

Abstract
A 39-year-old woman with autosomal dominant polycystic kidney disease (ADPKD) presented with acromegaly and a pituitary macroadenoma. There was a family history of this renal disorder. She had undergone surgery for pituitary adenoma 6 years prior. Physical examination disclosed bitemporal hemianopsia and elevation of both basal growth hormone (GH) 106 ng/mL (normal 0–5) and insulin-like growth factor (IGF-1) 811 ng/mL (normal 48–255) blood levels. A magnetic resonance imaging scan disclosed a 3.0 cm sellar and suprasellar mass with both optic chiasm compression and left cavernous sinus invasion. Histologic, immunohistochemical and ultrastructural studies of the lesion disclosed a sparsely granulated somatotroph adenoma. Standard chromosome analysis on the blood sample showed no abnormality. Sequence analysis of the coding regions of PKD1 and PKD2 employing DNA from both peripheral leukocytes and the tumor revealed the most common PKD1 mutation, 5014_5015delAG. Analysis of the entire SSTR5 gene disclosed the variant c.143C>A (p.L48M, rs4988483) change in the heterozygous state in both blood and tumor, while no pathogenic mutations were noted in the MEN1, AIP, p27Kip1 and SSTR2 genes. To our knowledge, this is the fourth reported case of a GH-producing pituitary adenoma associated with ADPKD, but the first subject to extensive morphological, ultrastructural, cytogenetic and molecular studies. The question arises whether the physical proximity of the PKD1 and SSTR5 genes on chromosome 16 indicates a causal relationship between ADPKD and the somatotroph adenoma.
Key terms: pituitary adenoma, acromegaly, inherited polycystic kidney disease, genetics
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is an inherited disorder caused by germline mutations in the PKD1 and PKD2 genes, presents with renal manifestations and, in some cases, hepatic and pancreatic cysts as well as vascular abnormalities (1, 2). Acromegaly typically results from excessive secretion of growth hormone (GH) by a somatotrophic pituitary adenoma, usually sporadic in nature (3, 4). The association of ADPKD with pituitary adenoma is rare (5, 6). We report here a 39-year old woman with ADPKD and acromegaly. Genetic studies were undertaken in an effort to determine whether the PKD1 and SSTR5 on chromosome 16 may be involved in the molecular pathogenesis of somatotroph adenomas in PKD1.
Materials and Methods
Pathology
The surgically removed tumor was formalin-fixed, paraffin-embedded, sectioned at 5 microns and stained with hematoxylin-eosin and the periodic acid-Schiff (PAS) method. Immunohistochemistry employed the streptavidin biotin peroxidase complex method. For transmission electron microscopy, glutaraldehyde-fixed, osmicated, Epon-embedded tissue was studied on a Hitachi 7650 transmission electron microscope.
Cytogenetics
G-banding at 500–600 band resolution was performed on cultured PHA-stimulated peripheral lymphocytes. In addition, replication banding (RBHG) was performed with a 6 hr terminal pulse of BrdU in order to study early and late replicating regions.
Molecular Analysis
Studies were based upon tumor frozen immediately after surgery as well as DNA from blood and the tumor. The entire coding and flanking intronic regions of PKD1 and PKD2 were screened for mutations by direct sequencing as previously described (7, 8). Also investigated were genes associated with familial pituitary tumor susceptibility. PCR amplification of the entire coding and promoter regions of each gene was performed using primers and experimental conditions described previously (9–11). The purified PCR products were sequenced using Big Dye Terminator v3.1 (Applied Biosystems, Foster City, CA) and an automated sequencer (ABI Prism 3130 ×l DNA Analyzer, Applied Biosystems, Foster City), according to the manufacturer’s recommendations. All variants were confirmed in independent experiments.
In silico analysis
Six different web-available mutation predictor softwares, Sort Intolerant From Tolerant, SIFT (http://sift.jcvi.org), Polymorphism Phenotyping, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), PMUT (http://mmb2.pcb.ub.es:8080/PMut), SNAP (http://rostlab.org/services/SNAP), Protein Analysis Through Evolutionary Relationships, PantherPSEC (http://pantherdb.org/tools/csnpScoreForm.jsp) and Align Grantham Variation and Grantham Deviation, Align-GVGD (http://agvgd.iarc.fr/agvgd_input.php), which utilize algorithms based on sequence homology, and two others that assess the protein structure, MU-Pro http://www.ics.uci.edu/~baldig/mutation.html and I-Mutant 2 http://gpcr.biocomp.unibo.it/cgi/predictors/I-Mutant2.0/I-Mutant2.0.cgi, were used to gain further information regarding the possible pathogenicity of the missense variants found in the studied genes.
Results
Case Report
A 39-year-old woman had undergone prior surgery for pituitary adenoma six years prior. At that time, there was no clinical evidence of acromegaly; aside from mild hyperprolactinemia, data regarding pituitary hormone blood levels was unavailable. Neither immunohistochemical nor ultrastructural studies had been undertaken. Postoperatively, the patient was treated with bromocriptine for five years. Of note was a family history of ADPKD, two uncles being affected.
Recently, the patient presented with headache and visual disturbance. Physical examination disclosed clubbing of fingers and enlargement of the jaw, as well as other features of acromegaly affecting the face, hands and feet. Visual field examination showed nearly complete right visual loss and left temporal hemianopsia. An MRI scan demonstrated a 3-cm sellar mass with suprasellar extension and chiasmal compression (Figure 1A, B). Abdominal computed tomography (CT) disclosed innumerable bilateral kidney and liver cysts (Figure 1E, F), but renal and hepatic function was normal. Blood hormone levels were as follows: GH 106 ng/mL (normal <5.0); 60 minutes post glucose GH 90 ng/mL (normal<1.0), insulin-like growth factor-1 (IGF-1) 811 ng/mL (48–255); prolactin (PRL) 23.1 ng/mL (0–15); luteinizing hormone (LH) 0.1 mIU/mL (2–12); follicle-stimulating hormone (FSH) 1.7 mIU/mL (1–8); thyroid-stimulating hormone (TSH) 1.11 mIU/mL (0.5–6.0), free thyroxin (T4) 1.0 ng/dL (0.8–1.8), and cortisol 19 mcg/dL (5–25). A second transsphenoidal surgery was undertaken. Resection was subtotal; minor residual tumor, affected the left cavernous sinus (Figure 1C, D). One month after surgery, blood hormone levels were as follows: GH 1.13 ng/mL (normal <5.0) and an insulin-like growth factor-1 (IGF-1) level of 393 ng/mL (48–255). At eight months, basal GH measured 2.5 ng/mL (post glucose level 2.2 ng/mL) and IGF-1 level was 365 ng/mL. Treatment with octreotide, a long-acting somatostatin analog, was begun at a dose of 20 mg per month.
Fig. 1.
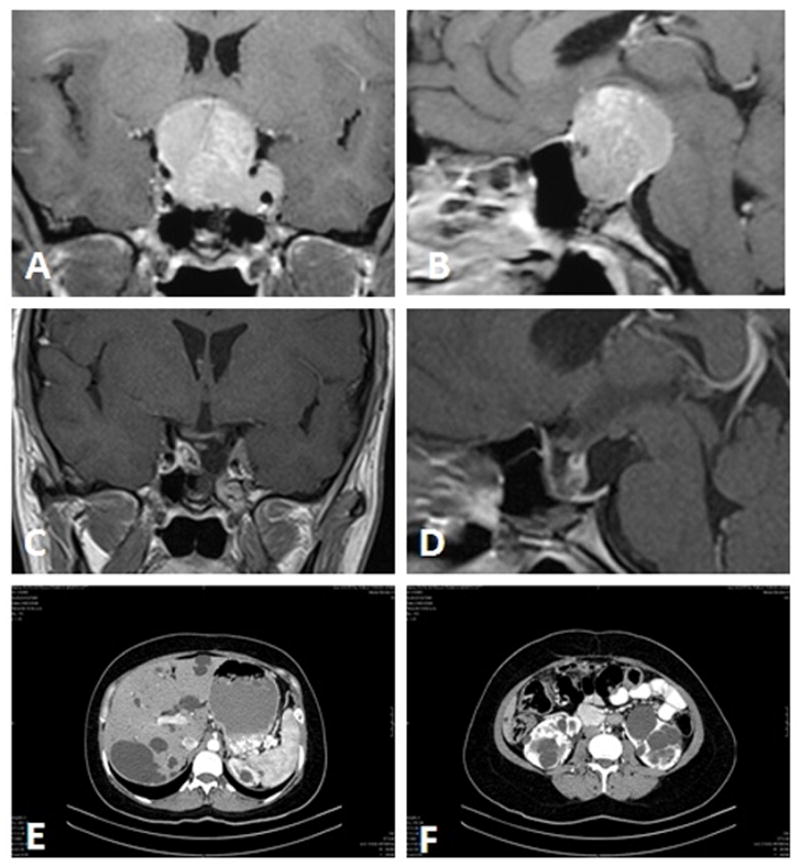
Coronal (A) and sagittal (B) T1-weighted MRI with gadolinium enhancement demonstrating the 3-cm, enhancing sellar and suprasellar tumor compressing the optic chiasm and invading the left cavernous sinus. Postoperative coronal (C) and sagittal (D) T1-weighted MRI scan with gadolinium enhancement demonstrating residual tumor within the left cavernous sinus. An abdominal CT scan showing countless hepatic (E) and bilateral renal cysts (F).
Pathology
By light microscopy, the slightly acidophilic, PAS-negative pituitary adenoma exhibited a diffuse growth pattern and mild cellular as well as nuclear pleomorphism, mitotic figures being rare. Immunohistochemistry demonstrated reactivity for GH in almost all cells and PRL staining in a few stains for ACTH, FSH, LH, TSH and alpha-subunit were negative. The nuclear labeling index using the MIB-1 antibody was estimated at 4–8% (Figure 2A–E). Ultrastructurally, the sparsely granulated growth hormone cell adenoma composed of medium to smaller size polyhedral to round cells. The uniformly ovoid to crescentic nuclei contained small to moderate size nucleoli and delicate heterochromatin. Rough endoplasmic reticulum was moderately developed. Polyribosomes were abundant. Golgi was not prominent, its flattened sacculi often being displaced by fibrous bodies which were noted in at least 30% of cells and consisted mainly of tubular smooth endoplasmic reticulum. Mitochondria were normal in number, size and ultrastructure. Secretory granules were sparse, randomly scattered and measured 90–236nm (mean 155nm) (Figure 2F). Based on the histologic, immunohistochemical and electron microscopic findings, a diagnosis of sparsely granulated GH cell adenoma was made.
Fig. 2.

(A) Light microscopic features of the pituitary adenoma. (B) Adenoma cells showing cytoplasmic immunopositivity for GH. (C) Few scattered adenoma cells show cytoplasmic immunopositivity for PRL. (D) Several tumor cell nuclei are immunopositive on MIB-1 stain. (E) Many adenoma cells contain cytoplasmic fibrous bodies immunopositive for low molecular weight keratin. (F) Electron microscopy reveals a sparsely granulated GH cell, adenoma with several large cytoplasmic fibrous bodies and few secretory granules. A, H&E 200x; B, 200x; C, 200x; D, 100x; E, 400x; F, 2500x.
Cytogenetics
Both G- and replication banding were normal, i.e., compatible with a 46, XX karyotype without detectable mosaicism or structural rearrangements.
Molecular Analysis
Molecular analysis of the PKD1 and PKD2 genes
Sequencing analysis of the patient and from the tumor itself revealed the most common PKD1 mutation, 5014_5015delAG (Figure 3). The normal allele was also present in the tumor, indicating no loss of heterozygosity of this region.
Fig. 3.
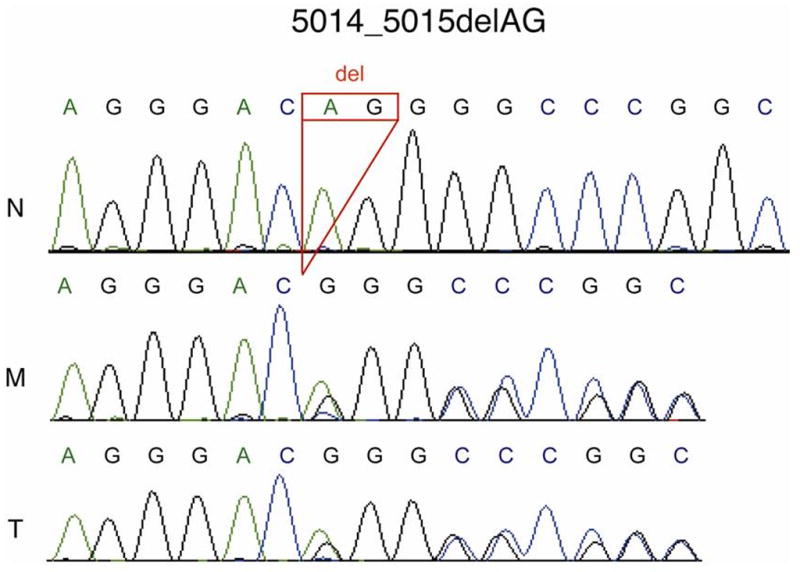
Sequence analysis of PKD1. N, normal; M, patient blood DNA and T, patient tumor DNA showing the common PKD1 mutation c.5014_15delAG in the two patient samples
Molecular analysis of the SSTR2, SSTR5, MEN1, p27Ki1p and AIP genes
Sequencing was carried out of the SSTR2 and SSTR5 genes, both composed of one exon that encodes either a 369 or a 364 amino acid transmembrane G-protein coupled receptors involved in somatostatin-mediated GH suppression. To verify whether the patient was at additional genetic risk of developing pituitary tumors, the MEN1, p27Kip1 and AIP genes were also analyzed. No germline pathological variants of these genes, as well as of the SSTR2 gene were found. However, the c.143C>A non-synonymous transversion (rs4988483) leading to the p.L48M substitution in the SSTR5 receptor was identified in heterozygous state in the patient’s blood DNA. Somatic analysis at the tumor level was also performed and showed the presence of both C/A alleles, indicating maintenance of heterozygosity at the SSTR5 locus (Figure 4). This variant is described in db SNP with a frequency of ~4% in Hispanic populations.
Fig. 4.

Sequence analysis of SSTR5 in blood (A) and tumor (B). The variant p.L48M is found in both patient samples.
The likelihood of pathogenicity of the p.L48M change in the SSTR5 receptor, which involves two amino acids with similar physicochemical properties (aliphatic hydrophobic side chains), was evaluated by six evolutionary-based softwares; all packages classified this missense variant as neutral/benign. However, the two softwares (MU-Pro and I-Mutant 2) that assessed the consequences to the protein structure of the SSTR5-L48M receptor indicated a possible decreased stability comparing to the wild-type SSTR5.
Discussion
To our knowledge, three previous cases of acromegaly associated with ADPKD have been reported (5, 6, 12). The first was published by Fajfr et al in 2002 (5). The patient, a 24-year-old woman with ADPKD undergoing an MRI study for aneurysm was found to have an incidental pituitary tumor. Three years later, the tumor had grown and was resected. Blood tests at that time showed high IFG-1 (569 ng/mL, normal 230–546 ng/mL) and elevation of GH after an oral glucose tolerance test (5.7 ng/mL), normal < 1 ng/mL). In 2005 Kannabiran (6) reported the second case, that of a 50-year-old woman with ADPKD, a 1.1 cm pituitary tumor, and high blood levels of GH, PRL and IGF-1. The GH level was not suppressed upon glucose tolerance testing. A transsphenoidal resection was undertaken. Although no physical signs of acromegaly were evident, the shoe size diminished. The third case, briefly mentioned by Ruggeneti et al. (12), involved a woman with ADPKD and acromegaly in whom the renal cysts showed no appreciable change after two years of long-acting somatostatin analogue treatment used to control GH excess. The authors suspected a potentially beneficial effect of octreotide in ADPKD patients. In our case, the patient presented with acromegaly and both chiasmal compression and visual disturbance. After surgery and at follow-up, persistence of GH excess and of acromegaly prompted octreotide therapy. Interestingly, only GH-producing adenomas have been reported to occur in association with ADPKD; all four patients were females. Among the three previously reported cases, histology was only available in one (5). Our case is the first with a detailed morphologic investigation.
Somatostatin exerts its inhibitory effect upon growth hormone, insulin and glucagon secretion by interaction with G-protein-coupled somatostatin receptors (4, 13–15). The two forms of endogenous somatostatin, composed of 14 and 28 amino acids, respectively, elicit cellular responses via five ubiquitously expressed somatostatin receptor subtypes, SSTR1 through 5. They are encoded by separate genes located on different chromosomes and are expressed in unique or partially overlapping distributions in various target organs. All the subtypes are expressed in the human fetal pituitary whereas the adult human pituitary expresses mainly SSTR1, 2, 3 and 5 (13–16). Pituitary SSTR5 and 2 are abundant in normal pituitary cells, whereas the other receptor subtypes are less represented. Their expression in pituitary tumors also differs (17–20); in human GH adenomas, SSTR2 and 5 are predominantly expressed. This has been the basis for using therapeutically long acting somatostatin analogs to reduce GH levels in acromegaly (21–23). Thus, such somatostatin receptor ligands as octreotide and lanreotide are used to treat acromegaly (24). It is of note that some patients with acromegaly do not respond to these long-acting somatostatin analogs. Resistance appears to be related to reduction of SSTR density, to differential expression of SSTR subtypes tumor (25–28), or to a SSTR5 mutation (29).
Ruggenenti et al (12) observed stabilization of renal cysts in an ADPKD patient with pituitary adenoma treated with octreotide for two years. The authors hypothesized that octreotide may have beneficial effects upon renal function in this disorder. Several recent randomized trials have shown a benefit of somatostatin analogue treatment upon renal and liver disease in ADPKD and autosomal dominant polycystic liver disease (ADPLD) (30–32).
Mutations in PKD1 and 2 cause ADPKD (1, 2). The PKD1 gene is located on the short (p) arm of chromosome 16 at position 13.3 (gene location 2,078,711–2,125,899 bp from the p-telomere). The SSTR5 gene is also located on chromosome 16 and in the same region of the PKD1 gene, its location being 1,062,757 to1,071,438, bp i.e., about 1 Mb (~1 cM) from it. Both genes, along with that of tuberous sclerosis 2 (TSC2), form part of a synteny group and are evolutionary conserved (33).
The association of GH-producing adenoma with ADPKD is of particular interest given the proximity of the two genes. In the present case, we considered the possibility that a macro-rearrangement involving both genes could be the basis of the concurrence of acromegaly and ADPKD. However, since a point mutation was evident in PKD1, not a large deletion, our findings do not indicate a contiguous gene syndrome. If PKD1 is involved in the regulation of other genes in its proximity, it may be possible that some interaction exists between the two genes.
On the NCBI database (http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=rs4988483), the SSTR5 p.L48M is a validated, non-synonymous variant (rs4988483) described in heterozygotes in 0 to 4% of control populations of diverse ethnicity[PCH1]. Among 14 different populations included in this database, only one (CAUC3) presented higher frequency of the p.L48M mutated allele (11.4%). Interestingly, there is some recent evidence indicating a potential role of SSTRs variants in determining hormone levels and tumor behavior (drug resistance). Ballarè et al. (29) reported SSTR5 p.R240W in a patient with pituitary tumor exhibiting somatostatin analog resistance, while SSTR5 variants were described in association with increased GH and IGF levels in patients with sporadic acromegaly (17). More recently, 4.600 subjects from Sweden (prostate cancer patients and controls) were subject to SSTR1 through 5 genotyping. A significant association was noted between the p.L48M-SSTR5 variant and IGF-1 and IGFBP3 serum levels (34). It has also been suggested that this polymorphism is associated with bipolar effective disorder (35).
Since SSTR5 and 2 are the main receptors controlling somatostatin-mediated GH suppression, our findings suggest that the p.L48M variant may somehow impair the proper function of the receptor and thereby play a role in adenomagenesis. The fact that Leucine-48 is an evolutional conserved amino acid and that our bioinformatics analyses indicate that this change could lead to decreased stability supports possible pathogenicity of the p.L48M variant[PCH2]. In fact, the adenoma subtype in this case, sparsely granulated GH cell adenoma, has been shown to be more aggressive than the densely granulated variant (4). A further possibility would be that linkage disequilibrium between this SNP and another causative genetic event is co-influential in the molecular etiology of concurrent kidney and pituitary disease. The proximity between the SSTR5 and PKD1 genes is of special interest in our patient. At present, the most likely explanation of a GH adenoma occurring in ADPKD is of two independent gene alterations in the same patient underlying the two phenotypes.
Acknowledgments
Authors are grateful to the Jarislowsky and Lloyd Carr Harris Foundations for their continuous support. RA Toledo is supported by a 2009 FAPESP (Sao Paulo State Research Foundation) postdoctoral fellowship. SAP Toledo is partially supported by a 2010 and 2011 CNPq (National Research Foundation) grants. The ADPKD gene analyses were funded by NIDDK grant DK058816 and the Mayo PKD Translational Center (DK090728). Lastly, the authors thank Mrs. Denise Chase of Mayo Clinic for her excellent secretarial support.
Footnotes
Disclosure: The authors have nothing to disclose.
References
- 1.Harris PC, Torres VE. Polycystic kidney disease. Annual review of medicine. 2009;60:321–337. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney international. 2009;76:149–168. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Melmed S. Medical progress: Acromegaly. The New England journal of medicine. 2006;355:2558–2573. doi: 10.1056/NEJMra062453. [DOI] [PubMed] [Google Scholar]
- 4.Melmed S. Acromegaly pathogenesis and treatment. The Journal of clinical investigation. 2009;119:3189–3202. doi: 10.1172/JCI39375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fajfr R, Muller B, Diem P. Hypophyseal incidentaloma in a patient with autosomal dominant polycystic kidney disease. Praxis. 2002;91:1123–1126. doi: 10.1024/0369-8394.91.25.1123. [DOI] [PubMed] [Google Scholar]
- 6.Kannabiran M, Singh V, Grewal S. Acromegaly presenting as psychotic disorder in a family with familial autosomal dominant polycystic kidney disease. German Psychiatry. 2006;9:136–138. [Google Scholar]
- 7.Rossetti S, Consugar MB, Chapman AB, Torres VE, Guay-Woodford LM, Grantham JJ, Bennett WM, Meyers CM, Walker DL, Bae K, Zhang QJ, Thompson PA, Miller JP, Harris PC. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18:2143–2160. doi: 10.1681/ASN.2006121387. [DOI] [PubMed] [Google Scholar]
- 8.Rossetti S, Kubly VJ, Consugar MB, Hopp K, Roy S, Horsley SW, Chauveau D, Rees L, Barratt TM, van’t Hoff WG, Niaudet P, Torres VE, Harris PC. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney international. 2009;75:848–855. doi: 10.1038/ki.2008.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Hofler H, Fend F, Graw J, Atkinson MJ. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A. 2006;103:15558–15563. doi: 10.1073/pnas.0603877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toledo RA, Lourenco DM, Jr, Liberman B, Cunha-Neto MB, Cavalcanti MG, Moyses CB, Toledo SP, Dahia PL. Germline mutation in the aryl hydrocarbon receptor interacting protein gene in familial somatotropinoma. The Journal of clinical endocrinology and metabolism. 2007;92:1934–1937. doi: 10.1210/jc.2006-2394. [DOI] [PubMed] [Google Scholar]
- 11.Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, Tuppurainen K, Ebeling TM, Salmela PI, Paschke R, Gundogdu S, De Menis E, Makinen MJ, Launonen V, Karhu A, Aaltonen LA. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006;312:1228–1230. doi: 10.1126/science.1126100. [DOI] [PubMed] [Google Scholar]
- 12.Ruggenenti P, Remuzzi A, Ondei P, Fasolini G, Antiga L, Ene-Iordache B, Remuzzi G, Epstein FH. Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney international. 2005;68:206–216. doi: 10.1111/j.1523-1755.2005.00395.x. [DOI] [PubMed] [Google Scholar]
- 13.Ben-Shlomo A, Melmed S. Pituitary somatostatin receptor signaling. Trends Endocrinol Metab. 2010;21:123–133. doi: 10.1016/j.tem.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel YC. Somatostatin and its receptor family. Front Neuroendocrinol. 1999;20:157–198. doi: 10.1006/frne.1999.0183. [DOI] [PubMed] [Google Scholar]
- 15.Reisine T, Bell GI. Molecular biology of somatostatin receptors. Endocr Rev. 1995;16:427–442. doi: 10.1210/edrv-16-4-427. [DOI] [PubMed] [Google Scholar]
- 16.Shimon I, Taylor JE, Dong JZ, Bitonte RA, Kim S, Morgan B, Coy DH, Culler MD, Melmed S. Somatostatin receptor subtype specificity in human fetal pituitary cultures. Differential role of SSTR2 and SSTR5 for growth hormone, thyroid-stimulating hormone, and prolactin regulation. The Journal of clinical investigation. 1997;99:789–798. doi: 10.1172/JCI119225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Filopanti M, Ronchi C, Ballare E, Bondioni S, Lania AG, Losa M, Gelmini S, Peri A, Orlando C, Beck-Peccoz P, Spada A. Analysis of somatostatin receptors 2 and 5 polymorphisms in patients with acromegaly. The Journal of clinical endocrinology and metabolism. 2005;90:4824–4828. doi: 10.1210/jc.2005-0132. [DOI] [PubMed] [Google Scholar]
- 18.Greenman Y, Melmed S. Expression of three somatostatin receptor subtypes in pituitary adenomas: evidence for preferential SSTR5 expression in the mammosomatotroph lineage. The Journal of clinical endocrinology and metabolism. 1994;79:724–729. doi: 10.1210/jcem.79.3.7521350. [DOI] [PubMed] [Google Scholar]
- 19.Thodou E, Kontogeorgos G, Theodossiou D, Pateraki M. Mapping of somatostatin receptor types in GH or/and PRL producing pituitary adenomas. J Clin Pathol. 2006;59:274–279. doi: 10.1136/jcp.2005.026914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tulipano G, Bonfanti C, Milani G, Billeci B, Bollati A, Cozzi R, Maira G, Murphy WA, Poiesi C, Turazzi S, Giustina A. Differential inhibition of growth hormone secretion by analogs selective for somatostatin receptor subtypes 2 and 5 in human growth-hormone-secreting adenoma cells in vitro. Neuroendocrinology. 2001;73:344–351. doi: 10.1159/000054651. [DOI] [PubMed] [Google Scholar]
- 21.Shimon I. Somatostatin receptors in pituitary and development of somatostatin receptor subtype-selective analogs. Endocrine. 2003;20:265–269. doi: 10.1385/ENDO:20:3:265. [DOI] [PubMed] [Google Scholar]
- 22.Taboada GF, Luque RM, Bastos W, Guimaraes RF, Marcondes JB, Chimelli LM, Fontes R, Mata PJ, Filho PN, Carvalho DP, Kineman RD, Gadelha MR. Quantitative analysis of somatostatin receptor subtype (SSTR1–5) gene expression levels in somatotropinomas and nonfunctioning pituitary adenomas. Eur J Endocrinol. 2007;156:65–74. doi: 10.1530/eje.1.02313. [DOI] [PubMed] [Google Scholar]
- 23.van der Hoek J, Lamberts SW, Hofland LJ. Preclinical and clinical experiences with the role of somatostatin receptors in the treatment of pituitary adenomas. Eur J Endocrinol. 2007;156(Suppl 1):S45–51. doi: 10.1530/eje.1.02350. [DOI] [PubMed] [Google Scholar]
- 24.Melmed S, Colao A, Barkan A, Molitch M, Grossman AB, Kleinberg D, Clemmons D, Chanson P, Laws E, Schlechte J, Vance ML, Ho K, Giustina A. Guidelines for acromegaly management: an update. The Journal of clinical endocrinology and metabolism. 2009;94:1509–1517. doi: 10.1210/jc.2008-2421. [DOI] [PubMed] [Google Scholar]
- 25.Bronstein MD. Acromegaly: molecular expression of somatostatin receptor subtypes and treatment outcome. Frontiers of hormone research. 2006;35:129–134. doi: 10.1159/000094315. [DOI] [PubMed] [Google Scholar]
- 26.Casarini AP, Jallad RS, Pinto EM, Soares IC, Nonogaki S, Giannella-Neto D, Musolino NR, Alves VA, Bronstein MD. Acromegaly: correlation between expression of somatostatin receptor subtypes and response to octreotide-lar treatment. Pituitary. 2009;12:297–303. doi: 10.1007/s11102-009-0175-1. [DOI] [PubMed] [Google Scholar]
- 27.van der Lely AJ, de Herder WW, Lamberts SW. New medical treatment for acromegaly. Pituitary. 1999;2:89–92. doi: 10.1023/a:1009930223314. [DOI] [PubMed] [Google Scholar]
- 28.Vitale G, Pivonello R, Ferone D, De Martino MC, Auriemma RS, Caraglia M, Abbruzzese A, Lombardi G, Colao A. The role of somatostatin receptors in the medical treatment of acromegaly. Dig Liver Dis. 2004;36(Suppl 1):S55–59. doi: 10.1016/j.dld.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 29.Ballare E, Persani L, Lania AG, Filopanti M, Giammona E, Corbetta S, Mantovani S, Arosio M, Beck-Peccoz P, Faglia G, Spada A. Mutation of somatostatin receptor type 5 in an acromegalic patient resistant to somatostatin analog treatment. The Journal of clinical endocrinology and metabolism. 2001;86:3809–3814. doi: 10.1210/jcem.86.8.7787. [DOI] [PubMed] [Google Scholar]
- 30.Caroli A, Antiga L, Cafaro M, Fasolini G, Remuzzi A, Remuzzi G, Ruggenenti P. Reducing polycystic liver volume in ADPKD: effects of somatostatin analogue octreotide. Clin J Am Soc Nephrol. 2010;5:783–789. doi: 10.2215/CJN.05380709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hogan MC, Masyuk TV, Page LJ, Kubly VJ, Bergstralh EJ, Li X, Kim B, King BF, Glockner J, Holmes DR, 3rd, Rossetti S, Harris PC, LaRusso NF, Torres VE. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol. 2010;21:1052–1061. doi: 10.1681/ASN.2009121291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Keimpema L, Nevens F, Vanslembrouck R, van Oijen MG, Hoffmann AL, Dekker HM, de Man RA, Drenth JP. Lanreotide reduces the volume of polycystic liver: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2009;137:1661–1668. e1661–1662. doi: 10.1053/j.gastro.2009.07.052. [DOI] [PubMed] [Google Scholar]
- 33.Sandford R, Sgotto B, Burn T, Brenner S. The tuberin (TSC2), autosomal dominant polycystic kidney disease (PKD1), and somatostatin type V receptor (SSTR5) genes form a synteny group in the Fugu genome. Genomics. 1996;38:84–86. doi: 10.1006/geno.1996.0596. [DOI] [PubMed] [Google Scholar]
- 34.Johansson M, McKay JD, Wiklund F, Rinaldi S, Hallmans G, Balter K, Adami HO, Gronberg H, Stattin P, Kaaks R. Genetic variation in the SST gene and its receptors in relation to circulating levels of insulin-like growth factor-I, IGFBP3, and prostate cancer risk. Cancer Epidemiol Biomarkers Prev. 2009;18:1644–1650. doi: 10.1158/1055-9965.EPI-08-0893. [DOI] [PubMed] [Google Scholar]
- 35.Nyegaard M, Borglum AD, Bruun TG, Collier DA, Russ C, Mors O, Ewald H, Kruse TA. Novel polymorphisms in the somatostatin receptor 5 (SSTR5) gene associated with bipolar affective disorder. Mol Psychiatry. 2002;7:745–754. doi: 10.1038/sj.mp.4001049. [DOI] [PubMed] [Google Scholar]