

Abstract
Rationale
Infusions of apoA-I, mimetic peptides or HDL remain a promising approach to treatment of atherosclerotic coronary disease. However, rapid clearance leads to a requirement for repeated administration of large amounts of material and limits effective plasma concentrations.
Objective
Since pegylation of purified proteins is commonly used as a method to increase their half-life in the circulation, we determined whether pegylation of apoA-I or HDL would increase its plasma half-life and in turn its anti-atherogenic potential.
Methods and Results
Initial pegylation attempts using lipid-poor apoA-I showed a marked tendency to form multi-pegylated (PEG) species with reduced ability to promote cholesterol efflux from macrophage foam cells. However, pegylation of human holo-HDL or reconstituted phospholipid/apoA-I particles (rHDL) led to selective N-terminal mono-pegylation of apoA-I with full preservation of cholesterol efflux activity. The plasma clearance of PEG-rHDL was estimated following injection into hypercholesterolemic Apoe−/− mice; the half-life of pegylated apoA-I following injection of PEG-rHDL was increased about 7-fold compared to apoA-I in non-pegylated rHDL. Compared to non-pegylated rHDL, infusion of PEG-rHDL (40 mg/kg) into hypercholesterolemic Apoe−/− mice led to more pronounced suppression of bone marrow myeloid progenitor cell proliferation and monocytosis, as well as reduced atherosclerosis and a stable plaque phenotype.
Conclusions
We describe a novel method for effective mono-pegylation of apoA-I in HDL particles, in which lipid binding appears to protect against pegylation of key functional residues. Pegylation of apoA-I in rHDL markedly increases its plasma half-life and enhances anti-atherogenic properties in vivo.
Keywords: HDL, apoA-I, atherosclerosis, hematopoiesis, cholesterol
INTRODUCTION
Extensive epidemiological data has shown an inverse relationship between HDL levels and coronary heart disease1,2. There is also convincing evidence for a protective role of HDL against atherogenesis in animal models3,4 notably following infusion of HDL or rHDL in hypercholesterolemic animal models5-7. However, the development of new therapies that increase plasma HDL levels has proven challenging. This was highlighted by the failure of torcetrapib and dalcetrapib, two different inhibitors of cholesteryl ester transfer protein, and of ER-niacin in recent clinical trials (AIM-HIGH and HPS2-THRIVE)8-12. In addition, carriers of the LIPG 396Ser allele in endothelial lipase had higher HDL cholesterol concentrations but this allele was not associated with an altered risk of myocardial infarction13, suggesting that raising HDL cholesterol by inhibition of endothelial lipase will not reduce atherosclerosis risk14. Ongoing clinical studies with more potent, apparently non-toxic CETP inhibitors9 will provide further evaluation of CETP inhibition but will not test the “HDL hypothesis” because these agents substantially lower LDL levels as well as increasing HDL.
A promising approach to reducing coronary atherosclerosis is the infusion of cholesterol-poor reconstituted HDL (rHDL)15-17. In two small-scale randomized clinical trials, infusions of wild type or the Milano variant of apoA-I in complexes with phospholipids into patients who had recently experienced acute coronary syndromes reduced atheroma volume in coronary arteries15-17. Infusion of rHDL in patients with peripheral vascular disease (PVD) also resulted in significant remodeling of the atheroma and suppression of inflammation16. These results most likely relate to the ability of cholesterol-poor rHDL particles to act as highly efficient acceptors of cholesterol from macrophage foam cells as well as other vascular cells involved in atherogenesis18,19 and to promote reverse cholesterol transport20. Animal models have revealed that raising HDL levels by genetic methods or infusion of rHDL inhibits vascular inflammation21, significantly remodels atherosclerotic lesions7,19,22-24 and improves endothelial function25.
Recent studies also indicate that dysregulated cholesterol homeostasis in hematopoietic stem and multipotent progenitor cells (HSPC) increases the risk of atherosclerosis as cholesterol accumulation in these cells promotes cell proliferation and causes monocytosis and neutrophilia, which contribute to accelerated atherosclerosis26,27. Accordingly, increasing HDL in hypercholesterolemic Apoe−/− mice reduced HSPC proliferation and monocytosis26, and decreased mobilization of HPSCs and extramedullary hematopoiesis28, suggesting a potential further application of rHDL infusions in the treatment of myeloproliferative neoplasms.
A major drawback of rHDL infusions in clinical practice is the need for repeated administration of a relatively large amount of material, due to the rapid clearance of HDL from the plasma17. The mechanisms responsible for turnover of plasma HDL are still poorly defined, though liver and kidney are the major organs taking up HDL29. One approach to reducing clearance has been to generate multimers of recombinant apoA-I. ApoA-I multimers had increased molecular size, decreased clearance relative to apoA-I monomers30 and caused reduced atherosclerosis in hypercholesterolemic mice30. Current methods to produce rHDL rely on the use of bile salts17. While the amphipathic nature of bile salt used in the preparation of rHDL facilitates rHDL particle formation, the residual bile salt in the rHDL preparation could cause adverse effects and therefore limit the quantity of the rHDL that can be administered17. These considerations suggest the need for novel strategies to improve the therapeutic efficacy of rHDL preparations.
Protein pegylation, a process of covalent attachment of polyethylene glycol polymer chain to the target protein, has been used extensively to increase the therapeutic efficacy of protein drugs31. However, protein pegylation has been used primarily for modification of purified proteins. It is not clear whether pegylation can also increase the plasma half-life of proteins complexed with other biological molecules such as lipids in lipoprotein particles and whether this modification affects their biological activities. The goal of this study was to determine if pegylation of lipid-poor apoA-I or rHDL particles could increase the biological half-life of the preparation while preserving anti-atherogenic functions.
METHODS
Animals
C57BL6/J or Apoe−/− mice were from Jackson Laboratory. For atherosclerosis studies, Apoe−/− mice were fed a Western type diet (WTD) (TD88137; Harlan Teklad) for the indicated period of time. Where indicated, vehicle (saline), rHDL, or PEG-rHDL was injected at the indicted dose into the mice via the tail vein. Purified human ApoA-I or rHDL (CSL-111) was provided by CSL Behring AG, Bern, Switzerland; CSL-111 is comprised of human apoA-I and phosphatidylcholine from soybean in a ratio of 1:150. The Columbia University animal ethics committee approved this study.
Pegylation of apoA-I or HDL
Holo-HDL was purified from human plasma as previously described using KBr density gradients32. Human apoA-I, HDL or rHDL were pegylated with M-PEG-ALD of MW 20000 or 40000. M-PEG-ALD was purchased from JenKem Technology USA (Allen, TX). After fully equilibrated to room temperature from −20°C storage and dissolved in an aliquot of 50 mM sodium acetate, pH 5.5, 10 mM sodium cyanoborahydride solution, M-PEG-ALD was immediately mixed at an indicated molar ratio with apoA-I, HDL or rHDL reconstituted in the same solution with gentle agitation. The final concentration of apoA-I, HDL or rHDL was ranging from 3 to 6 mg/ml. The mixture was incubated at 4°C for 16 to 72 hours. At the end of incubation, the reaction was quenched by addition of an aliquot of 1M Tris solution to the mixture to make the final concentration 100 mM Tris. The pegylated apoA-I, HDL or rHDL were subjected to SDS-PAGE and Coomassie Brilliant Blue staining for evaluation of the pegylation efficiency. The unmodified control was processed similarly without M-PEG-ALD. For cholesterol efflux assays or infusion of the pegylated or non-pegylated apoA-I, HDL or rHDL preparations into mice, same amounts of quenched, inactivated M-PEG-ALD were added to the non-pegylated apoA-I, HDL or rHDL preparations and the pegylated or non-pegylated preparations were dialyzed against phosphate buffered saline for final formulation. To determine the molecular mass of pegylated human apoA-I, we isolated pegylated apoA-I with a modified protocol of a method previously reported33. Briefly, pegylated apoA-I preparation was subjected to SDS-PAGE and the unfixed, unstained PEG-apoA-I band was excised from the gel after its location in the gel was estimated using a sample run in the same gel in parallel. After passive elution from the gel strip, the pegylated apoA-I sample was repeatedly diluted with phosphate buffered saline and concentrated with Amicon Centrifugal filters. The molecular mass of PEG-apoA-I was determined by MALDI-TOF.
Plasma clearance
An aliquot of unmodified or pegylated apoA-I, HDL or rHDL at the indicated dose was injected into the mice via tail vein. At the indicated time point, an aliquot of blood was collected from the mice. The blood samples were subjected to SDS-PAGE and Western analysis with anti-human apoA-I antibodies. Native or PEG-apoA-I was quantified by densitometry analysis with ImageJ.
Cholesterol efflux
Cholesterol efflux from mouse peritoneal macrophages or THP-1 cell derived macrophage like cells was performed as described previously32. Briefly, the cells were cholesterol loaded by incubation with the indicated amount of acetyl-LDL containing [3H]cholesterol for 16 hours in the presence of 1 μM TO901317. The cells were then washed and cholesterol efflux was initiated by addition of indicated amount of cholesterol acceptors before the media and cells were collected for analysis. Cholesterol efflux was expressed as the percentage of the radioactivity released from the cells into the medium relative to the total radioactivity in cells plus medium.
Analysis of blood leukocytes
Total while blood cell count in freshly collected mouse blood was performed using hematology cell counter (Oxford Science Inc.,). Monocytes and neutrophils were identified from whole blood as previously described26. Monocytes were identified as CD45hiCD115hi and neutrophils were identified as CD45hiCD115loLy6-C/Ghi (Gr-1).
Analysis of hematopoietic stem cells
Hematopoietic stem and progenitor cells from the BM were analyzed by flow cytometry as previously described26. Isolated bone marrow was incubated with a cocktail of antibodies against lineage-committed cells (B220, CD19, CD11b, CD3e, TER-119, CD2, CD8, CD4, Ly6-C/G: All FITC, eBioscience), Sca1-Pacific Blue and ckit-APC Cy7. HSPCs were identified as lin−, Sac1+ and ckit+ (LSK) while the hematopoietic progenitor subsets were separated by using antibodies to CD16/CD32 (FcγRII/III) and CD34. CMPs were identified as lin−, Sca1−, ckit+, CD34int, FcγRII/IIIint, GMPs as lin−, Sca1−, ckit+, CD34int, FcγRII/IIIhi. Cell cycle analysis was performed using DAPI (Sigma) in cells that had been stained with the above markers and then incubated in cytofix/cytoperm buffer (BD Biosciences).
Flow cytometry was performed using an LSRII running FACS DiVa software. Flow cytometry data was analyzed using FlowJo software (Tree Star Inc.).
Quantification of aortic atherosclerosis lesions
The lesions located in the aorta and aortic sinuses were analyzed using Oil-Red O staining. Quantification of Oil-Red O staining was performed off-line using Adobe Photoshop CS5 and presented as the percentage of the total surface are of the aorta. Lesion area in the aortic root was quantified by morphometric analysis of H&E stained sections as described34. Collagen staining was performed using picrosirius red as per the manufactures instructions (PolySciences, Inc). Macrophage staining was performed by staining with antibody against F4/80 (Abcam).
Statistical analysis
One-way ANOVA with post-hoc analysis and t tests were performed using GraphPad Prism version 5.00 for Mac OS X (GraphPad Software, San Diego California USA, www.graphpad.com) or STATVIEW 5.0 (Abacus Concepts, Inc). The post-test was Fisher's PLSD and the threshold for significance was p = 0.05. Data shown are mean ± SEM.
RESULTS
Characterization of N-terminally pegylated ApoA-I
Ideally, pegylation of apoA-I would not adversely affect apoA-I lipidation or its capacity to promote cholesterol efflux. ApoA-I structure and lipidation have been extensively studied. The carboxyl terminal amphipathic alpha-helix structure of apoA-I appears to be critical for lipid binding and its lipidation by ABCA1 activity35. In contrast, ABCA1-mediated cholesterol efflux is relatively insensitive to mutagenesis-induced changes in the N-terminal helix-bundle domain35. Thus, we reasoned that amino terminal pegylation of apoA-I might not disrupt the part of the structure of apoA-I necessary for mediating cholesterol efflux. In order to test this idea, we took advantage of an acidic reaction condition which favors selective pegylation of the amino terminal residue of the target protein36. When a reactive linear PEG molecule with average molecular size of 20 kDa was used for pegylation at 4°C, this strategy yielded a predominant single species of pegylated apoA-I with retarded migration in SDS polyacrylamide electrophoresis (SDS-PAGE) (Fig. 1A). The apparent Mr (~70 kDa) was higher than expected (i.e. human apoA-I: 28 kDa + PEG: 20 kDa = 48 kDa), likely reflecting aberrant migration, a common observation that has been attributed to PEG-SDS interactions when running pegylated proteins by SDS-PAGE37. We also used mass spectrometry to determine the molecular mass of the gel purified PEG-apoA-I band. This showed a molecular mass of approximately 48 kDa, as expected for mono-pegylated human apoA-I. Importantly, cholesterol efflux from macrophage foam cells was preserved using this method of pegylation (Fig. 1B). This indicated that mono-pegylation of apoA-I does not alter its function.
Figure 1. Characterization of pegylated human apoA-I.

(A) ApoA-I pegylated with M-PEG-ALD 20K at 4°C for 16 hours and analyzed by SDS-PAGE and Coomassie Blue staining. (B) Cholesterol efflux for 3 hours from cholesterol-loaded mouse peritoneal macrophages to apoA-I or PEG-apoA-I prepared as in (A). (C) ApoA-I pegylated with M-PEG-ALD 20K at 4°C for 40 and 72 hours and subjected to SDS-PAGE and Coomassie Blue staining. (D) Cholesterol efflux for 3 hours from cholesterol-loaded mouse peritoneal macrophages to apoA-I or PEG-apoA-I (10 μg protein/ml) prepared as in (C, 72 hour incubation).
However, the targeted pegylation of lipid-poor apoA-I at low temperature was suboptimal as the majority of the apoA-I was not pegylated. We attempted to increase the yield of pegylated apoA-I by adjusting variables such as the temperature and time of pegylation; however, this resulted in multi-pegylated species, suggesting pegylation of amino acid residues of apoA-I in addition to the N-terminus (Fig. 1C). Further, the multi-pegylated apoA-I had reduced capacity to promote cholesterol efflux from macrophage foam cells (Fig. 1D), indicating that unmodified amino acids of apoA-I other than the N-terminus are required to preserve functionality.
Characterization of pegylated HDL
In an attempt to reduce the pegylation of residues other than the N-terminus of apoA-I, we assessed the use of human holo-HDL particles as the substrate for protein pegylation. We reasoned that use of native HDL rather than lipid-poor apoA-I might increase the specificity and efficiency of targeted pegylation, as functionally important amino acid residues might be shielded from pegylation by lipid molecules in HDL particles. Under conditions where lipid-poor apoA-I gave rise to multi-pegylated species, pegylation of HDL gave rise mainly to mono-pegylated apoA-I species, as shown by SDS-PAGE and Coomassie Blue staining (Fig. 2A). After pegylation, >90% of human apoA-I in HDL particles migrated at the Mr of the mono-pegylated apoA-I species. The band that appears in the native HDL preparation of a similar size to PEG-HDL in Fig 2A is likely albumin, a common contaminant of HDL purified from plasma. We also performed a western analysis using an anti-apoA-I antibody and no band of that size was detected in the native HDL preparation confirming that it was not an apoA-I product (Fig 2B).
Figure 2. Characterization of pegylated human HDL.

(A) HDL with or without pegylation using M-PEG-ALD 20K at 4°C for 24 hours and analyzed by SDS-PAGE and Coomassie Blue staining. The minor higher Mr band in native HDL samples is likely albumin in the HDL preparations. (B) Western analysis using an anti-apoA-I antibody to HDL and PEG-HDL. (C) Cholesterol efflux for 3 hours from cholesterol-loaded macrophage cells derived from THP-1 cells to HDL or PEG-HDL prepared as in (A). (D) Plasma clearance of PEG-HDL following injection of 40mg/kg PEG-HDL prepared as in (A) and analyzed by Western analysis. N.S.= non-specific band.
We next carried out studies to determine if PEG-HDL promoted cholesterol efflux from macrophage foam cells. In order to rule out the potential effect of free PEG molecules in the efflux assay, the same amount of quenched PEG preparation was added to the unmodified HDL preparation during the efflux assay. The targeted pegylation of HDL particles did not affect the ability of HDL to promote cholesterol efflux. Cholesterol efflux from macrophages to the native HDL or PEG-HDL showed a similar dose response (Fig. 2C).
In order to evaluate the pharmacokinetics of PEG-HDL particles we infused PEG-HDL into chow-fed wild type mice. This preparation of PEG-HDL also contains a portion of non-pegylated human apoA-I which served as an internal control. The half-life of HDL protein (apoA-I) estimated by Western analysis of an aliquot of plasma was approximately 9.2 hours (lower Mr band in Fig. 2C), comparable to the estimated half life of human apoA-I infused into chow-fed wild type mice as reported by others30. Infusion of native human HDL at similar dose gave rise to a similar half-life of human apoA-I as determined from the internal standard (not shown). The turnover time of PEG-HDL, estimated by the clearance of the distinct PEG-apoA-I, was increased to ~48 hours as compared with that of unpegylated human HDL (Fig. 2D higher Mr band = PEG-apoA-I vs lower Mr band = non-PEG-apoA-I), indicating ~5-fold increase in circulation time. We also noted a non-specific band that was detected by western blot with anti-apoA-I antibody (Fig. 2D). However, we have confirmed this was not a modified form of human apoA-I as this band was also detected in plasma from mice that received a saline infusion (Fig. SI – lane 1).
Characterization of pegylated reconstituted HDL
For further characterization of properties of pegylated HDL, we used a preparation of rHDL that has been employed in clinical studies16,17. Targeted N-terminal pegylation of rHDL also yielded a largely homogeneous species of mono-pegylated apoA-I (Fig. 3A). In order to verify that apoA-I was pegylated at the N-terminus, the single species of pegylated apoA-I was isolated and subjected to Edman N-terminal protein sequencing. Only ~20% of the pegylated apoA-I could be sequenced by Edman degradation while, as a control, ~98% of apoA-I that was subjected to similar treatment but without pegylation could be sequenced (not shown). This suggests that the majority (~80%) of the mono-pegylated apoA-I was N-terminally pegylated. This sequencing result confirmed the method of preferential N-terminal protein pegylation, and suggests it can be applied to proteins complexed with other biological components so that amino acid residues that are essential for function are protected. In line with this idea cholesterol efflux assays showed that rHDL and PEG-rHDL promoted similar levels of cholesterol efflux from LXR activated human THP-1 macrophages (Fig. 3B). We next carried out studies to examine rHDL or PEG-rHDL clearance from plasma in a hypercholesterolemic mouse model. rHDL or PEG-rHDL at a dose of 60 mg/kg (based on rHDL protein content) was injected intravenously in Apoe−/− mice fed on a WTD. The clearance of human apoA-I or PEG-apoA-I from plasma was assessed by SDS-PAGE and Western blotting. Again as in Figure 2, some non-PEG-rHDL was present in the PEG-rHDL infused mice which served as an internal control. This along with the comparison to mice infused with rHDL clearly demonstrated the increased half-life of PEG-rHDL (Fig. 3C higher Mr band = PEG-HDL vs lower Mr band = non-PEG-HDL). Using densitometry we found that PEG-rHDL had a markedly reduced clearance rate and an approximately 7-fold prolonged half-life (t1/2=48.7 h for PEG-rHDL vs t1/2=7.7 h for rHDL and t1/2=7.5 h for non-PEG-rHDL internal control, n=5/group, p<0.001) (Fig. 3C,D). We also noted some intermediate bands in that reacted with the anti-apoA-I antibody in the mice infused with PEG-rHDL (Fig. 3C – right panel). It is possible that these could be partially degraded pegylated apoA-I molecules as it has previously been reported that apoA-I within HDL particles can be cleaved in vivo38,39. Again the non-specific band detected was not a by-product of PEG-rHDL preparation or a modification as this band was also detected in plasma from mice that received a saline infusion (Fig. SI).
Figure 3. Characterization of pegylated human rHDL.
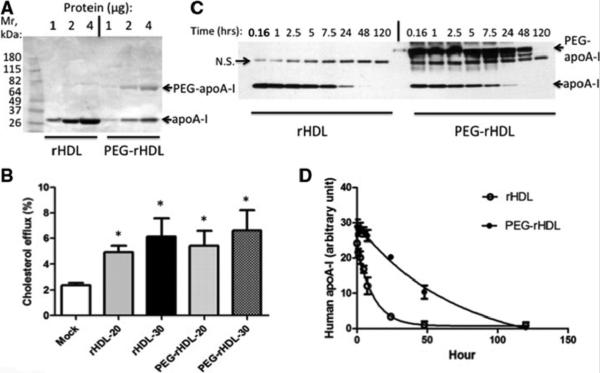
(A) rHDL pegylated with M-PEG-ALD 20K at 4°C for 16 hours and analyzed by SDS-PAGE and Coomassie Blue staining. (B) Cholesterol efflux for 3 hours from cholesterol-loaded LXR activated THP-1 macrophages to 20 or 30 μg rHDL or PEG-rHDL protein/ml. (C) Plasma clearance of rHDL or PEG-rHDL following injection of 60mg/kg rHDL or PEG-rHDL protein into circulation and analyzed by Western analysis. N.S.= non-specific band. (D) Densitometry quantification and analysis of (C), n=4.
PEG-rHDL is more efficient at reducing hematopoietic stem and progenitor cell proliferation and monocytosis
In order to test the idea that pegylation of rHDL would extend its half-life and thus promote a more prominent anti-atherogenic effect, we infused rHDL or PEG-rHDL into WTD-fed Apoe−/− mice. We and others have shown that hypercholesterolemic Apoe−/− mice develop monocytosis which appears to contribute to accelerated atherosclerosis26,40, and we showed that rHDL infusion could reduce monocyte levels in this mouse model26. Thus, monocyte counts could potentially act as a biomarker for the in vivo efficacy of rHDL infusions. In order to detect a potential difference in effectiveness of rHDL vs PEG-rHDL, we infused mice with two doses of 40 mg rHDL or PEG-rHDL protein/kg body weight separated by one week. This dosing schedule was chosen, as we had previously shown that 40mg/kg of rHDL was a submaximal dose, having a modest effect on inhibiting monocytosis in WTD-fed Apoe−/−26. At the 40mg/kg dose in the present study, rHDL infusion had no effect on the count of total white blood cells, monocytes or neutrophils 7 days after the second infusion (Fig. 4A, B). In contrast, PEG-rHDL significantly reduced these parameters relative to rHDL or the vehicle control. Our previous studies showed that effects of rHDL on monocyte counts reflected decreased proliferation of HSPCs and myeloid progenitors in the bone marrow26. Consistent with its effect of blood counts, PEG-rHDL infusion also more markedly decreased the count and proliferation of hematopoietic stem and progenitor cells (HSPC), common myeloid progenitors (CMPs) and granulocyte-macrophage progenitors (GMPs) (Fig. 4C, D).
Figure 4. Effects of rHDL or PEG-rHDL on leukocytosis and HSPC proliferation associated with ApoE deficiency.

Apoe−/− mice fed a WTD for 8 weeks (n=10/group) were given a single infusion of rHDL or PEG-rHDL (40 mg/kg) weekly for two weeks. 7 days post the second infusion, (A) peripheral blood WBC, (B) neutrophil and monocyte counts as well as (C) bone marrow progenitor cell abundance and (D) proliferation were determined by flow cytometry. Data are presented as mean ± SEM, n=10/group. *P<0.05 vs Saline.
PEG-rHDL is more effective at inhibiting atherosclerosis and stabilizing lesions
We initially conducted a dose response study using rHDL infusions in Apoe−/− mice fed a WTD for 4 wks to ascertain the amount of rHDL to use in our comparison to PEG-rHDL. Our goal was to find a dose of rHDL that failed to significantly improve atherosclerotic lesions. We administered two infusions of rHDL (40 mg, 80 mg or 120 mg protein/kg body weight) separated by one week. Doses of rHDL at 80 and 120, but not 40 mg/kg, significantly reduced the area of Oil-Red O staining of the aortic arch compare to mice treated with vehicle (Fig. SII). Therefore we decided to compare rHDL with PEG-rHDL at a dose of 40 mg/kg.
We examined the impact of rHDL or PEG-rHDL at a dose of 40 mg/Kg on the development of atherosclerosis in the hypercholesterolemic Apoe−/− mice. We observed no changes in total plasma or HDL cholesterol levels measure 7 days post the second infusion (Fig. SIII). Infusion of PEG-rHDL but not rHDL at equal doses (40mg/kg) significantly reduced the area of Oil-Red O staining of the aorta, indicating a reduction in aortic lipid content (Fig. 5A). Compared to our pilot experiment (Fig. SII) these results may suggest that PEG-rHDL administered at 40mg/kg has an efficacy similar to rHDL at 80mg/kg. Assessment of lesion formation in the proximal aorta showed no change in overall lesion area (Fig. 5B), which is consistent with a number of previous reports30. However, we observed a significant reduction in the macrophage content and cellularity of proximal aortic lesions of PEG-rHDL treated mice which was accompanied by a reduction in necrotic core size (Fig. 5 C,D,E). Lesions of PEG-rHDL treated mice also showed increased collagen (Fig. 5F). Taken together these results suggest that PEG-rHDL effectively remodeled lesions to what is considered a more stable phenotype, similar to that reported with rHDL infusions in humans with PVD16 and animal models where HDL levels were increased7,19,22-24.
Figure 5. Quantification of aortic atherosclerosis lesions.

Apoe−/− mice fed a WTD for 8 weeks were infused with rHDL or PEG-rHDL 40mg/kg weekly for two weeks. 7 days post the second infusion, the mice were euthanized lesion characteristics were analyzed. (A) The percentage of the aortic arch stained with Oil-Red O. (B) Lesion area in the proximal aorta. (C) Macrophage content as assessed by F4/80 staining in the proximal aorta. (D) Cellularity of the lesions in the proximal aorta determined by nuclei/area. (E) Necrotic core size from H&E stained sections of the proximal aorta. (F) Collagen staining in the proximal aorta by picrosirius red. Data are presented as mean ± SEM, n=10/group. *P<0.05 vs Saline.
DISCUSSION
We have developed a novel formulation of HDL prepared by targeted pegylation of HDL particles. While our initial studies focused on pegylation of lipid-poor apoA-I, attempts to scale up preparations by increasing temperature or PEG concentration were unsuccessful due to formation of multi-pegylated apoA-I species that were inefficient in cholesterol efflux assays. However, pegylation of plasma HDL or rHDL led to formation of functional, N-terminally, mono-pegylated apoA-I, likely because key amino acid residues were protected by binding to lipids. Following injection of this preparation, the half-life of apoA-I was increased about 7-fold. Most importantly, PEG-rHDL was significantly more effective than control rHDL at reducing myelopoiesis, monocyte counts, and atherosclerosis.
The apparent increase in efficiency of PEG-rHDL in reducing myelopoiesis and atherosclerosis was likely related to the increased half-life of apoA-I in the preparation. Since the half-life of apoA-I in HDL particles is several-fold more than that of HDL lipids, it is likely that apoA-I is recycled through several generations of HDL particles, probably being released by lipases that act on HDL, followed by lipidation of apoA-I via ABCA1 in various tissues41,42. However, it is important to note that we do not expect pegylation of rHDL to alter or enhance its cholesterol efflux ability on a molar basis compared to native rHDL. We speculate that compared to native apoA-I, mono-pegylated apoA-I may be less susceptible to glomerular filtration and degradation in the kidney or to catabolic uptake in the liver, allowing continued interaction with ABCA1 and ABCG1 in bone marrow progenitors, circulating leukocytes and macrophage foam cells.
The pegylation approach has several advantages. PEGs have been used to modify multiple protein drugs and their safety in humans has been rigorously examined43. The long-term, repeated use of some pegylated protein drugs, e.g. pegylated erythropoietin, is common in clinical practice44. A major use of protein pegylation is to increase hydrodynamic size of the target protein and reduce its glomerular filtration and degradation in kidney in order to increase its therapeutic concentration in the circulation43, desirable properties for apoA-I and HDL. While pegylation does not lead to conformational changes of the polypeptide chains in most cases43, it has the potential to reduce the immunogenicity of the target protein43.
Various formulations of cholesterol acceptors intended to promote cholesterol efflux from macrophage foam cells and RCT in vivo have been studied. In addition to rHDLs, infusions of synthetic amphipathic peptides complexed with phospholipids have been investigated45,46. However, on a molar basis the ability of synthetic peptides to promote cholesterol efflux from macrophage foam cells is much less than rHDL45,46. Infusion of a large amount of the peptide-phospholipid complexes is needed in order to achieve effective concentrations, which may increase the potential toxicity, for example in the kidney following glomerular filtration. In addition, the synthetic amphipathic peptides contain an artificial amino acid sequence, which has the potential to induce adverse immune responses.
Various strategies have been studied as a means of reducing the renal clearance of apoA-I. This included recombinant trimeric apoA-I, which showed an increased circulation time when injected into animals either as lipid-free or lipidated forms30. However, this approach has some intrinsic limitations. Cost-effective large scale manufacturing of recombinant proteins free of adverse foreign components remains a challenge, particularly for apoA-I with expected therapeutic doses higher than most recombinant protein drugs. Also, trimeric apoA-I, as a complex of recombinant fusion proteins of human apoA-I and the trimerization domain of human tetranectin, has the potential to be recognized as non-self protein and to induce adverse immune responses. The lipidated trimeric apoA-I also appears to be less efficient in promoting cholesterol efflux from macrophage foam cells relative to lipidated recombinant apoA-I monomers, at equal molecular mass30,47, however trimeric apoA-I preparations still retain many of the known anti-inflammatory functions of native apoA-I48.
Our study has several limitations. We compared the efficacy of rHDL and PEG-rHDL at a submaximal dose, which was chosen to assess a potential benefit of the pegylated preparation. For synthetic HDL preparations, the ability to use a lower dose represents a major advantage, both in terms of cost and convenience of preparations and also in terms of potential toxicities. We showed a beneficial effect of PEG-rHDL on aortic atherosclerosis as revealed by Oil-Red O staining, consistent with reduced macrophage foam cells content. However, lesion area in the proximal aorta was not reduced, similar to some previous studies using rHDL infusion strategies19. Nonetheless, macrophage content, lesion cellularity and necrotic core size were decreased and collagen content was increased, consistent with lesion stabilization7,19,22-24. PEG-rHDL, relative to rHDL, showed increased ability to suppress HSPC proliferation and monocytosis. This indicates that monocyte and leukocyte counts may be useful biomarkers to assess effectiveness of different HDL based therapies in vivo. Together, the improved pharmacokinetics and enhanced anti-atherogenic activity of PEG-rHDL suggest that it may be a useful preparation, with certain advantages such as lower dosing and decreased potential for toxicity.
Several recent clinical and genetic studies have led the generic “HDL hypothesis” to be called into question8-13. However, we believe this hypothesis should be refined to emphasize HDL functionality especially its ability to promote macrophage cholesterol efflux and reverse cholesterol transport49,50. While recent studies of HDL raising with niacin have not shown clinical benefit, the macrophage cholesterol efflux potential of HDL from subjects treated with niacin is at best marginally improved51. HDL raising with dalcetrapib also did not produce clinical benefit52 but lipoprotein changes may have been suboptimal due to incomplete CETP inhibition. Variants in some genes associated with increased HDL levels were not associated with expected effects on CAD13, but none of these variants are known to influence the production of apoA-I or HDL. Finally, a large body of preclinical evidence strongly supports the concept that interventions to increase macrophage cholesterol efflux and reverse cholesterol transport, as is likely to occur with rHDL infusions, are associated with reduced atherosclerosis49,50. Whether rHDL infusions succeed in improving clinical outcomes remains to be proven. If such therapies do prove successful, targeted N-terminal pegylation of various forms of apoA-I or related peptides, which may be accomplished by initial introduction into a lipid containing HDL particle prior to pegylation, may represent a novel method for extending the effectiveness, reducing the dose and decreasing toxicity of the therapeutic HDL preparation.
Supplementary Material
Novelty and Significance.
What Is Known?
Raising HDL by direct infusion of reconstituted HDL (rHDL) appears to be a promising therapeutic approach to treating cardiovascular disease (CVD).
However, the relatively short half-life of the current preparations may require frequent dosing.
Reconstituted HDL for clinical use has been difficult to manufacture, expensive, and toxicity has limited dosing and usefulness.
What New Information Does This Article Contribute?
We developed a method for efficient, N-terminal monopegylation of HDL, which greatly extends the in vivo half-life of HDL and increases its anti-atherogenic properties.
This extends the concept of protein pegylation to pegylation of a particle, with beneficial results.
The use of pegylated HDL may reduce the dosing requirement for reconstituted HDL preparations, with greater efficacy and reduced potential for side-effects.
Directly raising HDL levels by rHDL is thought to be a promising approach in the treatment of cardiovascular disease. However, rHDL is cleared fairly quickly from the circulation, limiting its bioavailability. We describe a technique to effectively mono-pegylate the N-terminus of apoA-I in HDL particles which extends its in vivo half-life. Pegylation of HDL particles can be applied to holo-HDL, rHDL and possibly peptide/lipid-complexes, increasing efficacy, decreasing dosage and potentially reducing side-effects. This approach could be applied to the treatment of atherosclerosis and myeloproliferative neoplasms.
Acknowledgments
SOURCES OF FUNDING
A.J.M. was supported by a post-doctoral fellowship from the American Heart Association (AHA; 12POST11890019). This project was funded by the NIH grants HL87123 and HL107653.
Nonstandard Abbreviations
- PEG
Pegylation
- HSPC
hematopoietic stem and multipotent progenitor cells
Footnotes
DISCLOSURES
A.R.T is a consultant for Merck, Pfizer, Roche, Amgen, CSL, and Arisaph. The other authors have no conflicts to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Castelli WP, Doyle JT, Gordon T, Hames CG, Hjortland MC, Hulley SB, Kagan A, Zukel WJ. HDL cholesterol and other lipids in coronary heart disease. The cooperative lipoprotein phenotyping study. Circulation. 1977;55:767–772. doi: 10.1161/01.cir.55.5.767. [DOI] [PubMed] [Google Scholar]
- 2.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. The American journal of medicine. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 3.Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N. HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 2008;7:365–375. doi: 10.1016/j.cmet.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Duffy D, Rader DJ. Update on strategies to increase HDL quantity and function. Nature reviews. 2009;6:455–463. doi: 10.1038/nrcardio.2009.94. [DOI] [PubMed] [Google Scholar]
- 5.Badimon JJ, Badimon L, Fuster V. Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J Clin Invest. 1990;85:1234–1241. doi: 10.1172/JCI114558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiesa G, Monteggia E, Marchesi M, Lorenzon P, Laucello M, Lorusso V, Di Mario C, Karvouni E, Newton RS, Bisgaier CL, Franceschini G, Sirtori CR. Recombinant apolipoprotein AI(Milano) infusion into rabbit carotid artery rapidly removes lipid from fatty streaks. Circ Res. 2002;90:974–980. doi: 10.1161/01.res.0000018422.31717.ee. [DOI] [PubMed] [Google Scholar]
- 7.Shah PK, Nilsson J, Kaul S, Fishbein MC, Ageland H, Hamsten A, Johansson J, Karpe F, Cercek B. Effects of recombinant apolipoprotein A-I(Milano) on aortic atherosclerosis in apolipoprotein E-deficient mice. Circulation. 1998;97:780–785. doi: 10.1161/01.cir.97.8.780. [DOI] [PubMed] [Google Scholar]
- 8.Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B, Investigators I. Effects of torcetrapib in patients at high risk for coronary events. The New England journal of medicine. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 9.Cannon CP, Shah S, Dansky HM, Davidson M, Brinton EA, Gotto AM, Stepanavage M, Liu SX, Gibbons P, Ashraf TB, Zafarino J, Mitchel Y, Barter P. Safety of anacetrapib in patients with or at high risk for coronary heart disease. The New England journal of medicine. 2010;363:2406–2415. doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
- 10.Group HTC HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. European heart journal. 2013 doi: 10.1093/eurheartj/eht055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Investigators A-H. Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. The New England journal of medicine. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 12.Kastelein JJ, van Leuven SI, Burgess L, Evans GW, Kuivenhoven JA, Barter PJ, Revkin JH, Grobbee DE, Riley WA, Shear CL, Duggan WT, Bots ML, Investigators R Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. The New England journal of medicine. 2007;356:1620–1630. doi: 10.1056/NEJMoa071359. [DOI] [PubMed] [Google Scholar]
- 13.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton-Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart A, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, Stoll M, Berger K, Martinelli N, Girelli D, McKeown PP, Patterson CC, Epstein SE, Devaney J, Burnett MS, Mooser V, Ripatti S, Surakka I, Nieminen MS, Sinisalo J, Lokki ML, Perola M, Havulinna A, de Faire U, Gigante B, Ingelsson E, Zeller T, Wild P, de Bakker PI, Klungel OH, Maitland-van der Zee AH, Peters BJ, de Boer A, Grobbee DE, Kamphuisen PW, Deneer VH, Elbers CC, Onland-Moret NC, Hofker MH, Wijmenga C, Verschuren WM, Boer JM, van der Schouw YT, Rasheed A, Frossard P, Demissie S, Willer C, Do R, Ordovas JM, Abecasis GR, Boehnke M, Mohlke KL, Daly MJ, Guiducci C, Burtt NP, Surti A, Gonzalez E, Purcell S, Gabriel S, Marrugat J, Peden J, Erdmann J, Diemert P, Willenborg C, Konig IR, Fischer M, Hengstenberg C, Ziegler A, Buysschaert I, Lambrechts D, Van de Werf F, Fox KA, El Mokhtari NE, Rubin D, Schrezenmeir J, Schreiber S, Schafer A, Danesh J, Blankenberg S, Roberts R, McPherson R, Watkins H, Hall AS, Overvad K, Rimm E, Boerwinkle E, Tybjaerg-Hansen A, Cupples LA, Reilly MP, Melander O, Mannucci PM, Ardissino D, Siscovick D, Elosua R, Stefansson K, O'Donnell CJ, Salomaa V, Rader DJ, Peltonen L, Schwartz SM, Altshuler D, Kathiresan S. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishida T, Choi SY, Kundu RK, Spin J, Yamashita T, Hirata K, Kojima Y, Yokoyama M, Cooper AD, Quertermous T. Endothelial lipase modulates susceptibility to atherosclerosis in apolipoprotein-E-deficient mice. J Biol Chem. 2004;279:45085–45092. doi: 10.1074/jbc.M406360200. [DOI] [PubMed] [Google Scholar]
- 15.Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA : the journal of the American Medical Association. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 16.Shaw JA, Bobik A, Murphy A, Kanellakis P, Blombery P, Mukhamedova N, Woollard K, Lyon S, Sviridov D, Dart AM. Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ Res. 2008;103:1084–1091. doi: 10.1161/CIRCRESAHA.108.182063. [DOI] [PubMed] [Google Scholar]
- 17.Tardif JC, Gregoire J, L'Allier PL, Ibrahim R, Lesperance J, Heinonen TM, Kouz S, Berry C, Basser R, Lavoie MA, Guertin MC, Rodes-Cabau J. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA : the journal of the American Medical Association. 2007;297:1675–1682. doi: 10.1001/jama.297.15.jpc70004. [DOI] [PubMed] [Google Scholar]
- 18.Weibel GL, Alexander ET, Joshi MR, Rader DJ, Lund-Katz S, Phillips MC, Rothblat GH. Wild-type ApoA-I and the Milano variant have similar abilities to stimulate cellular lipid mobilization and efflux. Arterioscler Thromb Vasc Biol. 2007;27:2022–2029. doi: 10.1161/ATVBAHA.107.148403. [DOI] [PubMed] [Google Scholar]
- 19.Shah PK, Yano J, Reyes O, Chyu KY, Kaul S, Bisgaier CL, Drake S, Cercek B. High-dose recombinant apolipoprotein A-I(milano) mobilizes tissue cholesterol and rapidly reduces plaque lipid and macrophage content in apolipoprotein e-deficient mice. Potential implications for acute plaque stabilization. Circulation. 2001;103:3047–3050. doi: 10.1161/hc2501.092494. [DOI] [PubMed] [Google Scholar]
- 20.Eriksson M, Carlson LA, Miettinen TA, Angelin B. Stimulation of fecal steroid excretion after infusion of recombinant proapolipoprotein A-I. Potential reverse cholesterol transport in humans. Circulation. 1999;100:594–598. doi: 10.1161/01.cir.100.6.594. [DOI] [PubMed] [Google Scholar]
- 21.Nicholls SJ, Dusting GJ, Cutri B, Bao S, Drummond GR, Rye KA, Barter PJ. Reconstituted high-density lipoproteins inhibit the acute pro-oxidant and proinflammatory vascular changes induced by a periarterial collar in normocholesterolemic rabbits. Circulation. 2005;111:1543–1550. doi: 10.1161/01.CIR.0000159351.95399.50. [DOI] [PubMed] [Google Scholar]
- 22.Choudhury RP, Rong JX, Trogan E, Elmalem VI, Dansky HM, Breslow JL, Witztum JL, Fallon JT, Fisher EA. High-density lipoproteins retard the progression of atherosclerosis and favorably remodel lesions without suppressing indices of inflammation or oxidation. Arterioscler Thromb Vasc Biol. 2004;24:1904–1909. doi: 10.1161/01.ATV.0000142808.34602.25. [DOI] [PubMed] [Google Scholar]
- 23.Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, Rayner K, Moore K, Garabedian M, Fisher EA. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7166–7171. doi: 10.1073/pnas.1016086108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rong JX, Li J, Reis ED, Choudhury RP, Dansky HM, Elmalem VI, Fallon JT, Breslow JL, Fisher EA. Elevating high-density lipoprotein cholesterol in apolipoprotein E-deficient mice remodels advanced atherosclerotic lesions by decreasing macrophage and increasing smooth muscle cell content. Circulation. 2001;104:2447–2452. doi: 10.1161/hc4501.098952. [DOI] [PubMed] [Google Scholar]
- 25.Kaul S, Coin B, Hedayiti A, Yano J, Cercek B, Chyu KY, Shah PK. Rapid reversal of endothelial dysfunction in hypercholesterolemic apolipoprotein E-null mice by recombinant apolipoprotein A-I(Milano)-phospholipid complex. Journal of the American College of Cardiology. 2004;44:1311–1319. doi: 10.1016/j.jacc.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 26.Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo CL, Wang M, Sanson M, Abramowicz S, Welch C, Bochem AE, Kuivenhoven JA, Yvan-Charvet L, Tall AR. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest. 2011;121:4138–4149. doi: 10.1172/JCI57559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, Tall AR. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–1693. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Westerterp M, Gourion-Arsiquaud S, Murphy AJ, Shih A, Cremers S, Levine RL, Tall AR, Yvan-Charvet L. Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell stem cell. 2012;11:195–206. doi: 10.1016/j.stem.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glass CK, Pittman RC, Keller GA, Steinberg D. Tissue sites of degradation of apoprotein A-I in the rat. J Biol Chem. 1983;258:7161–7167. [PubMed] [Google Scholar]
- 30.Graversen JH, Laurberg JM, Andersen MH, Falk E, Nieland J, Christensen J, Etzerodt M, Thogersen HC, Moestrup SK. Trimerization of apolipoprotein A-I retards plasma clearance and preserves antiatherosclerotic properties. J Cardiovasc Pharmacol. 2008;51:170–177. doi: 10.1097/FJC.0b013e31815ed0b9. [DOI] [PubMed] [Google Scholar]
- 31.Milla P, Dosio F, Cattel L. PEGylation of proteins and liposomes: a powerful and flexible strategy to improve the drug delivery. Curr Drug Metab. 2012;13:105–119. doi: 10.2174/138920012798356934. [DOI] [PubMed] [Google Scholar]
- 32.Wang N, Ranalletta M, Matsuura F, Peng F, Tall AR. LXR-induced redistribution of ABCG1 to plasma membrane in macrophages enhances cholesterol mass efflux to HDL. Arterioscler Thromb Vasc Biol. 2006;26:1310–1316. doi: 10.1161/01.ATV.0000218998.75963.02. [DOI] [PubMed] [Google Scholar]
- 33.Brace RJ, Sorrenson B, Sviridov D, McCormick SP. A gel-based method for purification of apolipoprotein A-I from small volumes of plasma. Journal of lipid research. 2010;51:3370–3376. doi: 10.1194/jlr.D008300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seidelmann SB, Kuo C, Pleskac N, Molina J, Sayers S, Li R, Zhou J, Johnson P, Braun K, Chan C, Teupser D, Breslow JL, Wight TN, Tall AR, Welch CL. Athsq1 is an atherosclerosis modifier locus with dramatic effects on lesion area and prominent accumulation of versican. Arterioscler Thromb Vasc Biol. 2008;28:2180–2186. doi: 10.1161/ATVBAHA.108.176800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vedhachalam C, Liu L, Nickel M, Dhanasekaran P, Anantharamaiah GM, Lund-Katz S, Rothblat GH, Phillips MC. Influence of ApoA-I structure on the ABCA1-mediated efflux of cellular lipids. J Biol Chem. 2004;279:49931–49939. doi: 10.1074/jbc.M406924200. [DOI] [PubMed] [Google Scholar]
- 36.Lee H, Jang IH, Ryu SH, Park TG. N-terminal site-specific mono-PEGylation of epidermal growth factor. Pharm Res. 2003;20:818–825. doi: 10.1023/a:1023402123119. [DOI] [PubMed] [Google Scholar]
- 37.Zheng C, Ma G, Su Z. Native PAGE eliminates the problem of PEG-SDS interaction in SDS-PAGE and provides an alternative to HPLC in characterization of protein PEGylation. Electrophoresis. 2007;28:2801–2807. doi: 10.1002/elps.200600807. [DOI] [PubMed] [Google Scholar]
- 38.Lee M, Kovanen PT, Tedeschi G, Oungre E, Franceschini G, Calabresi L. Apolipoprotein composition and particle size affect HDL degradation by chymase: effect on cellular cholesterol efflux. Journal of lipid research. 2003;44:539–546. doi: 10.1194/jlr.M200420-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Liz MA, Gomes CM, Saraiva MJ, Sousa MM. ApoA-I cleaved by transthyretin has reduced ability to promote cholesterol efflux and increased amyloidogenicity. Journal of lipid research. 2007;48:2385–2395. doi: 10.1194/jlr.M700158-JLR200. [DOI] [PubMed] [Google Scholar]
- 40.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res. 2005;96:1221–1232. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- 42.Rye KA, Barter PJ. Formation and metabolism of prebeta-migrating, lipid-poor apolipoprotein A-I. Arterioscler Thromb Vasc Biol. 2004;24:421–428. doi: 10.1161/01.ATV.0000104029.74961.f5. [DOI] [PubMed] [Google Scholar]
- 43.Bhadra D, Bhadra S, Jain P, Jain NK. Pegnology: a review of PEG-ylated systems. Pharmazie. 2002;57:5–29. [PubMed] [Google Scholar]
- 44.Weinreich T, Leistikow F, Hartmann HG, Vollgraf G, Dellanna F. Monthly continuous erythropoietin receptor activator treatment maintains stable hemoglobin levels in routine clinical management of hemodialysis patients. Hemodial Int. 2012;16:11–19. doi: 10.1111/j.1542-4758.2011.00608.x. [DOI] [PubMed] [Google Scholar]
- 45.Remaley AT, Thomas F, Stonik JA, Demosky SJ, Bark SE, Neufeld EB, Bocharov AV, Vishnyakova TG, Patterson AP, Eggerman TL, Santamarina-Fojo S, Brewer HB. Synthetic amphipathic helical peptides promote lipid efflux from cells by an ABCA1-dependent and an ABCA1-independent pathway. Journal of lipid research. 2003;44:828–836. doi: 10.1194/jlr.M200475-JLR200. [DOI] [PubMed] [Google Scholar]
- 46.Amar MJ, D'Souza W, Turner S, Demosky S, Sviridov D, Stonik J, Luchoomun J, Voogt J, Hellerstein M, Remaley AT. 5A apolipoprotein mimetic peptide promotes cholesterol efflux and reduces atherosclerosis in mice. J Pharmacol Exp Ther. 2010;334:634–641. doi: 10.1124/jpet.110.167890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohnsorg PM, Mary JL, Rohrer L, Pech M, Fingerle J, von Eckardstein A. Trimerized apolipoprotein A-I (TripA) forms lipoproteins, activates lecithin: cholesterol acyltransferase, elicits lipid efflux, and is transported through aortic endothelial cells. Biochim Biophys Acta. 2011;1811:1115–1123. doi: 10.1016/j.bbalip.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 48.Murphy AJ, Hoang A, Aprico A, Sviridov D, Chin-Dusting J. Anti-inflammatory functions of apolipoprotein a-I and high-density lipoprotein are preserved in trimeric apolipoprotein a-I. J Pharmacol Exp Ther. 2013;344:41–49. doi: 10.1124/jpet.112.199257. [DOI] [PubMed] [Google Scholar]
- 49.Hewing B, Moore KJ, Fisher EA. HDL and cardiovascular risk: time to call the plumber? Circ Res. 2012;111:1117–1120. doi: 10.1161/CIRCRESAHA.112.280958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rader DJ, Tall AR. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nature medicine. 2012;18:1344–1346. doi: 10.1038/nm.2937. [DOI] [PubMed] [Google Scholar]
- 51.Yvan-Charvet L, Kling J, Pagler T, Li H, Hubbard B, Fisher T, Sparrow CP, Taggart AK, Tall AR. Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler Thromb Vasc Biol. 2010;30:1430–1438. doi: 10.1161/ATVBAHA.110.207142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS, dal OI. Effects of dalcetrapib in patients with a recent acute coronary syndrome. The New England journal of medicine. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.