


Abstract
Topoisomerase IB (Top1) inhibitors, such as camptothecin (CPT), stabilize the Top1-DNA cleavage complex in a DNA sequence-dependent manner. The sequence selectivity of Top1 inhibitors is important for targeting specific genomic sequences of therapeutic value. However, the molecular mechanisms underlying this selectivity remain largely unknown. We performed molecular dynamics simulations to delineate structural, dynamic and energetic features that contribute to the differential sequence selectivity of the Top1 inhibitors. We found the sequence selectivity of CPT to be highly correlated with the drug binding energies, dynamic and structural properties of the linker domain. Chemical insights, gained by per-residue binding energy analysis revealed that the non-polar interaction between CPT and nucleotide at the +1 position of the cleavage site was the major (favorable) contributor to the total binding energy. Mechanistic insights gained by a potential of mean force analysis implicated that the drug dissociation step was associated with the sequence selectivity. Pharmaceutical insights gained by our molecular dynamics analyses explained why LMP-776, an indenoisoquinoline derivative under clinical development at the National Institutes of Health, displays different sequence selectivity when compared with camptothecin and its clinical derivatives.
INTRODUCTION
Topoisomerase IB (Top1) is an essential enzyme required for DNA replication and RNA transcription. Top1 is composed of 765 amino acids and has four distinct domains: the NH2-terminal domain (1-214), the core domain (215-635), the linker (636-712) and the COOH-terminal domain (713-765). The Top1 enzyme catalyzes the relaxation of DNA superhelicity by reversibly nicking one strand of DNA to generate a Top1-DNA cleavage complex that allows controlled DNA swiveling. Top1 inhibitors act as interfacial inhibitors (1). They intercalate into the DNA base pairs at the cleavage site and form specific hydrogen bonds with Top1 to stabilize the Top1-DNA-inhibitor ternary complex (Figure 1A). The stabilized ternary complex produces collisions with replication and transcription forks leading to DNA double strand breaks and cell death (Supplementary Scheme S1) (2). The discovery and development of Top1 inhibitors as anticancer drugs is one of the significant achievements in cancer chemotherapy (3,4). Topotecan and irinotecan, analogs of the natural alkaloid camptothecin (CPTs, Supplementary Chart S1), have been approved by the US Food and Drug Administration for the treatment of ovarian and colon cancers, respectively. The CPT class of anticancer drugs targets only Top1 and the poisoning of Top1-DNA cleavage complexes occurs only at specific cleavage sites along the DNA chain. This sequence selectivity is a key step in preferentially directing the action of drugs onto specific genomic sequences of therapeutic interest (5–8). The sequence selectivity exhibited by Top1 inhibitors is ‘intrinsically’ dependent on the base sequences immediately preceding (−1) and following (+1) the cleavage site. CPT binding has a strict requirement for T at the −1 site and G at the +1 site (9). Analysis of sequence selectivity around Top1 cleavage sites in SV40 DNA and 30mer duplex oligonucleotides indicated that CPT action is strongly dependent on the base pair at the +1 position (9–12). Recent reports showed that CPT interfered with transcription regulation in a sequence-selective manner, leading to alterations in gene expression that may be relevant for cancer therapy (13,14). However, the biochemical and biophysical determinants underlying the sequence selectivity remains unclear.
Figure 1.
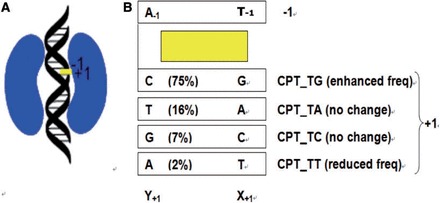
(A) Schematic diagram of a Top1-DNA-inhibitor ternary complex. Top1, DNA and CPT are shown in kidney-, helix- and rectangle-shape, respectively. (B) Schematic diagram of the CPT at the cleavage site, with its flanking base pairs. The frequencies of base sequences observed in CPT-induced DNA cleavage in SV40 DNA are given in parenthesis. Experimental base frequencies revealed that base frequencies at DNA cleavage sites was significantly (99.9% confidence interval) enhanced at the TG cleavage site in the presence of CPT, but no significant preference was seen for the TA and TC cleavage sites. The action of CPT was almost abolished at the TT cleavage site. Its base frequency was significantly (99.9% confidence interval) reduced in the presence of CPT.
In the present study, we investigated the impact of various local DNA sequences on the energetic, dynamic and structural properties of Top1-DNA-drug ternary complexes. This is the first molecular dynamics (MD) study on the sequence selectivity of Top1 inhibitors. We first examined four ternary complexes namely, CPT_TG, CPT_TA, CPT_TC and CPT_TT (Figure 1B). Experimental base frequencies (9–12) revealed that base frequencies at DNA cleavage sites was significantly (99.9% confidence interval) enhanced at the TG cleavage site in the presence of CPT, but no significant preference was seen for the TA and TC cleavage sites. The action of CPT was almost abolished at the TT cleavage site; its base frequency was significantly (99.9% confidence interval) reduced in the presence of CPT. In other words, sequence selectivity of CPT is in the following order of preference (Figure 1B): CPT_TG (frequency enhanced, compared with the frequency of the cleavage site in the absence of CPT) > CPT_TA (no change) ∼ CPT_TC (no change) > CPT_TT (frequency reduced). Potential of mean force (PMF) is defined as the free-energy changes along an arbitrarily defined path (15). In the present study, PMF study is performed to mimic the drug dissociation process. This is the first PMF studies on Top1-DNA-drug ternary complexes. We further demonstrate that MD approach could be used to study the sequence selectivity of an indenoisoquinoline derivative NSC725776 (LMP-776, Supplementary Chart S1). LMP-776 produces Top1 cleavage at unique genomic position compared with those resulting from CPT treatment, causes cell cycle arrest in both S and G2/M, exerts part of their anti-tumor effect through antiangiogenesis. In contrast to CPT, overexpression of ABCG2 and ABCB1 pumps do not induce resistance for LMP-776. These favorable pharmaceutical properties render LMP-776 a promising anticancer drug candidate (2,3). LMP-776 is currently under clinical development at the National Institutes of Health (NIH).
MATERIALS AND METHODS
MD simulations
The initial geometries of the Top1-DNA-drug ternary complexes were modeled from the crystallographic structures of ternary complexes (PDB 1K4T, 1SC7) (16,17). The CPT-bound and LMP-776-bound ternary complexes were modeled using atomic positions from the 1K4T and 1SC7 structures, respectively (18–22). The topotecan and MJ-III-65 were changed to CPT and LMP-776, respectively; the 5′-sulfhydryl group was replaced by a 5′-hydroxyl group at the cleavage site in the ternary complexes using the Accelrys DS Visualizer. The lactone form of CPT was assumed (23–25). All MD simulations were carried out with the Particle Mesh Ewald molecular dynamics (PMEMD) module in AMBER12 using the ff10 variant and general AMBER force field (GAFF) force fields (26,27). Although MD simulations are known to be limited by force field approximations, AMBER-based force fields have been successfully used to reflect key features of the structural dynamics of DNA and protein molecules (28–30). Counter ions (20 Na+) were added to keep the whole simulated system neutral. The whole system was immersed in the rectangular box of TIP3P water molecules. The water box extended 8 Å away from any solute atom. This resulted in ∼28 270 water molecules used for solvation. Electrostatic interactions were taken into account using the Particle Mesh Ewald method with a cutoff distance of 10 Å (31). We used SHAKE to constrain all bonds involving hydrogen atoms (32). Energy-minimization was executed by the steepest descent method for the first 1000 steps and by the conjugated gradient method for the subsequent 2500 steps. Relaxation of solvent molecules and Na+ ions was initially performed keeping solute atoms restrained to their initial positions for 200ps. The temperature was controlled by Langevin dynamics and was kept constant at 300 K. Each model was simulated for 100 ns. Over 100 000 frames were uniformly extracted over the 100 ns of simulations (33), unless otherwise specified.
Analysis of trajectories
The trajectories were analyzed using the AMBER12 packages (26), the VMD program (34) and the GROMACS MD package v4.5.5 (35).
Root-mean-square deviations
The cpptraj module in AMBER12 (26) was used to calculate the root-mean-square deviations (RMSD). RMSD is defined as the root-mean-square-average distance between protein Cα atoms of two optimally superimposed macromolecules, i.e. superimposed macromolecules at each frame (Si) and the starting geometry (S0). After a mass-weighted least square fitting to the starting geometry, the RMSD were calculated using Equation (1):
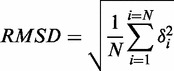 |
(1) |
where N is the number of equivalent protein Cα atoms in each macromolecules, δ is the distance between N pairs of equivalent atoms (i.e. protein Cα atoms). The RMSD values reach an average value of ∼3 Å (Supplementary Figure S1A). Excluding the linker domain from the analysis, the RMSD values reduce to ∼2 Å (Supplementary Figure S1B). Stable plateaus were reached in all the systems well before 30 ns of the simulation time.
Cosine content (ci)
Based on the production MD trajectories (the last 70 ns), non-weighted covariance matrix of Cα atoms of the models were calculated to obtain cosine content (ci) using the GROMACS MD package v4.5.5 (35). The cosine content (ci) of a principal component is used as an negative indicator for bad sampling (36). It ranges between 0 (no cosine) and 1 (perfect cosine), when the first of three principal components is similar to a cosine with half a period; the sampling is far from converged. The cosine contents of the first three principal components are far from a perfect cosine (ci < 0.1), indicating that our systems are not bad sampling. Thus, all the analyses listed later in the text were calculated over the production MD trajectories (i.e. the last 70 ns).
Dynamic cross-correlation coefficients (Ci,j)
The dynamic cross-correlation coefficients were done with cpptraj module in the AMBER package (26), taking into account only the coordinates of the protein Cα, nucleotide N1, N1@CPT, N1@LMP-776 atom (Supplementary Chart S1), as they contain enough information to describe the largest system motions (37). The dynamic cross-correlation coefficients were computed based on Equation (2):
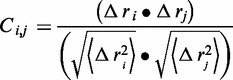 |
(2) |
where Δri is the displacement from the mean position of the ith atom and <>represents the time average over the whole trajectory. Positive Ci,j values represent a correlated motion between residues i and j (i.e. the residues move in the same direction). Negative values of Ci,j represent an anti-correlated motion between residues i and j (i.e. they move in opposite directions).
Root-mean-square fluctuations
The per-residue root-mean-square fluctuations (RMSF) is defined as a root-mean-square-average distance between a protein Cα atom (xi) and its time-averaged position of the equivalent atom (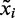 ) in a set of S structures that were extracted from the frames of the production MD trajectories. RMSF were computed using Equation (3):
) in a set of S structures that were extracted from the frames of the production MD trajectories. RMSF were computed using Equation (3):
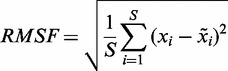 |
(3) |
Clustering
The clustering was calculated using the cpptraj module in AMBER12 (26). Structures from the frames of the production trajectories were used to perform average-linkage hierarchical clustering, based on their mass-weighted RMSD distance matrix.
DNA-linker angle
The DNA-linker angle is defined as the angle between N1@A+12−N1@X+1−Cα@ASP677 (Supplementary Scheme S2). X+1 and A+12 are the nucleotides at the +1 and +12 positions of the intact DNA strand.
Hydrogen bond
Hydrogen bonds were defined as the default setting at the cpptraj program. Hydrogen bonds are determined via the distance between the heavy atoms using a cutoff of 3.5 Å and the angle between the acceptor, hydrogen, and donor atoms using a cutoff of 135 degrees, unless otherwise specified.
Molecular mechanics-generalized born surface area
The drug binding energies were calculated using the mmGBSA.py module (igb=2) in AMBER12 (26). The per-residue decomposition energies reflect the relative strength of each specific residue, i.e. its importance to the binding. The post-processing molecular mechanics-generalized born surface area (MM-GBSA) method, using the modified generalized Born model (38), is known to be good at qualitative accuracy (39–43). We herein focused our discussions on qualitative comparison. The total binding energy (ΔGtot) was decomposed into contributions from electrostatic energies (ΔEelec), van der Waals interaction energies (ΔEvdw), electrostatic solvation (ΔGGB) and non-polar solvation (ΔGnon-polar) free energies as shown in Equation (4):
| (4) |
The entropy was not considered because of its high computational demand and relatively low accuracy of prediction (44). The per-residue decomposition with 1–4 VDW and 1–4 EEL terms were added to internal potential terms.
PMF
PMF was calculated to study the free-energy profiles for drug dissociation. The PMF is a function of the distance between the CPT and the ternary complex (χ), which can be used to monitor the drug dissociation process (Supplementary Scheme S3). The distance (χ) is defined as the distance between P@A−1 and N1@CPT (Supplementary Chart S1).
Umbrella sampling was performed by calculating a series of dynamic simulations with biased potential in a format of K(χ − χi)2, where K = 5 or 10 or 20 kcal mol−1 and χi ranged from 0 to 50 Å. Umbrella sampling windows were separated by 1 Å. Simulations with K = 5 were carried out for at least 0.2 ns; the rest of the simulations were carried out with K = 10 and 20 for at least 0.4 ns each, until convergence is reached. All the simulations were combined to construct the final PMF. Multiple starting structures were extracted from the all-atom simulations. Each simulation was started from the end point of the previous one. Adjacent histograms were found to show significant overlaps, indicating that all values of a distance within the desired range had been appropriately sampled. The unbiased free energy was obtained using the weight histogram analysis method (http://membrane.urmc.rochester.edu/content/wham) with convergence criterion of 10−4 and a bin width of 0.2 Å (45). The convergence of final PMF was tested by comparing the free energy change calculated using the full sampling to that calculated using shorter sampling time. For each PMF models, at least 50 ns of simulations were carried out until the PMF profile converged.
RESULTS AND DISCUSSION
Binding energies of CPT to the Top1-DNA complex
The total binding energies of CPT to the Top1-DNA complex ranged from −54.9 kcal mol−1 for CPT_TG to −49.9 kcal mol−1 for CPT_TT (Table 1), which were considered to be reasonable for two reasons. First, in the previous studies by Cushman et al. (46) (−32 kcal mol−1) and Siu et al. (47) (−26 to −31 kcal mol−1), the reported binding energies of a Top1 inhibitor to its flanking base pairs were in the same order of magnitude. The more favorable binding energies obtained in the present study could be attributed to contributions from protein residues and other DNA bases, which were not considered in the two previous studies (46,47). Second, the experimental frequencies of cleavage sites correlate strongly with the total binding energies (R2 = 0.8, Table 1). To probe properties that underpin the sequence selectivity, we identified their correlations with various per-residue decomposition energies (Table 1). A strong correlation between the experimental frequencies and the binding energy between CPT and the nucleotide at the +1 position of the scissile strand (X+1) was observed (R2 = 0.9, Table 1). The non-polar interaction (ΔEvdw +ΔGnon-polar, Table 2) was the major favorable contributor to the total binding energy. Taken together, our results indicate that the non-polar interaction between CPT and the X+1 is a predominant, if not exclusive, factor governing the sequence selectivity of CPT. We would like to emphasize that given there are only four data points for the correlation analyses, no quantitative analysis were discussed.
Table 1.
The per-residue decomposition energies (kcal mol−1)a
| Decomp. En.a | CPT_TG | CPT_TA | CPT_TC | CPT_TT |
|---|---|---|---|---|
| T−1 | −4.8 | −5.0 | −5.0 | −5.0 |
| X+1 | −9.1 | −6.6 | −5.8 | −6.4 |
| Y+1 | −2.4 | −3.2 | −5.0 | −3.3 |
| A−1 | −5.4 | −4.5 | −5.6 | −5.2 |
| Arg364 | −2.5 | −2.5 | −2.2 | −2.4 |
| Lys532 | −1.8 | −1.7 | −1.1 | −0.9 |
| CPT | −27.2 | −26.0 | −25.4 | −24.5 |
| Others | −1.7 | −2.0 | −2.2 | −2.2 |
| Total | −54.9 | −51.5 | −52.3 | −49.9 |
aThe total binding energies are highlighted in bold. The per-residue decomposition energies reflect the relative importance of each specific residue, including flanking base pairs at the cleavage site, Arg364, Lys532, CPT and other residues, to the total drug binding energies.
Table 2.
The MM-GBSA per residue decomposition energies (kcal mol−1)a for the interaction between CPT and the X+1 base at the cleavage site
| Decomp. En. | CPT_TG | CPT_TA | CPT_TC | CPT_TT |
|---|---|---|---|---|
| ΔEelec | −2.9 | −1.5 | −0.9 | −1.8 |
| ΔEvdw | −7.8 | −6.9 | −5.6 | −5.7 |
| ΔGGB | 2.2 | 2.4 | 1.2 | 1.5 |
| ΔGnon-polar | −0.6 | −0.6 | −0.5 | −0.4 |
| Electrostb | −0.7 | 0.9 | 0.3 | −0.3 |
| Non-polarc | −8.4 | −7.5 | −6.1 | −6.1 |
| ΔGtot | −9.1 | −6.6 | −5.8 | −6.4 |
aThe major favorable contributor is highlighted in bold.
bElectrost = ΔEelec + ΔGGB.
cNon-polar = ΔEvdw + ΔGnon-polar.
Flexibility of the linker domain and its orientation with respect to the DNA
The per-residue RMSF calculated from the production trajectories showed that the four complexes underwent similar fluctuations (Figure 2). As observed in previous studies (24,48), the linker domain (residues 636–712) was the most flexible region. Moreover, the flexibility of the linker domain is qualitatively correlated with the experimental frequencies of the sequence selectivity. First, for the linker domain, the RMSF maximum value for the preferred CPT_TG complex (2.1 Å) is lower than those of the unfavorable CPT_TA/TC/TT complexes (2.4–4.7 Å, Figure 2). The linker and core domains are connected by residues 633–643, whose overall mobility increases as the number of orientations of the linker domain increases (24). Consistently, our analysis revealed that the RMSF maximum value of residues 633–643 is the lowest for the preferred CPT_TG complex (1.2 Å) when compared with those of the unfavorable CPT_TA/TC/TT complexes (1.5–2.1 Å, Figure 2). Cluster analysis further confirmed that the conformational space visited by the preferred CPT_TG complex (nine clusters) is less than that of the unfavorable complexes (CPT_TA/TC/TT complexes, 11–27 clusters). Collectively, our study revealed that the sequence selectivity increases as the flexibility of the linker domain decreases.
Figure 2.
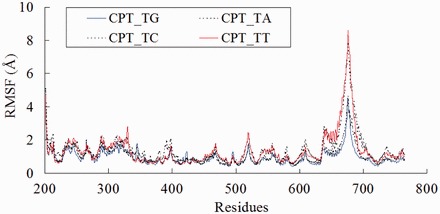
RMSF of CPT-bound ternary complexes. The solid lines represent the ternary complex with highest observed frequency (CPT_TG) and lowest observed frequency (CPT_TT).
Comparing the average structures of the CPT-bound ternary complexes (Figure 3), a notable difference was observed in the linker domain. The angles formed between the DNA and the linker domain (θ) in these complexes were determined (Supplementary Scheme S2). The linker domain of the preferred CPT_TG complex adopts a position that is closer to the DNA when compared with that of the unfavorable CPT_TA/TC/TT complexes (Figure 3). The experimental frequencies of cleavage sites correlate strongly with the DNA-linker angle (R2 = 0.9). We postulate that interaction(s) between the linker domain and the DNA duplex is needed to stabilize the ternary complexes, which leads to the higher observed frequencies.
Figure 3.
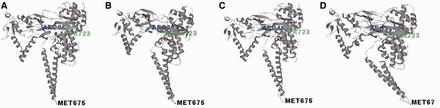
Average structures of (A) CPT_TG, (B) CPT_TA, (C) CPT_TC and (D) CPT_TT complexes. The DNA and CPT are not shown, and the ARG364, PTR723 and MET675 residues are labeled.
Long-range interactions between CPT and protein residues
To examine why the orientation of the linker domain changed dramatically, we studied the hydrogen bonds (Supplementary Figure S2) and the correlative motion of the CPT-bound ternary complexes. The correlative motion of CPT with protein residues (Figure 4) were investigated by means of the dynamic cross-correlation coefficients (CCPT,j), with positive and negative CCPT,j values signify correlated and anti-correlated motions, respectively (37).
Figure 4.
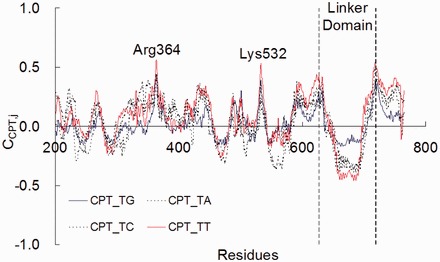
Dynamic cross-correlation coefficients (CCPT,j) between CPT and protein residues in Top1.
The CCPT,j (Figure 4) values are highly positive for the Arg364 and Lys532 residues. Single amino acid mutations at these residues are known to confer CPT resistance (49). The highly positive CCPT,Arg364 values for the four CPT-bound ternary complexes revealed that CPT moves along the same direction with the Arg364 residue. A hydrogen bond is observed between N1 of CPT and the side-chain of Arg364 (Supplementary Figure S2). This hydrogen bond was also observed in the X-ray crystallographic structure of a Top1-DNA-CPT complex (17). The highly positive CCPT,Lys532 values also revealed that motion of CPT correlate with that of the Lys532 residue (Figure 4). The side-chain of Lys532 forms hydrogen bond with the lactone ring of CPT (O21@CPT, Supplementary Chart S1) for >80% of the simulation time (Supplementary Figure S2). Lys532 is a known catalytic residue making a hydrogen bond with carboxylate O21 of topotecan in the toptoecan-Top1-DNA ternary X-ray crystallographic structure (16).
In contrast, CPT moves in an anti-correlated way with the linker domain (residues 636–712). The CCPT,linker values (Figure 4) indicates that the strength of the anti-correlated motion is in the order of CPT_TT (most negative) > CPT_TA ∼ CPT_TC > CPT_TG (least negative). As there is no direct hydrogen bond between CPT and the linker domain, the anti-correlated motion is expected to be mediated through DNA-linker interactions. Hydrogen bonds are observed between the backbone nitrogen of the Arg634 and Gly717 residues (linker domain) and the phosphodiester groups of G+2 and A+3 bases at the scissile strand, respectively. In addition, the free 5′-OH group of the +1 base at the scissile strand (X+1) forms intermolecular hydrogen bond with the lateral chain of the Asn722 residue. This hydrogen bond presents for >80% of the simulation time for all the complexes. The Arg634 forms a loop (residues 634–640) connecting the linker to the core domain. This loop is known to have a direct impact on the flexibility of the linker domain (24), as the loop and the linker domain are also absent in the X-ray crystallographic structure of the Top1-DNA binary complex (PDB: 1A31), but observable in the drug-bound ternary complexes (PDB: 1K4T) (Supplementary Figure S3) (50). Gly717Val mutated Top1 was isolated in a CPT-resistant cell line (51). In Top1, the Asn722 amino acid residue is immediately NH2-terminal to the catalytic Tyr723. The mutation Asn722Ser in Top1 leads to CPT resistance (52,53). Collectively, these findings indicate that there are long-range interactions between the cleavage site and the linker domain.
The linker flexibility has been shown to affect enzyme catalytic activity, Top1 sensitivity to CPT and long-range effects on the flexibility of the linker domain. In the absence of CPT, Lys681Ala and Thr718Ala mutations render the flexibility of the linker domain reduces. The Lys681Ala mutant has a CPT-like behavior: the stability of the cleavage complex is enhanced and the distance between Lys532 and the 5′OH of the +1 base phosphates is increased (54). The Thr718Ala mutant shows a decreased efficiency in DNA relaxation (55). We note with interest that although the Ala653Pro mutation in the linker domain confers CPT resistance and renders the linker domain more flexible (56), the mutations in Asp677Gly, Val703Ile and Ile728Thr renders the mutant more sensitive to CPT and higher binding affinity to DNA (57). It is also well-known that the reconstituted top1 lacking the linker domain has a reduced affinity for DNA (58). X-ray crystallographic (50) and MD studies (24) revealed that the linker domain is highly flexible in the drug-free binary Top1-DNA complexes, when compared with that of the CPTs-bound ternary complexes (Supplementary Figure S3). Collectively, the CPT-induced or mutation-mediated inhibitory effects on Top1 increase as the mobility of the linker domain decreases. Accordingly, we found the sequence selectivity of CPT is correlated with the flexibility of the linker domains. We conceive that both the inhibitory and sequence selectivity effects are associated with the stability of the cleavage complexes, which has long-range effects on the flexibility of the linker domain.
Drug development prospective
Having established the driving force for the site selectivity of CPT, we extended our study to non-CPTs, with an aim to evaluate the potential application of MD approach for studying the site selectivity of non-CPT Top1 poisons (59). CPTs have several undesirable properties, including E-ring inactivation by lactone ring opening, affinity for cellular efflux transporters and relatively high toxicity, which limits their therapeutic index. Non-CPT Top1 inhibitors could have distinct pharmacological activities because they target distinct genomic sites due to their different sequence selectivity; they also overcome the chemical instability of CPT and lead to distinct pharmacological activity (59–62). An indenoisoquinoline NSC725776 (LMP-776, Supplementary Chart S1) is currently in clinical trial at the National Institutes of Health (USA). The DNA cleavage pattern induced by LMP-776 is different from that of CPT. Notably, LMP-776 prefers a cleavage site at the CG site [site 44 in the original article (3,63)], rather than the TG site (the preferred site of CPT).
In the present study, we compared the ternary complexes of LMP-776 bound with Top1-DNA complexes with CG versus TG as the cleavage sites (referred to as LMP_CG and LMP_TG, Figure 5). In agreement with the trend observed in CPT, the drug binding energy of the LMP_CG complex (−60.6 kcal mol−1) was more favorable than the binding energy of the LMP_TG complex (−54.3 kcal mol−1). The flexibility of the linker domain of LMP_CG (2.7 Å) was also lower than that of LMP_TG (2.9 Å, Figure 5B). In addition, the DNA-linker angle (θ) of the LMP_CG (14°) complex was significantly smaller (t-test: P < 1E-5) than that of the LMP_TG complex (29°, Figure 5C). Collectively, LMP-776 is consistent with CPT that sequence selectivity was governed by drug binding energies, flexibility of the linker domain and the DNA-linker angle. To target specific genomic sequences of therapeutic value, developing top1 inhibitors with different preference on the cleavage site is of high importance. Determining the base frequencies of various top1 inhibitors experimentally is time-consuming and tremendous. We have illustrated that the MD approach reported in the present study can be used to study the sequence selectivity of CPT and non-CPTs. We envision MD approach can be used to in silico screen compounds that target specific genomic sequences.
Figure 5.
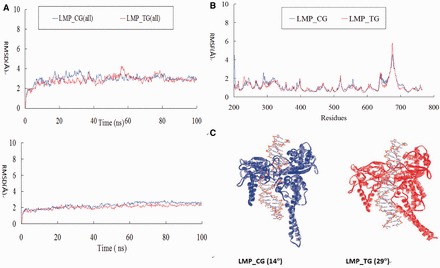
Data for LMP-776 (NSC725776). (A) RMSD with and without the linker domain; (B) RMSF; (C) Average Structures for the LMP-bound ternary complex.
Mechanistic perspective
The perturbed mobility of the linker domain has been reported to be associated with alteration in religation rate and drug resistance (56,64–67). Drug dissociation is the rate determining step when compared with DNA religation. To investigate the underlying mechanism for the sequence selectivity of the drugs, we modeled the drug dissociation process using the PMF approach. PMF is defined as the free energy changes along the drug dissociation distance (χ) (Supplementary Scheme S3). Using multiple starting geometries, we calculated the PMFs and found that CPT dissociates towards to the NH2-terminal domain (Figure 6A). The residues 190–210 at the NH2-terminal domain are known to be important for Top1 activity (68–72): The physical contact of Trp203 with His346 and of Trp205, Trp206 with the hinge region of the core domain controls the DNA rotation process (73). Site mutations at these tryptophan residues are known to confer CPT resistance (68). Using MD approach, Chillemi et al. (71) found that the residues 203–207 at the NH2-terminal domain and the linker domain participate in a network of correlated movements, involved in the Top1 catalytic cycle and associated with the DNA relaxation activity. Our results shed light on the potential role of the NH2-terminal domain in the drug dissociation process that may warrant further study.
Figure 6.
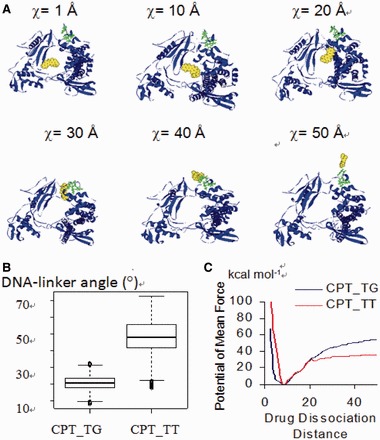
(A) Snapshots of the ternary complex as the drug dissociate from the cleavage complex. The CPT and the three Trp203/205/206 residues in the NH2-terminal of Top1 are shown in CPK and ball and stick styles, respectively; the DNA strands are not shown for the ease of visualization. (B) Angles formed between DNA and the linker domain of the complexes during the drug dissociation process. (C) The PMF of CPT_TG complex is higher than that of the CPT_TT complex.
A closer inspection of the PMFs of the CPT_TG and CPT_TT complexes revealed that the DNA-linker angle in the CPT_TG complex is smaller than that of the CPT_TT complex (Figure 6B), and the drug dissociation energy barrier for CPT_TG is significantly higher than that of the CPT_TT complex (Figure 6C). The smaller DNA-linker angle in the CPT_TG complexes, when compared with that of CPT_TT, implied that the linker domain and DNA remain in close contact during the drug dissociation process in the former complex. The linker domain is known to play important roles in stabilizing the cleavage complex. The stabilized CPT_TG complex renders drug dissociation more difficult, as represented by a higher drug dissociation barrier. We therefore conclude that the DNA-linker interaction also play a role in impeding drug dissociation process. In the absence of CPT, the DNA sequence does not affect the dissociation rate constants or the rate of association between DNA and Top1 (74), as the four sequences were similarly cleaved (9). DNA sequence does not appear to play a dominating role in the rate of drug-mediated DNA uncoiling (75). Top1-mediated DNA breakage is enhanced the greatest at those sites where closure of the break is the slowest and that the CPT-mediated religation rate is sequence dependent (76–78). Collectively, previous experiments together with our findings imply that the drug dissociation step plays a critical role in the sequence selectivity of Top1 inhibitors.
Final remarks
Our study provides novel insights into the mechanistic determinants for the sequence selectivity of Top1 inhibitors. The sequence selectivity of CPT was revealed to be correlated with energetic (drug binding energy), dynamic (flexibility of the linker domain) and structural (angle formed between the linker domain and DNA) properties of the drug-bound ternary complexes. Our structural analysis and PMF study further reveal that the hydrogen bonding network between DNA and the linker domain and the drug dissociation step plays a role in the sequence selectivity induced by CPT. We also demonstrated the use of MD approaches to study the sequence selectivity of non-CPTs Top1 inhibitor. The same approach can be envisioned to aid the development of additional Top1 inhibitors.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Center of Cancer Research, National Cancer Institute Intramural Program at the National Institutes of Health (US Government); National Science Foundation of China (NSFC), [21203237] and Shenzhen Basic Research Grant; [JCYJ20130401170306875] seed funding from the Committee on Research and Conference Grants (CRCR) of the University of Hong Kong; the Areas of Excellence Scheme administrated by the University Grants Committee of the Hong Kong Special Administrative Region, China [AoE/P-10/01], Innovation and Technology Fund [ITF-Tier 2, ITS/134/09FP]; National Key Basic Research Program of China [2013CB834802]; Special Equipment Grant [SEG HKU09]; University Development Fund and Strategic Research Fund. Funding for open access charge: Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences; NSFC [21203237]; and Shenzhen Basic Research Grant [JCYJ20130401170306875].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The Visiting Scientist position granted to F-M.S. by the National Institutes of Health (US Government) and professional leave granted to F-M.S. by Professor CM Che and the University of Hong Kong are gratefully acknowledged. This study used the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, Maryland (http://biowulf.nih.gov) and the high-performance computational capabilities of the Gridpoint Linux cluster at the University of Hong Kong, and the National Supercomputing Center at Shenzhen.
REFERENCES
- 1.Pommier Y, Marchand C. Interfacial inhibitors: targeting macromolecular complexes. Nat. Rev. Drug. Discov. 2012;11:25–36. doi: 10.1038/nrd3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010;17:421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antony S, Agama KK, Miao ZH, Takagi K, Wright MH, Robles AI, Varticovski L, Nagarajan M, Morrell A, Cushman M, et al. Novel indenoisoquinolines NSC 725776 and NSC 724998 produce persistent topoisomerase I cleavage complexes and overcome multidrug resistance. Cancer Res. 2007;67:10397–10405. doi: 10.1158/0008-5472.CAN-07-0938. [DOI] [PubMed] [Google Scholar]
- 4.Wang X, Zhou X, Hecht SM. Role of the 20-hydroxyl group in camptothecin binding by the topoisomerase I-DNA binary complex. Biochemistry. 1999;38:4374–4381. doi: 10.1021/bi982708k. [DOI] [PubMed] [Google Scholar]
- 5.Oussedik K, Francois JC, Halby L, Senamaud-Beaufort C, Toutirais G, Dallavalle S, Pommier Y, Pisano C, Arimondo PB. Sequence-specific targeting of IGF-I and IGF-IR genes by camptothecins. FASEB J. 2010;24:2235–2244. doi: 10.1096/fj.09-132324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arimondo PB, Laco GS, Thomas CJ, Halby L, Pez D, Schmitt P, Boutorine A, Garestier T, Pommier Y, Hecht SM, et al. Activation of camptothecin derivatives by conjugation to triple helix-forming oligonucleotides. Biochemistry. 2005;44:4171–4180. doi: 10.1021/bi048031k. [DOI] [PubMed] [Google Scholar]
- 7.Baranello L, Bertozzi D, Fogli MV, Pommier Y, Capranico G. DNA topoisomerase I inhibition by camptothecin induces escape of RNA polymerase II from promoter-proximal pause site, antisense transcription and histone acetylation at the human HIF-1α gene locus. Nucleic Acids Res. 2010;38:159–171. doi: 10.1093/nar/gkp817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vekhoff P, Duca M, Guianvarc'h D, Benhida R, Arimondo PB. Sequence-specific base pair mimics are efficient topoisomerase IB inhibitors. Biochemistry. 2012;51:43–51. doi: 10.1021/bi2012959. [DOI] [PubMed] [Google Scholar]
- 9.Jaxel C, Capranico G, Kerrigan D, Kohn KW, Pommier Y. Effect of local DNA sequence on topoisomerase I cleavage in the presence or absence of camptothecin. J. Bio. Chem. 1991;266:20418–20423. [PubMed] [Google Scholar]
- 10.Jaxel C, Kohn KW, Pommier Y. Topoisomerase I interaction with SV40 DNA in the presence and absence of camptothecin. Nucleic Acids Res. 1988;16:11157–11170. doi: 10.1093/nar/16.23.11157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kjeldsen E, Mollerup S, Thomsen B, Bonven BJ, Bolund L, Westergaard O. Sequence-dependent effect of camptothecin on human topoisomerase I DNA cleavage. J. Mol. Biol. 1988;202:333–342. doi: 10.1016/0022-2836(88)90462-7. [DOI] [PubMed] [Google Scholar]
- 12.Porter SE, Champoux JJ. The basis for camptothecin enhancement of DNA breakage by eukaryotic topoisomerase I. Nucleic Acids Res. 1989;17:8521–8532. doi: 10.1093/nar/17.21.8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solier S, Barb J, Zeeberg BR, Varma S, Ryan MC, Kohn KW, Weinstein JN, Munson PJ, Pommier Y. Genome-wide analysis of novel splice variants induced by topoisomerase I poisoning shows preferential occurrence in genes encoding splicing factors. Cancer Res. 2010;70:8055–8065. doi: 10.1158/0008-5472.CAN-10-2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Capranico G, Marinello J, Baranello L. Dissecting the transcriptional functions of human DNA topoisomerase I by selective inhibitors: implications for physiological and therapeutic modulation of enzyme activity. Biochim. Biophys. Acta. 2010;1806:240–250. doi: 10.1016/j.bbcan.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Roux B. The calculation of the potential of mean force using computer simulations. Comp. Phys. Commun. 1995;91:275–282. [Google Scholar]
- 16.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Jr, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl Acad. Sci. USA. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J. Med. Chem. 2005;48:2336–2345. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 18.Ivanova B, Spiteller M. Structure and properties of camptothecin derivatives, their protonated forms, and model interaction with the topoisomerase I-DNA complex. Biopolymers. 2012;97:134–144. doi: 10.1002/bip.21714. [DOI] [PubMed] [Google Scholar]
- 19.Laco GS. Evaluation of two models for human topoisomerase I interaction with dsDNA and camptothecin derivatives. PLoS One. 2011;6:e24314. doi: 10.1371/journal.pone.0024314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drwal MN, Agama K, Wakelin LP, Pommier Y, Griffith R. Exploring DNA topoisomerase I ligand space in search of novel anticancer agents. PLoS One. 2011;6:e25150. doi: 10.1371/journal.pone.0025150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lauro G, Romano A, Riccio R, Bifulco G. Inverse virtual screening of antitumor targets: pilot study on a small database of natural bioactive compounds. J. Nat. Prod. 2011;74:1401–1407. doi: 10.1021/np100935s. [DOI] [PubMed] [Google Scholar]
- 22.Lauria A, Ippolito M, Almerico AM. Molecular docking approach on the Topoisomerase I inhibitors series included in the NCI anti-cancer agents mechanism database. J. Mol. Model. 2007;13:393–400. doi: 10.1007/s00894-006-0159-2. [DOI] [PubMed] [Google Scholar]
- 23.Chillemi G, Coletta A, Mancini G, Sanna N, Desideri A. An amber compatible molecular mechanics force field for the anticancer drug topotecan. Theor. Chem. Acc. 2010;127:293–302. [Google Scholar]
- 24.Mancini G, D'Annessa I, Coletta A, Sanna N, Chillemi G, Desideri A. Structural and dynamical effects induced by the anticancer drug topotecan on the human topoisomerase I - DNA complex. PLoS One. 2010;5:e10934. doi: 10.1371/journal.pone.0010934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanna N, Chillemi G, Grandi A, Castelli S, Desideri A, Barone V. New hints on the Ph-driven tautomeric equilibria of the topotecan anticancer drug in aqueous solutions from an integrated spectroscopic and quantum-mechanical approach. J. Am. Chem. Soc. 2005;127:15429–15436. doi: 10.1021/ja052637u. [DOI] [PubMed] [Google Scholar]
- 26.Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, III, Laughton CA, Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys. J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perez A, Lankas F, Luque FJ, Orozco M. Towards a molecular dynamics consensus view of B-DNA flexibility. Nucleic Acids Res. 2008;36:2379–2394. doi: 10.1093/nar/gkn082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao ZW, Xue Y, Han LY, Xie B, Zhou H, Zheng CJ, Lin HH, Chen YZ. MoViES: molecular vibrations evaluation server for analysis of fluctuational dynamics of proteins and nucleic acids. Nucleic Acids Res. 2004;32:W679–W685. doi: 10.1093/nar/gkh384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maffeo C, Luan B, Aksimentiev A. End-to-end attraction of duplex DNA. Nucleic Acids Res. 2012;40:3812–3821. doi: 10.1093/nar/gkr1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Darden T, York D, Pedersen LG. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 32.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-integration of cartesian equations of motion of a system with constraints: molecular dynamics of N-alkanes. J. Comp. Phys. 1977;23:327–341. [Google Scholar]
- 33.Chu WT, Zhang JL, Zheng QC, Chen L, Zhang HX. Insights into the folding and unfolding processes of wild-type and mutated SH3 domain by molecular dynamics and replica exchange molecular dynamics simulations. PLoS One. 2013;8:e64886. doi: 10.1371/journal.pone.0064886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38, 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 35.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for high efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 36.Hess B. Convergence of sampling in protein simulations. Phys. Rev. E. 2002;65:031910. doi: 10.1103/PhysRevE.65.031910. [DOI] [PubMed] [Google Scholar]
- 37.Chillemi G, D'Annessa I, Fiorani P, Losasso C, Benedetti P, Desideri A. Thr729 in human topoisomerase I modulates anti-cancer drug resistance by altering protein domain communications as suggested by molecular dynamics simulations. Nucleic Acids Res. 2008;36:5645–5651. doi: 10.1093/nar/gkn558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 39.Gohlke H, Kiel C, Case DA. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J. Mol. Biol. 2003;330:891–913. doi: 10.1016/s0022-2836(03)00610-7. [DOI] [PubMed] [Google Scholar]
- 40.Hou T, Zhang W, Case DA, Wang W. Characterization of domain-peptide interaction interface: a case study on the amphiphysin-1 SH3 domain. J. Mol. Biol. 2008;376:1201–1214. doi: 10.1016/j.jmb.2007.12.054. [DOI] [PubMed] [Google Scholar]
- 41.Balius TE, Rizzo RC. Quantitative prediction of fold resistance for inhibitors of EGFR. Biochemistry. 2009;48:8435–8448. doi: 10.1021/bi900729a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGillick BE, Balius TE, Mukherjee S, Rizzo RC. Origins of resistance to the HIVgp41 viral entry inhibitor T20. Biochemistry. 2010;49:3575–3592. doi: 10.1021/bi901915g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zoete V, Michielin O. Comparison between computational alanine scanning and per-residue binding free energy decomposition for protein-protein association using MM-GBSA: application to the TCR-p-MHC complex. Proteins. 2007;67:1026–1047. doi: 10.1002/prot.21395. [DOI] [PubMed] [Google Scholar]
- 44.Lee VS, Tue-ngeun P, Nangola S, Kitidee K, Jitonnom J, Nimmanpipug P, Jiranusornkul S, Tayapiwatana C. Pairwise decomposition of residue interaction energies of single chain Fv with HIV-1 p17 epitope variants. Mol. Immunol. 2010;47:982–990. doi: 10.1016/j.molimm.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 45. Grossfield,A. WHAM: The Weighted Histogram Analysis Method, version 2.0.7.
- 46.Xiao X, Cushman M. An ab initio quantum mechanics calculation that correlates with ligand orientation and DNA cleavage site selectivity in camptothecin-DNA-topoisomerase I ternary cleavage complexes. J. Am. Chem. Soc. 2005;127:9960–9961. doi: 10.1021/ja042485n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siu FM, Che CM. Persistence of camptothecin analog-topoisomerase I-DNA ternary complexes: a molecular dynamics study. J. Am. Chem. Soc. 2008;130:17928–17937. doi: 10.1021/ja806934y. [DOI] [PubMed] [Google Scholar]
- 48.Redinbo MR, Stewart L, Champoux JJ, Hol WG. Structural flexibility in human topoisomerase I revealed in multiple non-isomorphous crystal structures. J. Mol. Biol. 1999;292:685–696. doi: 10.1006/jmbi.1999.3065. [DOI] [PubMed] [Google Scholar]
- 49.Pommier Y, Pourquier P, Urasaki Y, Wu J, Laco GS. Topoisomerase I inhibitors: selectivity and cellular resistance. Drug Resist. Updat. 1999;2:307–318. doi: 10.1054/drup.1999.0102. [DOI] [PubMed] [Google Scholar]
- 50.Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WG. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science. 1998;279:1504–1513. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- 51.Wang LF, Ting CY, Lo CK, Su JS, Mickley LA, Fojo AT, Whang-Peng J, Hwang J. Identification of mutations at DNA topoisomerase I responsible for camptothecin resistance. Cancer Res. 1997;57:1516–1522. [PubMed] [Google Scholar]
- 52.Fujimori A, Harker WG, Kohlhagen G, Hoki Y, Pommier Y. Mutation at the catalytic site of topoisomerase I in CEM/C2, a human leukemia cell line resistant to camptothecin. Cancer Res. 1995;55:1339–1346. [PubMed] [Google Scholar]
- 53.Sirikantaramas S, Yamazaki M, Saito K. Mutations in topoisomerase I as a self-resistance mechanism coevolved with the production of the anticancer alkaloid camptothecin in plants. Proc. Natl Acad. Sci. USA. 2008;105:6782–6786. doi: 10.1073/pnas.0801038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fiorani P, Tesauro C, Mancini G, Chillemi G, D'Annessa I, Graziani G, Tentori L, Muzi A, Desideri A. Evidence of the crucial role of the linker domain on the catalytic activity of human topoisomerase I by experimental and simulative characterization of the Lys681Ala mutant. Nucleic Acids Res. 2009;37:6849–6858. doi: 10.1093/nar/gkp669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chillemi G, Fiorani P, Castelli S, Bruselles A, Benedetti P, Desideri A. Effect on DNA relaxation of the single Thr718Ala mutation in human topoisomerase I: a functional and molecular dynamics study. Nucleic Acids Res. 2005;33:3339–3350. doi: 10.1093/nar/gki642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fiorani P, Bruselles A, Falconi M, Chillemi G, Desideri A, Benedetti P. Single mutation in the linker domain confers protein flexibility and camptothecin resistance to human topoisomerase I. J. Bio. Chem. 2003;278:43268–43275. doi: 10.1074/jbc.M303899200. [DOI] [PubMed] [Google Scholar]
- 57.Scaldaferro S, Tinelli S, Borgnetto ME, Azzini A, Capranico G. Directed evolution to increase camptothecin sensitivity of human DNA topoisomerase I. Chem. Biol. 2001;8:871–881. doi: 10.1016/s1074-5521(01)00059-x. [DOI] [PubMed] [Google Scholar]
- 58.Stewart L, Ireton GC, Champoux JJ. Reconstitution of human topoisomerase I by fragment complementation. J. Mol. Biol. 1997;269:355–372. doi: 10.1006/jmbi.1997.1056. [DOI] [PubMed] [Google Scholar]
- 59.Pommier Y, Cushman M. The indenoisoquinoline noncamptothecin topoisomerase I inhibitors: update and perspectives. Mol. Cancer Ther. 2009;8:1008–1014. doi: 10.1158/1535-7163.MCT-08-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fukasawa K, Komatani H, Hara Y, Suda H, Okura A, Nishimura S, Yoshinari T. Sequence-selective DNA cleavage by a topoisomerase I poison, NB-506. Int. J. Cancer. 1998;75:145–150. doi: 10.1002/(sici)1097-0215(19980105)75:1<145::aid-ijc22>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 61.Teicher BA. Next generation topoisomerase I inhibitors: Rationale and biomarker strategies. Biochem. Pharm. 2008;75:1262–1271. doi: 10.1016/j.bcp.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 62.Marco E, Laine W, Tardy C, Lansiaux A, Iwao M, Ishibashi F, Bailly C, Gago F. Molecular determinants of topoisomerase I poisoning by lamellarins: comparison with camptothecin and structure-activity relationships. J. Med. Chem. 2005;48:3796–3807. doi: 10.1021/jm049060w. [DOI] [PubMed] [Google Scholar]
- 63.Antony S, Jayaraman M, Laco G, Kohlhagen G, Kohn KW, Cushman M, Pommier Y. Differential induction of topoisomerase I-DNA cleavage complexes by the indenoisoquinoline MJ-III-65 (NSC 706744) and camptothecin: base sequence analysis and activity against camptothecin-resistant topoisomerases I. Cancer Res. 2003;63:7428–7435. [PubMed] [Google Scholar]
- 64.Stewart L, Ireton GC, Champoux JJ. A functional linker in human topoisomerase I is required for maximum sensitivity to camptothecin in a DNA relaxation assay. J. Bio. Chem. 1999;274:32950–32960. doi: 10.1074/jbc.274.46.32950. [DOI] [PubMed] [Google Scholar]
- 65.Losasso C, Cretaio E, Palle K, Pattarello L, Bjornsti MA, Benedetti P. Alterations in linker flexibility suppress DNA topoisomerase I mutant-induced cell lethality. J. Bio. Chem. 2007;282:9855–9864. doi: 10.1074/jbc.M608200200. [DOI] [PubMed] [Google Scholar]
- 66.Losasso C, Cretaio E, Fiorani P, D'Annessa I, Chillemi G, Benedetti P. A single mutation in the 729 residue modulates human DNA topoisomerase IB DNA binding and drug resistance. Nucleic Acids Res. 2008;36:5635–5644. doi: 10.1093/nar/gkn557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gongora C, Vezzio-Vie N, Tuduri S, Denis V, Causse A, Auzanneau C, Collod-Beroud G, Coquelle A, Pasero P, Pourquier P, et al. New Topoisomerase I mutations are associated with resistance to camptothecin. Mol. Cancer. 2011;10:64. doi: 10.1186/1476-4598-10-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Laco GS, Pommier Y. Role of a tryptophan anchor in human topoisomerase I structure, function and inhibition. Biochem. J. 2008;411:523–530. doi: 10.1042/BJ20071436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frohlich RF, Andersen FF, Westergaard O, Andersen AH, Knudsen BR. Regions within the N-terminal domain of human topoisomerase I exert important functions during strand rotation and DNA binding. J. Mol. Biol. 2004;336:93–103. doi: 10.1016/j.jmb.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 70.Vassallo O, Castelli S, D'Annessa I, della Rocca BM, Stella L, Knudsen BR, Desideri A. Evidences of a natively unfolded state for the human topoisomerase IB N-terminal domain. Amino Acids. 2011;41:945–953. doi: 10.1007/s00726-010-0794-x. [DOI] [PubMed] [Google Scholar]
- 71.Chillemi G, Redinbo M, Bruselles A, Desideri A. Role of the linker domain and the 203-214 N-terminal residues in the human topoisomerase I DNA complex dynamics. Biophys J. 2004;87:4087–4097. doi: 10.1529/biophysj.104.044925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Christensen MO, Barthelmes HU, Boege F, Mielke C. Residues 190-210 of human topoisomerase I are required for enzyme activity in vivo but not in vitro. Nucleic Acids Res. 2003;31:7255–7263. doi: 10.1093/nar/gkg927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Redinbo MR, Champoux JJ, Hol WG. Novel insights into catalytic mechanism from a crystal structure of human topoisomerase I in complex with DNA. Biochemistry. 2000;39:6832–6840. doi: 10.1021/bi992690t. [DOI] [PubMed] [Google Scholar]
- 74.Pond CD, Holden JA, Schnabel PC, Barrows LR. Surface plasmon resonance analysis of topoisomerase I-DNA binding: effect of Mg2+ and DNA sequence. Anticancer Drugs. 1997;8:336–344. doi: 10.1097/00001813-199704000-00005. [DOI] [PubMed] [Google Scholar]
- 75.Koster DA, Czerwinski F, Halby L, Crut A, Vekhoff P, Palle K, Arimondo PB, Dekker NH. Single-molecule observations of topotecan-mediated TopIB activity at a unique DNA sequence. Nucleic Acids Res. 2008;36:2301–2310. doi: 10.1093/nar/gkn035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tanizawa A, Kohn KW, Kohlhagen G, Leteurtre F, Pommier Y. Differential stabilization of eukaryotic DNA topoisomerase I cleavable complexes by camptothecin derivatives. Biochemistry. 1995;34:7200–7206. doi: 10.1021/bi00021a035. [DOI] [PubMed] [Google Scholar]
- 77.Kjeldsen E, Svejstrup JQ, Gromova II, Alsner J, Westergaard O. Camptothecin inhibits both the cleavage and religation reactions of eukaryotic DNA topoisomerase I.J. Mol. Biol. 1992;228:1025–1030. doi: 10.1016/0022-2836(92)90310-g. [DOI] [PubMed] [Google Scholar]
- 78.Tanizawa A, Fujimori A, Fujimori Y, Pommier Y. Comparison of topoisomerase I inhibition, DNA damage, and cytotoxicity of camptothecin derivatives presently in clinical trials. J. Natl Cancer Inst. 1994;86:836–842. doi: 10.1093/jnci/86.11.836. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.