

Abstract
Isocitrate lyase (ICL) catalyzes the reversible retro-aldol cleavage of isocitrate to generate glyoxylate and succinate. ICL is the first enzyme of the glyoxylate shunt, which allows for the anaplerosis of citric acid cycle intermediates under nutrient limiting conditions. In Mycobacterium tuberculosis, the source of ICL for these studies, ICL is vital for the persistence phase of the bacteria’s life cycle. Solvent kinetic isotope effects (KIEs) in the direction of isocitrate cleavage of D2OV = 2.0 ± 0.1 and D2O[V/Kisocitrate] = 2.2 ± 0.3 arise from the initial deprotonation of the C2 hydroxyl group of isocitrate or the protonation of the aci-acid of succinate product of the isocitrate aldol cleavage by a solvent-derived proton. This KIE suggested that an equilibrium mixture of all protiated isocitrate, glyoxylate and succinate prepared in D2O, would undergo transient changes in equilibrium concentrations as a result of the solvent KIE and solvent-derived deuterium incorporation into both succinate and isocitrate. No change in the isotopic composition of glyoxylate was expected or observed. We have directly monitored the changing concentrations of all isotopic species of all reactants and products using a combination of NMR spectroscopy and mass spectrometry. Continuous monitoring of glyoxylate by 1H NMR spectroscopy shows a clear equilibrium perturbation in D2O. The final equilibrium isotopic composition of reactants in D2O revealed di-deuterated succinate, protiated glyoxylate, and mono-deuterated isocitrate, with the transient appearance and disappearance of mono-deuterated succinate. A model for the equilibrium perturbation of substrate species, and their time-dependent isotopic composition is presented.
Keywords: isocitrate lyase, equilibrium perturbation, isotope effects, tuberculosis
Introduction
Isocitrate lyase (ICL) is present in a number of bacteria, fungi, and plants, but not in humans.(1) The retro-aldol cleavage of isocitrate to succinate and glyoxylate catalyzed by ICL is the first step of the glyoxylate shunt, a two enzyme pathway that converts AcCoA and isocitrate into the four carbon citric acid cycle intermediates succinate and malate. These steps bypass the isocitrate and β-ketoglutarate dehydrogenase steps of the citric acid cycle, preventing the loss of the two carbon units of acetyl coenzyme A (AcCoA) from being released as CO2, and allowing for the incorporation of the two carbon units, via gluconeogenesis, into triose and hexose pools. M. tuberculosis requires ICL to maintain metabolite pools when restricted to oxidation of fatty acids as a food source during the persistence phase of the bacterial life cycle. The persistence phase is particularly difficult to treat, and a knockout of ICL in M. tuberculosis has shown attenuated virulence in mice(2).
The original identification of the enzyme activity of ICL was reported in 1953,(3) and was followed by a series of reports that demonstrated that threo-Ds-isocitrate was the true substrate and the enzyme was induced when organisms were grown on acetate.(4-6) Studies of the stereochemistry of the reaction revealed that cleavage of isocitrate in D2O yielded [2-2H]L-succinate as the product, a net inversion of configuration, and was associated with a small solvent kinetic isotope effect of ~1.2.(7, 8) In these studies, it was demonstrated that the incorporation of solvent-derived deuterium into succinate was absolutely dependent upon glyoxylate being present, suggesting that glyoxylate bound to the active site metal, followed by succinate. Crystal structures of the M. tuberculosis ICL where the active site magnesium ion is coordinated by the aldehyde and carboxylate of glyoxylate and nitroproprionate, an isosteric mimic of the carbanion of succinate, support this interpretation.(9)
Solvent isotopic composition can influence both the rates of enzyme catalyzed reactions (solvent kinetic isotope effects) and in the case of solvent derived deuterium incorporation into non-exchangeable C-H bonds in products, solvent equilibrium isotope effects. The former reveal potential rate-limiting proton transfer steps in the chemical reaction, while the latter reflect the stiffness of the C-H bond in the deuterated product compared to water. These two effects are not mutually exclusive, and one of the first reports suggesting such coupling was reported by Cardinale and Abeles for proline racemase. By monitoring the racemization of L- to D-proline by optical rotation they found that the reaction in D2O approached and “overshot” the equilibrium concentrations of the reactants, and then returned to equilibrium. This is in contrast to the reaction in H2O, which approached equilibrium concentrations but never overshot them. The authors proposed that this was due to the incorporation of deuterium from solvent into the [β-2H]D-proline product and the slower reisomerization of D- to L-proline as a result of a solvent kinetic isotope effect.(10) Subsequently, Schimerlik et al. demonstrated that when an equilibrium mixture of enzyme substrates was prepared with hydrogen-containing substrates and deuterium-containing products, and enzyme was added, a transient perturbation of the concentrations of substrates/products was observed due to the KIEs, and the magnitude of the KIE could be determined from the magnitude of the displacement, or isotopically-induced “perturbation”.(11) This method was sensitive enough to measure heavy atom KIEs, and in the original report 2H, 13C, and 15N substituted substrates were used to determine primary KIEs on malate dehydrogenase, malic enzyme and glutamate dehydrogenase.(11)
In a case more related to the present report, primary and secondary deuterium, solvent deuterium and 18O kinetic and equilibrium isotope effects were reported for pig heart fumarase.(12) Solvent derived deuterium is incorporated into the C3-HR bond of 2S-malate, and the measured primary solvent kinetic and equilibrium isotope effects were inverse (0.92 and 0.98, respectively). The latter equilibrium isotope effect is likely to be similar to the effect for solvent deuterium incorporation into succinate (and isocitrate) in the ICL reaction.
All of the foregoing examples relied on the continuous measurement of changes in absorbance or optical rotary dispersion. Our initial attempts to measure the ICL reaction and potential perturbation using optical rotary dispersion due to changes in the concentration of [2R, 3S]isocitrate revealed that this method would not be sensitive enough to perform the desired experiments. Rather, we posited that 1H NMR spectroscopy and small molecule mass spectrometry could be used to semi-continuously measure both the concentrations and isotopic composition of all substrates and products simultaneously.
Materials and Methods
Materials
All kinetic experiments used isocitrate from Sigma Aldrich as the DL-isocitric acid trisodium salt hydrate. All mass spectrometry, NMR spectroscopy, and Keq experiments used [2R, 3S]isocitrate enzymatically synthesized with ICL and purified as described previously(13). In both cases the concentration of isocitrate was determined enzymatically with isocitrate dehydrogenase.
Cloning, Expression, and Purification
M. tuberculosis icl1 (Rv0467) was PCR amplified from H37Rv M. tuberculosis DNA with Nde1 and HindIII restriction sites. The PCR product was ligated into a pET-28a(+) vector, containing a N-terminal His6 tag. After positive sequencing results, the plasmid was transformed into T7 express competent E. coli. A single colony was used to generate a starter culture, of which 10 mL was added to each of (6) 2 L flasks containing 1 L Luria Broth and 30 μg/mL kanamycin. The E. coli were grown, shaking at 200 RPM at 37°C until the A600 reached 0.6. After cooling, 0.5 mM IPTG was added to each flask. The E. coli continued to grow, shaking, overnight at 18°C. Cells were collected by centrifugation and resuspended in 50 mM Tris, pH 8.0 containing 250 mM NaCl, 1 mM imidazole, one tablet of EDTA-free protease inhibitor cocktail, 800 units of DNase, and 10 mg lysozyme. After 30 min cells were lysed by sonication, and cellular debris were removed by centrifugation. Batch nickel affinity purification was performed using 1 mL of Ni-NTA resin slurry for every 4 mL of lysate. After a 1 hour incubation the slurry mixture was poured into a column, and elution was performed stepwise with a 0 – 500 mM imidazole gradient. ICL-containing fractions were dialyzed into a storage buffer of 50 mM HEPES, pH 7.0 containing 150 mM NaCl, and 50% glycerol, and stored at −20°C. Protein concentration was determined by absorbance with ε=71,975 M−1.(14)
Measurement of ICL Activity
Isocitrate cleavage was monitored by coupling glyoxylate formation to lactate dehydrogenase (LDH), which can convert glyoxylate to glycolate with the concomitant oxidation of NADH. A typical 1 mL assay contained 50 mM HEPES, pH 7.5 containing 5 mM MgCl2, 50 μM NADH, 25 units LDH and varying amounts of isocitrate. The reaction was initiated with 20 nM ICL, from an intermediate dilution in 50 mM HEPES, pH 7.5 containing 150 mM NaCl, 5 mM MgCl2 and 5 mg/mL BSA. In the opposite direction, isocitrate formation was coupled to isocitrate dehydrogenase, which catalyzes the reduction of NADP+ with isocitrate conversion to β-ketoglutarate. A typical 1 mL assay contained 50 mM HEPES, pH 7.5 containing 5 mM MgCl2, 5 mM MnCl2, 500 μM NADP+, 5 mM (saturating) glyoxylate, 80 nM M. tuberculosis ICDH-1, and varying amounts of succinate or [2,2,3,3-2H]succinate. The reactions were initiated with 20 nM ICL from the same intermediate dilution as the cleavage reactions.
Steady-State Data Analysis
Initial kinetic parameters were obtained by fitting to the Michaelis-Menten equation. Primary KIEs and solvent KIEs were fit to eq 1 where Fi = 0 or 1 in the primary KIE experiment, and Fi = 0 or 0.945 in the solvent KIE experiment.
| (1) |
Keq Determination
Keq was determined by starting a reaction in 50 mM HEPES, pH 7.5 containing 5 mM MgCl2, 480 μM isocitrate, and 1.5 μM ICL. The reaction was determined to reach equilibrium when the amount of glyoxylate was no longer changing, as judged by the LDH coupled assay. Keq = 0.00107 ± 0.00004 M was determined in order to estimate equilibrium concentrations of the substrates and products in the ICL reaction. Keq is reported in the direction of isocitrate cleavage and was performed in triplicate (footnote 1).
NMR Equilibrium Perturbation Experiments
The determined equilibrium concentrations of substrates from the Keq determination experiments were scaled up 30× in 50 mM sodium phosphate buffer or 50 mM ammonium acetate buffer, pH 7.5 ([isocitrate] = 3.9 mM, [succinate] = 10.5 mM and [glyoxylate] = 10.5 mM). ICL, which had been exchanged into the appropriate buffer, was then added, and left 48 hours in H2O, to assure exact equilibrium conditions. ICL was removed with a 10K MWCO Amicon filter and the mixture was frozen and lyophilized prior to reconstitution in deuterated solvent. Since ammonium acetate is volatile 50 mM deuterated ammonium acetate buffer (pD 7.8) was used to reconstitute the mixture after lyophilization, while D2O was used for the sodium phosphate buffered experiments. All of the 1H NMR spectra for equilibrium perturbation experiments were acquired at 25°C on a Bruker DRX 600 MHz spectrometer and 1D proton spectra were collected using 64 scans with a 30 degree pulse width, a sweep width of 20 ppm sampled with 64k points and a recycle delay of 2 s for a total of about 5 minutes per spectrum. All spectra were processed with a −0.20 Hz exponential window function. For each experiment, 600 μL of the reconstituted equilibrium mixture was transferred to a NMR tube and a spectrum was acquired. ICL, exchanged into the appropriate deuterated buffer, was then added to the reaction (final concentration of 0.3, 1, or 5 μM) and mixed thoroughly. NMR spectra of the ICL-containing equilibrium mixture were collected continuously until the reaction was deemed to have reached equilibrium. NMR spectra were analyzed using MestReNova. For initial identification of the deuterated species present in the mixture, an aliquot of an in-progress NMR equilibrium perturbation experiment (0.3 μM ICL) was diluted into methanol and centrifuged to remove precipitated enzyme. The sample was analyzed in negative mode by mass spectrometry using a 12 T Fourier transform ion cyclotron resonance mass spectrometer (Agilent).
Mass Spectrometry Time Course Experiment
For mass spectrometry experiments, an equilibrium mixture in ammonium acetate buffer was prepared in an identical manner as described above for NMR spectroscopy experiments. After reconstitution of the lyophilized mixture in 50 mM deuterated ammonium acetate buffer (pD 7.0), ICL was added (1 μM final concentration) to initiate the reaction. Individual time points were taken by quenching samples of the reaction mixture in methanol in a 1:100 dilution. The methanol used as the quenching agent contained 103.95 μM malate as an internal standard. Samples were centrifuged to remove precipitated ICL and were transferred to vials for analysis by mass spectrometry. Time course mass spectrometry was performed in negative mode on a Waters Synapt G2 ESI mass spectrometer and all data were processed using MassLynx and TargetLynx (Waters). For each time point, three replicate measurements were obtained and each of the metabolites of interest was quantified. Using malate as the internal standard, the peak area of each compound was obtained.
Graphing and Fitting
Graphing and fitting were performed with GraphPad Prism 5.0d. The error bars are the standard deviation of duplicates or triplicates and the error reported with the fitted values is the error of the fit. A.u. is an abbreviation for arbitrary units.
Results and Discussion
Steady State Kinetics
Initial kinetic parameters were determined by fitting the data to the Michaelis-Menten equation. In the direction of isocitrate cleavage: kcat = 5.2 ± 0.2 sec−1 and Kisocitrate = 45 ± 7 μM. In the direction of isocitrate formation, kcat = 3.6 ± 0.2 sec−1, Kglyoxylate = 400 ± 70 μM and Ksuccinate = 240 ± 40 μM. Solvent KIEs on isocitrate cleavage of D2OV = 2.0 ± 0.1 and D2O[V/Kisocitrate] = 2.2 ± 0.3 were observed (Figure 1A). A small viscosity effect was detected when the reaction in water was compared to a glycerol control (8.5% w/v) of the same viscosity as D2O and the solvent KIEs were calculated in comparison to the glycerol control. The observation of a solvent KIE could indicate that initiation of the isocitrate cleavage reaction by proton removal from the C2 hydroxyl group of isocitrate is partially rate-limiting or that the protonation of the succinate carbanion by a solvent derived proton from Cys191 is partially rate-limiting (see below).(15) If proton removal from the C2 hydroxyl is rate-limiting, this would suggest that in the reverse reaction that alkoxide protonation would be more energetically difficult than the C-C bond forming reaction, which is unlikely. Intimate metal chelation could stabilize the alkoxide in the condensation reaction, but would likely destabilize the alcohol in the isocitrate cleavage reaction. In support of protonation of the succinate anion as being the source of the solvent kinetic isotope effect and the rate-limiting step, a large primary kinetic isotope effect is observed when isocitrate formation from either succinate or tetradeutero-succinate are compared (DV= 1.7 ± 0.1 and DV/Ksuccinate= 2.9 ± 0.2, Figure 1B). This is also supported by studies reported by Moynihan and Murkin(15).
Figure 1.

A) “Solvent Kinetic Isotope Effect in comparison to glycerol control.” D2OV = 2.0 ± 0.1 and D2O[V/Kisocitrate] = 2.2 ± 0.3. B) “Primary Kinetic Isotope Effect” DV= 1.7 ± 0.1 and DV/Ksuccinate= 2.9 ± 0.2.
It was therefore necessary to account for solvent-derived deuterium incorporation into the reactants. As shown in Scheme 1, the species in B represent the isotopic composition of the reactants at equilibrium in H2O. When the mixture is reconstituted in D2O and ICL is added, the isocitrate cleavage reaction yields a succinate molecule with a solvent derived deuterium at the C2 position (Scheme 1C). Since succinate is symmetric, the mono-deuterated molecule can enter the active site in two different orientations, allowing chemistry to occur by cleavage of the C-D bond (Scheme 1C) or cleavage of the C-H bond (Scheme 1D) generating fully protiated isocitrate or isocitrate bearing a deuterium on C4. The ratio of the two reaction fluxes will be subject to a substantial intramolecular isotope effect due to the large primary DV/Ksuccinate= 2.9 ± 0.2 observed in the condensation direction. Finally cleavage of [4-2H]isocitrate in D2O would yield di-deuterated succinate ([2,3-2H2]threo-Ls-succinate, Scheme 1E).
Scheme 1.

Chemical Mechanism (A) and Distribution oflsotopologues Before, During and After Incubation with D10 (B, C&D and E).
Isotopic Changes in Reactants Caused by D2O
An initial comparison of the 1H NMR spectra before and after ICL addition indicated that deuterium incorporation into isocitrate and succinate had occurred (Figure 2). The initial mixture in D2O, before the addition of ICL is representative of the equilibrium compositions and concentrations of substrates and products in H2O (blue spectrum, Figure 2). After ICL addition and incubation of the mixture at room temperature, a new equilibrium in D2O is reached (red spectrum, Figure 2). The two doublet of doublets in the region of the 1H NMR spectrum between 2.48 and 2.34 ppm collapse to a single doublet that is shifted slightly upfield after incubation with ICL (5 μM final concentration). This can be attributed to the stereospecific incorporation of a deuterium atom at the C4 position of isocitrate. The reduced complexity of the peak at 2.90 ppm corresponding to the C3 hydrogen of isocitrate is also consistent with the incorporation of a deuterium at C4. Deuterium incorporation also occurs enzymatically into succinate during the ICL catalyzed reaction. The peak at 2.32 ppm due to the succinate hydrogens undergoes an upfield shift and a decrease in area corresponding to the loss of 2 hydrogens after incubation with enzyme. This result explains the previous report that 1.4 deuterium atoms were found in succinate isolated from the cleavage of isocitrate by the enzyme in D2O, suggesting that in this experiment, isotopic equilibrium had failed to be achieved.(8)
Figure 2.

“NMR Spectra Before and After ICL addition.” Before ICL addition (blue spectrum) and 23.6 hours after the addition of 5 μM ICL, when isotopic equilibrium has been reached (red spectrum).
Equilibrium Perturbation
As revealed above, there is no solvent exchange into glyoxylate, suggesting that this reactant would be ideal to monitor the potential solvent deuterium-induced equilibrium perturbation. The glyoxylate hydrate (Keq (hydrate/aldehyde) = 163)(16) hydrogen peak at 4.95 ppm is in a clear spectral window and was integrated over the time course of the experiment. Shown in Figure 3 is the absolute integration of the glyoxylate peak in the 1H NMR spectrum over 23.7 h after the addition of 5 μM ICL. After addition of ICL to the equilibrium mixture, the concentration of glyoxylate decreases rapidly due to the higher rate of protiated succinate and glyoxylate condensation to form isocitrate relative to the rate of the isocitrate cleavage reaction, which is slowed in D2O by the solvent KIE. After this initial decrease, the glyoxylate concentration then increases and eventually reaches the new isotopic equilibrium that is experimentally indistinguishable from the original (Figure 3). Since two solvent-derived protons are incorporated into the C2 and C3 sp3 C-H bonds in succinate, and one is incorporated into the C4 sp3 C-H bond of isocitrate, the solvent equilibrium effect is predicted to be 0.98 or close to one.(12)
Figure 3.

“ Equilibrium Perturbation of Glyoxylate.” The main panel is continuous NMR monitoring after the addition of 5 μM ICL. Inset is monitoring the addition of 1 μM ICL.
In order to better observe and examine the spectral changes due to the incorporation of deuterium into succinate, we performed an additional equilibrium perturbation experiment at 1 μM ICL (Figure 4A). The initial peak, at 2.32 ppm corresponding to all protiated succinate decreases with the formation of a peak at 2.31 ppm that transiently increases, then decreases. A peak at 2.30 ppm slowly appears thereafter, representing the final product, di-deuterated succinate. The shift in peak resonances is due to the incorporation of deuterium, and we tentatively assign the middle peak to mono-deuterated succinate, produced from cleavage of isocitrate in D2O. We integrated each peak over the course of a similar experiment performed with 5 μM ICL (Figure 4B). It was not possible to clearly integrate the peaks corresponding to the all protiated isocitrate and mono-deuterated isocitrate due to peak overlap (Figure 2).
Figure 4.

“Time Course of Succinate 1H Resonances.” A) The red scan was taken before 1 μM ICL was added, the yellow 10 min after ICL addition, light green after 104 min, dark green after 329 min, blue after 659 min, and purple after 1313 min. B) Integration of each isotopic form of succinate after the addition of 5 μM ICL over 23.7 hours. Time zero is before ICL addition.
In the original report on the phenomenon of isotopically induced perturbations to the equilibrium by Schimerlik et al.,(11) the ability to observe the perturbation, and its size, was shown to depend on, and be useful in determining, the magnitude of the kinetic isotope effect. In that original treatment, only the situation in which one reactant in an equilibrium mixture was isotopically labeled and transferred to a product without exchange of labeled solvent into, or washout of label from, either reactant, was examined. Clearly, that is not the situation with isocitrate lyase, where both products and reactants exchange with solvent isotope, and in the case of succinate, multiple times. However, in the original treatment, it was shown that the magnitude of the perturbation could be correlated with the kinetic isotope effect. Isotope effects determined by the equilibrium perturbation method, termed D(Eq.Pert.), are most similar to the corresponding DV/K effects. Although we probed the perturbation with a substrate that does not become isotopically labeled, we have used it as a surrogate for the solvent kinetic isotope effect on isocitrate cleavage, since this is where solvent isotope is incorporated into the first formed product, monodeuterosuccinate (Scheme 1). Using equation 2 from the original reference (11)
| (2) |
where [Rmax] – [R0] / [R’0] represents the relative magnitude of the equilibrium perturbation, and β represents the estimated kinetic isotope effect on V/K, we calculate a displacement of 0.259, which corresponds to an isotope effect of between 2.0 and 2.1. This can be compared to the experimentally determined value in the steady-state of D2O[V/Kisocitrate] = 2.2 ± 0.3.
Mass Spectrometry
Using mass spectrometry we examined the conversions of isocitrate to mono-deuterated isocitrate and succinate to mono- and di-deuterated succinate from the point of ICL addition until the return to equilibrium (Figure 5). Immediately following ICL additions, we observe a decrease in the amount of protiated isocitrate and an increase in the amount of mono-deuterated isocitrate (Figure 5A). This continues until all the isocitrate in the reaction is present in the mono-deuterated form. We can also observe a slight equilibrium perturbation in the first three points where the isocitrate concentrate briefly increases before the more marked decrease. This confirms our results observed with glyoxylate (Figure 3). If glyoxylate initially decreases, isocitrate must initially increase. For succinate, we see a rapid decrease in the protiated species that is accompanied by an increase in the mono-deuterated species and a slower increase in the di-deuterated species (Figure 5B). After approximately 2 h the concentration of the mono-deuterated succinate peaks begins to decrease, and eventually all the succinate is di-deuterated. This pattern of formation and depletion of each of the succinate species confirms what we found when the reaction was monitored by 1H NMR spectroscopy (Figure 4B). These data could be fitted to equations describing single exponential behavior, and rate constants from the formation of each species were determined (Figure S2).
Figure 5.

“The Change in Isotopic Composition Over Time by Mass Spectrometry.” A) Conversion of isocitrate to mono-deuterated isocitrate. B) Conversion of succinate to di-deuterated succinate through a transient mono-deuterated succinate species.
Modeling and Simulation
With these data in hand, we queried whether we could generate a model and set of rate constants that could qualitatively simulate the mass spectrometric data for the time course for the appearance and disappearance of the various isotopic species. We used a chemically reasonable model shown in Scheme 2 corresponding to the isotopologues identified in Scheme 1, where the identity of the isotopic species of isocitrate and succinate are noted.
Scheme 2.
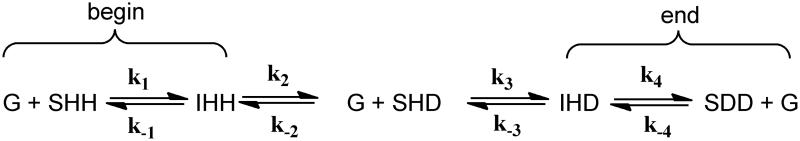
"Model for the Equilibrium Perturbation of the ICL-catalyzed reaction by D20." SHH, fully protiated succinate; G, glyoxylate ; IHH, fully protiated isocitrnte ; SHD, mono-deuterated succinate; IHD, mono-deuterated isocitrnte; SDD, di-deuterated succinate.
Initial estimates for the rates of all protio-succinate disappearance were obtained by fitting the mass spectrometric data to a single exponential. All calculated rate constants were in the range of 3.0 − 0.7 × 10−4 s−1. There are several simulation constraints in the model. The rate constant for conversion of isocitrate to glyoxylate and mono-deuterated succinate, k2, should be similar to the rate constant for conversion of mono-deuterated isocitrate to glyoxylate and di-deuterated succinate, k4, perhaps with a very small difference as a result of a secondary kinetic isotope effect. Similarly, the rate constant for conversion of mono-deuterated succinate to protio isocitrate, k−2, should be similar to the rate constant for conversion of di-deuterated succinate to mono-deuterated isocitrate, k−4, again with a possible small difference due to a secondary kinetic isotope effect. These two sets of rate constants were held equal during the simulation.
As seen in Figure 7, reasonable values for all eight rate constants could be obtained which qualitatively fit the time course of appearance and disappearance for all isotopically labeled substrate and products. This includes an initial increase in protio isocitrate concentration due to the equilibrium perturbation, which is equal and opposite to that observed for glyoxylate (Figure 3), and the predicted increase then decrease in mono-deuterated succinate concentration as it is initially formed in D2O from isocitrate cleavage and then ultimately converted into di-deuterated succinate from cleavage of deuterated isocitrate. The largest deviations are observed when the concentrations of species are small, and this is likely due to the larger errors in the measurement of the absolute amounts of these species by mass spectrometry. Finally, although the reverse fluxes of species via steps with associated rate constants k−1 and k−3 are likely to be small, the data could not be easily fit when these rate constants were set to zero.
Figure 7.

“Global Simulation of Mass Spectrometry Data” The Model shown in Scheme 2 was used as was the KinTek Global Kinetic Explorer (Ver. 3.0.2362). Isotopic species are: all protio isocitrate (red), all protio succinate (blue), mono-deutero isocitrate (green), mono-deuterated succinate (yellow) and di-deutero succinate (light blue). The rate constants yielding the curves through the experimental points are: k1 = 9.53 × 10−5 s−1; k−1 = 2.33 x10−4 s−1; k2 = 3.83 × 10−4 s−1; k−2 = 2.83 × 10−4 s−1; k3 = 3.43 × 10−5 s−1; k−3 = 4.05 × 10−5 s−1; k4 = 3.83 × 10−5 s−1; k−4 = 2.83 × 10−4 s−1.
Conclusions
In this study, we report the solvent isotope-induced transient change in the concentration and isotopic composition of the reactants. These effects were determined using a combination of 1H NMR spectroscopy and mass spectrometry, since no absorbance changes are manifest during the reaction. The normal solvent kinetic isotope effect for isocitrate cleavage generated an equilibrium perturbation whose size and direction were predicted by the solvent kinetic isotope effect. Mass spectrometry of the reactants as a function of time revealed the incorporation of solvent-derived deuterium into isocitrate and succinate; the initial formation of monodeuterosuccinate was followed by the eventual formation of dideuterosuccinate. A chemically reasonable model was generated and rate constants were obtained that could qualitatively simulate the experimental results.
Isocitrate lyase catalyzes the cleavage of isocitrate in D2O to form monodeuterosuccinate, a symmetric molecule that is asymmetrically isotopically labeled. This aspect of the reaction is similar to the reaction of diaminopimelate (DAP) epimerase, in which D,L-DAP is converted into monodeutero-L,L-DAP, which is similarly a symmetric molecule with asymmetric isotope labeling. This results in a double overshoot for which a mechanism was proposed and modeled (17). We report here the first determination of a solvent kinetic isotope effect-induced equilibrium perturbation by 1H NMR spectroscopy. The idea was suggested by the large steady-state solvent kinetic isotope effect on isocitrate cleavage, performed by NMR spectroscopy because of the lack of any spectroscopic change associated with the reaction and confirmed by mass spectrometry. Further investigation is required to determine whether this method can be used to quantitatively determine the kinetic isotope effect in the isocitrate lyase system; however, there are a large number of non-redox enzyme-catalyzed chemistries, including the large class of aldol and Claisen-type cleavage reactions and isomerases, for which this method may be broadly applicable to estimate kinetic isotope effects. The widespread availability and stability of modern NMR instruments and mass spectrometers should make its implementation routine.
Figure 8.

“Determination of Rate Constants from the Mass Spectrometric data.” A) The decrease of SHH was fitted to (y = (y0 – a)e(-kt)+ a) yielding k = 2.81 × 10−4 ± 8 × 10−6 s−1. B) The increase of SDD was fitted to y = y0 + (a−yo)(1−e(−kt)) yielding k = 8.4 × 10−5 ± 3 × 10−6 s−1. C) The decrease of IHH was fitted to y = y0 + (a−yo)(1−e(−kt)) (the first two time points were excluded from this fit) yileding k = 1.01 × 10−4 ± 8 × 10−6 s−1. D) The increase in IHD was fitted to y = y0 + (a−yo)(1−e(−kt)) yielding k = 7.5 × 10−5 ± 3 × 10−6 s−1.
Acknowledgements
We would like to thank Dr. William R. Jacobs Jr. for allowing us to use his mass spectrometer for the equilibrium perturbation experiments, and Drs. Brian Weinrick and Travis Hartman for training and insightful discussions. We would like to thank Dr. Hui Xiao for performing the initial mass spectrometric identification of the deuterated species, and Dr. Syun-Ru Yeh and Dr. Ariel Lewis for use and instruction on their circular dichroism machine. We thank Mary Thompson for performing the purification of ICL and efforts in initial assay development.
Dedication: This work is dedicated to the memory of W.W. Cleland
This work was supported by the National Institutes of Health Grant AI60899 to J.S.B. The instrumentation in the Structural NMR Resource is supported by the Albert Einstein College of Medicine.
Footnotes
Footnote
The Haldane relationship represents the equality of the equilibrium constant and the kinetic parameters for the reaction catalyzed. For the isocitrate lyase reaction which exhibits a Uni-Bi kinetic mechanism (isocitrate > glyoxylate + succinate), the relevant Haldane relationship (An Analysis of Haldane Relationships, Cleland, W.W. (1982) Methods Enzymol. 87, 366-369) is Keq = (V1•Kip•Kq)/(V2•Kia), where V1 and V2 are the maximum velocities in the isocitrate cleaving reaction and the aldol condensation reaction, respectively. There is strong evidence to support glyoxylate binding to the active site metal ion prior to succinate binding and therefore Kia is the dissociation constant for isocitrate, Kip is the dissociation constant for succinate and Kq the Michaelis constant for glyoxylate. Assuming that the determined Michaelis constants for isocitrate and succinate are reasonable approximations of the dissociation constants, our experimentally determined kinetic constants (see Results and Discussion) can be used to calculate an equilibrium constant of 0.0003. This is within a factor of 3.5 of the experimentally determined value of 0.0011, which considering the standard errors for the determined kinetic parameters and the assumption that the Michaelis and dissociation constants for the substrates are similar, is in good agreement. It also argues that glyoxylate hydration/dehydration is fast, since the total concentration of glyoxylate was used in the calculation of Keq.
References
- 1.Kondrashov FA, Koonin EV, Morgunov IG, Finogenova TV, Kondrashova MN. Evolution of glyoxylate cycle enzymes in Metazoa: evidence of multiple horizontal transfer events and pseudogene formation. Biol Direct. 2006;1:31. doi: 10.1186/1745-6150-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, Swenson D, Sacchettini JC, Jacobs WR, Jr., Russell DG. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–738. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- 3.Campbell JJ, Smith RA, Eagles BA. A deviation from the conventional tricarboxylic acid cycle in Pseudomonas aeruginosa. Biochimica et biophysica acta. 1953;11:594. doi: 10.1016/0006-3002(53)90107-3. [DOI] [PubMed] [Google Scholar]
- 4.Smith RA, Guncalus IC. Isocitritase: A New Tricarboxylic Acid Cleavage System. Journal of the American Chemical Society. 1954;76:5002–5003. [Google Scholar]
- 5.Smith RA, Gunsalus IC. Distribution and formation of isocitritase. Nature. 1955;175:774–775. doi: 10.1038/175774b0. [DOI] [PubMed] [Google Scholar]
- 6.Smith RA, Gunsalus IC. Isocitritase; enzyme properties and reaction equilibrium. The Journal of biological chemistry. 1957;229:305–319. [PubMed] [Google Scholar]
- 7.Sprecher M, Berger R, Sprinson DB. Stereochemistry of the isocitrate lyase reaction. Biochemical and biophysical research communications. 1964;16:254–257. doi: 10.1016/0006-291x(64)90335-3. [DOI] [PubMed] [Google Scholar]
- 8.Sprecher M, Berger R, Sprinson DB. Stereochemical Course of the Isocitrate Lyase Reaction. The Journal of biological chemistry. 1964;239:4268–4271. [PubMed] [Google Scholar]
- 9.Sharma V, Sharma S, Hoener zu Bentrup K, McKinney JD, Russell DG, Jacobs WR, Jr., Sacchettini JC. Structure of isocitrate lyase, a persistence factor of Mycobacterium tuberculosis. Nat Struct Biol. 2000;7:663–668. doi: 10.1038/77964. [DOI] [PubMed] [Google Scholar]
- 10.Cardinale GJ, Abeles RH. Purification and mechanism of action of proline racemase. Biochemistry. 1968;7:3970–3978. doi: 10.1021/bi00851a026. [DOI] [PubMed] [Google Scholar]
- 11.Schimerlik MI, Rife JE, Cleland WW. Equilibrium perturbation by isotope substitution. Biochemistry. 1975;14:5347–5354. doi: 10.1021/bi00695a020. [DOI] [PubMed] [Google Scholar]
- 12.Blanchard JS, Cleland WW. Use of isotope effects to deduce the chemical mechanism of fumarase. Biochemistry. 1980;19:4506–4513. doi: 10.1021/bi00560a019. [DOI] [PubMed] [Google Scholar]
- 13.Quartararo CE, Hazra S, Hadi T, Blanchard JS. Structural, Kinetic and Chemical Mechanism of Isocitrate Dehydrogenase-1 from Mycobacterium tuberculosis. Biochemistry. 2013;52:1765–1775. doi: 10.1021/bi400037w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.ExPASy http://ca.expasy.org/tools/protparam.html (15 June 2011) ProtParam Tool.
- 15.Moynihan MM, Murkin AS. Personal Communication. 2013 [Google Scholar]
- 16.Meany JE, Pocker Y. The Dehydration of Glyoxylate Hydrate: General-Acid, General-Base, Metal Ion, and Enzymatic Catalysis. Journal of the American Chemical Society. 1991;113:6155–6161. [Google Scholar]
- 17.Koo CW, Blanchard JS. The Chemical Mechanism of Haemophilus influenzae Diaminopimelate Epimerase. Biochemistry. 1999;38:4416–4422. doi: 10.1021/bi982911f. [DOI] [PubMed] [Google Scholar]