT was used for the collection of 1H, 15N-HSQC titration spectra. All NMR samples were prepared in 30 mM HEPES, pH 7.3-7.5, 100 mM NaCl, 1 mM DTT, and 10% D2O. All NMR spectra were processed using NMRPipe/NMRDraw 42 and further analyzed using NMRView 43. Backbone resonance assignments for human ubiquitin 44 and Rbx1 25 have been reported previously. In all peak intensity analyses, HSQC peak heights were used. Binding affinity between CC0651 and Cdc34ACAT in the absence or presence of ubiquitin was determined based on chemical shift changes. Chemical shift perturbations (CSP) were calculated as a weighted average using the formula CSP=[(δHN)2+(δN/5)2]1/2. The relative affinities between Cdc34ACAT and CC0651 in the presence and absence of ubiquitin were determined based on chemical shift changes. EC50 values were obtained by fitting CSPs of the HSQC titration spectra 45 using GraphPad Prism.
X-ray crystallography
Crystals of a CC0651:Cdc34ACAT:ubiquitin complex were grown in hanging drops by mixing 1 μL protein solution containing 1.2 mM CC0651 , 900 μM Cdc34ACAT and 900 μM ubiquitin with 1 μL well solution (28% PEG3350, 0.1 M HEPES pH 7.0) at 20°C. For cryoprotection, a single crystal was soaked in well solution supplemented with 20% glycerol. Diffraction data was collected at 100°K at 0.97919 Å wavelength on beamline NE-CAT 24-ID-E (APS. Chicago, IL) and processed with HKL2000 46. Molecular replacement was performed using Phaser 47 using the crystal structure of Cdc34A (PDB 2OB4) and ubiquitin (PDB 1FXT 22). Model building and refinement were performed using Coot 48 and Refmac5 49. The final refined structure showed Ramachandran statistics of 98.2, 1.8 and 0.0% in the preferred, allowed and outlier regions, respectively. All protein structure figures were generated using PyMOL (www.pymol.org).
Chemical synthesis of CC0651
Starting materials and reagents were obtained from Aldrich, except for (S)-3-(4-Bromophenyl)-2-((tert-butoxycarbonyl)amino)propanoic acid (Boc-p-BrPhe) which was obtained from Chem-Impex International. The synthetic schemes and spectroscopic data for CC0651 and analogs are summarized in Supplementary Fig. 11 and Supplementary Table 2, respectively. The reported synthesis of CC0651 is a new route to the same compound described previously 19. The reduction of Boc-p-BrPhe to (S)-Tert-butyl (1-(4-bromophenyl)-3-hydroxypropan-2-yl)carbamate (7) was conducted via literature methods50. Purification by medium-pressure liquid chromatography (MPLC) was performed on a Combiflash Rf instrument (Teledyne Isco Inc., Lincoln, NE). Analytical reverse phase HPLC was conducted as follows. Method A: Agilent POROSHELL® 120 EC-C18, 2.7um, 2.1×30 mm column with mobile phase of solvent A (5% MeOH, 95% H2O, 0.1% AcOH) and solvent B (95% MeOH, 5% H2O, 0.1% AcOH) in a gradient of 100% solvent A at t=0 to 100% solvent B at t=1 with stop time of 4 min and flow rate of 1.0m L/min. Method B: Zorbax SB-C18 3.5um, 4.6×30 mm column with mobile phase of solvent A (5% MeOH, 95% H2O, 0.1% AcOH) and solvent B (95% MeOH, 5% H2O, 0.1% AcOH) in a gradient of 100% solvent A at t=0 and 100% solvent B at t=1 with stop time of 4 min and flow rate of 1.0mL/min. Assignments of 1H NMR and 13C NMR signals were made by correlation spectroscopy (COSY) and HSQC. Optical rotations were determined irradiating with the sodium D line (λ= 589 nm) using a Perkin Elmer 341 polarimeter, with specific rotation “[α]D” and concentration “c” values given in units (deg·mL)/(g·dm) and g/100 mL, respectively. Abbreviations: AcOH, acetic acid; MeCN, acetonitrile; MeOH, methanol; EtOAc, ethyl acetate; TFA, 2,2,2-trifluoroacetic acid; DBU, 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine, Dess martin periodinane: 1,1,1-Triacetoxy-1,1-dihydro-1,2-benziodoxol-3(1H)-one; (DHQ)2Pyr, (1S,1′S,3R,3′R,4S,4′S,6S,6′S)-6,6′-((1R,1′R)-((2,5-diphenylpyrimidine-4,6-diyl)bis(oxy))bis((6-methoxyquinolin-4-yl)methylene))bis(3-ethylquinuclidine); DIEA, N,N-Diisopropylethylamine; DMAP, 4-dimethylaminopyridine; EDC, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide; HATU, 2-(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yl)-1,1,3,3-tetramethylisouronium hexafluorophosphate(V).
(S)-Tert-butyl (1-(3′,5′-dichloro-[1,1′-biphenyl]-4-yl)-3-hydroxypropan-2-yl)carbamate (8)
A mixture of (S)-tert-butyl (1-(4-bromophenyl)-3-hydroxypropan-2-yl)carbamate (7) (1.0 g, 3.0 mmol), (3,5-dichlorophenyl)boronic acid (751 mg, 3.94 mmol), Na2CO3 (0.642 g, 6.06 mmol), and tetrakistriphenylphosphine palladium(0) (105 mg, 0.091 mmol) in a sealed tube was treated with 50% MeCN in H2O (12 mL, nitrogen degassed) and heated at 80 °C for 3 h. The reaction was cooled to room temperature, diluted with water and EtOAc, and the layers were separated. The organic phase was washed with saturated NaHCO3, brine, dried over MgSO4, filtered, and concentrated to a solid brown residue (1.5 g), which was purified by normal-phase MPLC (40 g silica gel cartridge, EtOAc in hexane) to give 1.17 g of 8, in 93% yield.
(S)-Tert-butyl (1-(3′,5′-dichloro-[1,1′-biphenyl]-4-yl)-3-oxopropan-2-yl)carbamate (9)
A CH2Cl2 (6 mL) solution of Dess-Martin periodate (1.122 g, 2.65 mmol) was slowly treated with a CH2Cl2 (6 mL) solution of (S)-tert-butyl (1-(3′,5′-dichloro-[1,1′-biphenyl]-4-yl)-3-hydroxypropan-2-yl)carbamate (8) (953 mg, 2.41 mmol) and stirred at room temperature for 2 h. The reaction was poured into a mixture of 20% KHCO3/H2O (7 mL) and 10% Na2S2O3/H2O (7 mL), stirred for 30 min, and the phases separated. The organic phase was washed with 20% KHCO3/H2O, water, dried over MgSO4, filtered, and concentrated to give 875 mg of 9 as an amorphous pale yellow solid in 92% yield, which was subsequently used without further purification.
(S,E)-Methyl 4-((tert-butoxycarbonyl)amino)-5-(3′,5′-dichloro-[1,1′-biphenyl]-4-yl)pent-2-enoate (10)
A dry MeCN (20 mL) solution of (S)-tert-butyl (1-(3′,5′-dichloro-[1,1′-biphenyl]-4-yl)-3-oxopropan-2-yl)carbamate (9) (0.875 g, 2.22 mmol) at 5 °C was slowly treated with LiCl (141 mg, 3.33 mmol) and methyl 2-(dimethoxyphosphoryl)acetate (0.539 mL, 3.33 mmol) at 5 °C followed by dropwise addition of DBU (0.332 mL, 2.22 mmol). After 30 min at 5 °C, the reaction was quenched with saturated NH4Cl, diluted with water (20 mL), and extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated. The crude product (1.34 g) was purified by normal-phase MPLC (40 g silica gel cartridge, 5→15% EtOAc gradient in hexane over 15 min, 40 mL/min) to give 776 mg of 10 as a white solid in 78% yield.
(2R,3S,4S)-Methyl 4-((tert-butoxycarbonyl)amino)-5-(3′,5′-dichloro-[1,1′-biphenyl]-4-yl)-2,3-dihydroxypentanoate (11)
A mixture of potassium hexacyanoferrate(II) (1.705 g, 4.63 mmol), K2CO3 (0.64 g, 4.63 mmol), potassium tetrahydroxydioxidoosmium (11 mg, 0.031 mmol), methanesulfonamide (147 mg, 1.543 mmol), and (DHQ)2Pyr (41.4 mg, 0.046 mmol) was dissolved in 50% tBuOH/H2O (30 mL) by stirring at room temperature for 5 min and subjecting to sonication. The mixture was subsequently added to (S,E)-methyl 4-((tert-butoxycarbonyl)amino)-5-(3′,5′-dichloro-[1,1′-biphenyl]-4-yl)pent-2-enoate (10) (695 mg, 1.54 mmol) and vigorously stirred for 16 h at room temperature. The reaction was cooled to 5 °C, treated with Na2SO3 (2 g) powder, stirred for 30 min at 5 °C, 1.5 h at room temperature, and extracted with EtOAc (3 × 30 mL). The combined organic extracts were washed with brine, dried over MgSO4, filtered, and concentrated. The crude product was purified by normal-phase MPLC (40 g silica gel, 0 → 5% MeOH gradient in CH2Cl2, 30 mL/min) to give 414 mg of 11 as a white solid in 54% yield.
(2R,3S,4S)-Methyl 5-(3′,5′-dichloro-[1,1′-biphenyl]-4-yl)-2,3-dihydroxy-4-(2-methoxyacetamido)pentanoate (12)
A CH2Cl2 (2 mL) solution of (2R,3S,4S)-Methyl 4-((tert-butoxycarbonyl)amino)-5-(3′,5′-dichloro-[1,1′-biphenyl]-4-yl)-2,3-dihydroxypentanoate (11) (100 mg, 0.206 mmol) was treated with TFA (0.200 mL, 2.69 mmol), stirred at room temperature for 1.5 h, and concentrated to dryness to afford the crude deprotected amine product, (2S,3S,4R)-1-(3′,5′-dichloro-[1,1′-biphenyl]-4-yl)-3,4-dihydroxy-5-methoxy-5-oxopentan-2-aminium 2,2,2-trifluoroacetate, which was set aside for use in the next reaction and assumed to contain the theoretical yield. For liquid chromatography (Method A) retention time was 2.63 min and for LC-MS (APCI) m/z: [M + H]+ was calculated for C18H20Cl2NO4 at 384.1 and 386.1 and determined as 384.0and 386.2, with the secondary mass peaks due to the natural abundance of heavy chlorine isotopes. A MeCN (1 mL) solution of 2-methoxyacetic acid (20.4 mg, 0.227 mmol) at 0 °C was treated with HATU (157 mg, 0.412 mmol), triethylamine (43 μL, 0.309 mmol), followed by slow addition of a MeCN (2 mL) solution of the above deprotected amine (0.103g, 0.206 mmol). The reaction was stirred at 5 °C for 1h 15 min, allowed to reach room temperature, stirred 15 min, and diluted with EtOAc. The mixture was washed with 1 N HCl, saturated NaHCO3, and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified by normal-phase MPLC (12 g silica gel cartridge, MeOH in CH2Cl2, 20 mL/min) to give 76 mg of 12 as a white solid in 81% yield.
(2R,3S,4S)-5-(3′,5′-Dichloro-[1,1′-biphenyl]-4-yl)-2,3-dihydroxy-4-(2 methoxyacetamido)pentanoic acid (1, CC0651)
(2R,3S,4S)-Methyl 5-(3′,5′-dichloro-[1,1′-biphenyl]-4-yl)-2,3-dihydroxy-4-(2-methoxyacetamido)pentanoate (12) (69 mg, 0.151 mmol) was dissolved in THF (1 mL) and water (1 mL), cooled to 5 °C, treated with 1 N lithium hydroxide (0.166 mL, 0.166 mmol), and stirred at 5 °C for 30 min. Once LC-MS analysis indicated reaction completion, 1 N HCl (0.300 mL, 0.300 mmol) was slowly added at 5 °C and the volatiles were removed. The residue was dissolved in DMSO and purified on Prep HPLC (Zorbax column, 5 um, 21.2 × 100 mm, MeCN gradient in water (0.05% TFA), 20 mL/min) to give 42 mg of CC0651 as a fluffy white solid after lyophilization in 63% yield.
Synthesis of CC0651 analogs
Biaryl derivatives were synthesized by a modification of the synthetic scheme in which the chiral amino alcohol (7) was subjected directly to oxidation, Wittig olefination, and dihydroxylation conditions to give p-bromo intermediates. Boc deprotection was followed by acylation using excess EDC-activated 2-methoxyacetic acid with DMAP as a mediator. The resulting triply acylated aryl bromide was Suzuki-coupled with the appropriate aryl boronic acids to directly afford the target biaryl analogues (2-6).
Supplementary Material
Acknowledgments
We thank Andreas Ernst, Ray Deshaies, Almer van der Sloot, Raik Grunberg, Jean-François Lavallée, Clint James, Kyle Chan and Frank Mercurio for helpful discussions. We also thank I. Kourinov and staff at Argonne National Laboratory for assistance with microdiffraction experiments at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, as supported by award RR-15301 from the National Center for Research Resources at the National Institutes of Health and contract DE-AC02-06CH11357 from the U.S. Department of Energy. G.K. and A.Z. are funded by grants from the National Center for Research Resources (5P20RR016464-11) and the National Institute of General Medical Sciences (8 P20 GM103440-11) from the National Institutes of Health. This work was supported by a grant to F.S. and M.T. from the Canadian Institutes of Health Research (MOP-126129), by an award from the Ministère de l’enseignement supérieur, de la recherche, de la science et de la technologie du Québec through Génome Québec to M.T, by a Canadian Institutes of Health Research Postdoctoral Fellowship to D. S-C., by a Canada Research Chair in Structural Biology to F.S. and by a Canada Research Chair in Systems and Synthetic Biology to M.T.
Footnotes
Author Contributions H.H. performed NMR; D.C. performed X-ray crystallography; S.O. and A.Z. performed ubiquitination assays; P.G. generated protein reagents; S.P. and D.S-C. synthesized CC0651 and its analogs; M.A., S.S, A.M., G.K., M.T, and F.S. designed experiments and interpreted results; M.T. and F.S. wrote the manuscript with contributions from all other authors.
Competing Financial Interests A provisional patent application for E2 enzyme inhibition has been filed, with M.T. and F.S. as inventors.
Accession codes. Final coordinates and structure factors for the CC0651:Cdc34ACAT:ubiquitin complex were deposited to the PDB with accession codes 4MDK and RCSB081775.
References
- 1.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 2.Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458:438–444. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- 3.Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE. The NEDD8 conjugation pathway and its relevance in cancer biology and therapy. Genes Cancer. 2011;1:708–716. doi: 10.1177/1947601910382898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacGurn JA, Hsu PC, Emr SD. Ubiquitin and membrane protein turnover: from cradle to grave. Annu Rev Biochem. 2012;81:231–259. doi: 10.1146/annurev-biochem-060210-093619. [DOI] [PubMed] [Google Scholar]
- 5.Matyskiela ME, Martin A. Design principles of a universal protein degradation machine. J Mol Biol. 2012 doi: 10.1016/j.jmb.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009;10:755–764. doi: 10.1038/nrm2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wenzel DM, Stoll KE, Klevit RE. E2s: structurally economical and functionally replete. Biochem. J. 2011;433:31–42. doi: 10.1042/BJ20100985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saha A, Lewis S, Kleiger G, Kuhlman B, Deshaies RJ. Essential role for ubiquitin-ubiquitin-conjugating enzyme interaction in ubiquitin discharge from Cdc34 to substrate. Mol. Cell. 2011;42:75–83. doi: 10.1016/j.molcel.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wickliffe KE, Lorenz S, Wemmer DE, Kuriyan J, Rape M. The mechanism of linkage-specific ubiquitin chain elongation by a single-subunit E2. Cell. 2011;144:769–781. doi: 10.1016/j.cell.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pruneda JN, et al. Structure of an E3:E2-Ub complex reveals an allosteric mechanism shared among RING/U-box ligases. Mol Cell. 2012;47:933–942. doi: 10.1016/j.molcel.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 12.Emanuele MJ, et al. Global identification of modular cullin-RING ligase substrates. Cell. 2011;147:459–474. doi: 10.1016/j.cell.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liao H, et al. Quantitative proteomic analysis of cellular protein modulation upon inhibition of the NEDD8-activating enzyme by MLN4924. Mol Cell Proteomics. 2011;10:M111 009183. doi: 10.1074/mcp.M111.009183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleiger G, Saha A, Lewis S, Kuhlman B, Deshaies RJ. Rapid E2-E3 assembly and disassembly enable processive ubiquitylation of cullin-RING ubiquitin ligase substrates. Cell. 2009;139:957–968. doi: 10.1016/j.cell.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duda DM, et al. Structure of a glomulin-RBX1-CUL1 complex: inhibition of a RING E3 ligase through masking of its E2-binding surface. Mol Cell. 2012;47:371–382. doi: 10.1016/j.molcel.2012.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duda DM, et al. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134:995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soucy TA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 18.Brownell JE, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol. Cell. 2010;37:102–111. doi: 10.1016/j.molcel.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 19.Ceccarelli DF, et al. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell. 2011;145:1075–1087. doi: 10.1016/j.cell.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 20.Spratt DE, Shaw GS. Association of the disordered C-terminus of CDC34 with a catalytically bound ubiquitin. J Mol Biol. 2011;407:425–438. doi: 10.1016/j.jmb.2011.01.047. [DOI] [PubMed] [Google Scholar]
- 21.Choi YS, et al. The human Cdc34 carboxyl terminus contains a non-covalent ubiquitin binding activity that contributes to SCF-dependent ubiquitination. J Biol Chem. 2010;285:17754–17762. doi: 10.1074/jbc.M109.090621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamilton KS, et al. Structure of a conjugating enzyme-ubiquitin thiolester intermediate reveals a novel role for the ubiquitin tail. Structure. 2001;9:897–904. doi: 10.1016/s0969-2126(01)00657-8. [DOI] [PubMed] [Google Scholar]
- 23.Plechanovova A, Jaffray EG, Tatham MH, Naismith JH, Hay RT. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature. 2012 doi: 10.1038/nature11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dou H, Buetow L, Sibbet GJ, Cameron K, Huang DT. BIRC7-E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat Struct Mol Biol. 2012;19:876–883. doi: 10.1038/nsmb.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spratt DE, Wu K, Kovacev J, Pan ZQ, Shaw GS. Selective recruitment of an E2~ubiquitin complex by an E3 ubiquitin ligase. J Biol Chem. 2012;287:17374–17385. doi: 10.1074/jbc.M112.353748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Capili AD, Lima CD. Structure and analysis of a complex between SUMO and Ubc9 illustrates features of a conserved E2-Ubl interaction. J Mol Biol. 2007;369:608–618. doi: 10.1016/j.jmb.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knipscheer P, van Dijk WJ, Olsen JV, Mann M, Sixma TK. Noncovalent interaction between Ubc9 and SUMO promotes SUMO chain formation. EMBO J. 2007;26:2797–2807. doi: 10.1038/sj.emboj.7601711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seol JH, et al. Cdc53/cullin and the essential Hrt1 RING-H2 subunit of SCF define a ubiquitin ligase module that activates the E2 enzyme Cdc34. Genes Dev. 1999;13:1614–1626. doi: 10.1101/gad.13.12.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell. 2008;32:21–31. doi: 10.1016/j.molcel.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bracher PJ, Snyder PW, Bohall BR, Whitesides GM. The relative rates of thiolthioester exchange and hydrolysis for alkyl and aryl thioalkanoates in water. Orig Life Evol Biosph. 2011;41:399–412. doi: 10.1007/s11084-011-9243-4. [DOI] [PubMed] [Google Scholar]
- 31.Song J, et al. Stability of thioester intermediates in ubiquitin-like modifications. Protein Sci. 2009;18:2492–2499. doi: 10.1002/pro.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pierce NW, Kleiger G, Shan SO, Deshaies RJ. Detection of sequential polyubiquitylation on a millisecond timescale. Nature. 2009;462:615–619. doi: 10.1038/nature08595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burroughs AM, Jaffee M, Iyer LM, Aravind L. Anatomy of the E2 ligase fold: implications for enzymology and evolution of ubiquitin/Ub-like protein conjugation. J. Struct. Biol. 2008;162:205–218. doi: 10.1016/j.jsb.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thiel P, Kaiser M, Ottmann C. Small-molecule stabilization of protein-protein interactions: an underestimated concept in drug discovery? Angew Chem Int Ed Engl. 2012;51:2012–2018. doi: 10.1002/anie.201107616. [DOI] [PubMed] [Google Scholar]
- 35.Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol. 2009;10:319–331. doi: 10.1038/nrm2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pashkova N, et al. WD40 repeat propellers define a ubiquitin-binding domain that regulates turnover of F box proteins. Mol Cell. 2010;40:433–443. doi: 10.1016/j.molcel.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Husnjak K, Dikic I. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem. 2012;81:291–322. doi: 10.1146/annurev-biochem-051810-094654. [DOI] [PubMed] [Google Scholar]
- 39.Ernst A, et al. A strategy for modulation of enzymes in the ubiquitin system. Science. 2013;339:590–595. doi: 10.1126/science.1230161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell. 2000;102:533–539. doi: 10.1016/s0092-8674(00)00057-x. [DOI] [PubMed] [Google Scholar]
- 41.Tang X, et al. Suprafacial orientation of the SCFCdc4 dimer accommodates multiple geometries for substrate ubiquitination. Cell. 2007;129:1165–1176. doi: 10.1016/j.cell.2007.04.042. [DOI] [PubMed] [Google Scholar]
- 42.Delaglio F, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 43.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 44.Wang AC, Grzesiek S, Tschudin R, Lodi PJ, Bax A. Sequential backbone assignment of isotopically enriched proteins in D2O by deuterium-decoupled HA(CA)N and HA(CACO)N. J Biomol NMR. 1995;5:376–382. doi: 10.1007/BF00182281. [DOI] [PubMed] [Google Scholar]
- 45.Fielding L. NMR methods for the determination of protein–ligand dissociation constants. Prog Nucl Mag Res Spect. 2007;51:219–242. doi: 10.2174/1568026033392705. [DOI] [PubMed] [Google Scholar]
- 46.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 47.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 49.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qian X, et al. Discovery of the First Potent and Selective Inhibitor of Centromere-Associated Protein E: GSK923295. ACS Medicinal Chemistry Letters. 2010;1:30–34. doi: 10.1021/ml900018m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.