


Abstract
The two-step process of selection and counter-selection is a standard way to enable genetic modification and engineering of bacterial genomes using homologous recombination methods. The tetA and sacB genes are contained in a DNA cassette and confer a novel dual counter-selection system. Expression of tetA confers bacterial resistance to tetracycline (TcR) and also causes sensitivity to the lipophillic chelator fusaric acid; sacB causes sensitivity to sucrose. These two genes are introduced as a joint DNA cassette into Escherichia coli by selection for TcR. A medium containing both fusaric acid and sucrose has been developed, in which, coexpression of tetA-sacB is orders of magnitude more sensitive as a counter-selection agent than either gene alone. In conjunction with the homologous recombination methods of recombineering and P1 transduction, this powerful system has been used to select changes in the bacterial genome that cannot be directly detected by other counter-selection systems.
INTRODUCTION
Genetic and functional genome analyses demand precise manipulation and modification of regulatory elements and the genes they control. To this end, recombineering has been shown to be a powerful method to modify genetic targets in vivo by the process of homologous recombination (1–4). This recombination is carried out by phage recombinases, like those of the λ Red system, which are functions that mediate precise targeted insertion of linear double-strand DNAs (dsDNAs) or single-strand DNA oligonucleotides (oligos) into the DNA of the bacterial genome and its episomes (1,2,5,6). These Red-promoted recombination events require only limited, ∼50 base segments of homology to the target. λ Exo binds dsDNA ends and removes bases from the 5′-end, and λ Beta binds single strands and enhances annealing to complementary target strands during the recombination event (3). Recombination with oligos requires only the Beta protein to bind the single-strand DNA and anneal it to the target (6). A third λ Red function, Gam, inhibits the host RecBCD and SbcCD nucleases and thereby enhances linear dsDNA recombination 10-fold (7).
Genetic modifications engineered by the process of recombineering often involve one or more selection steps. For example, replacing genes with drug markers generates gene knockouts, and adding genetic tags to genes by co-selection with a linked drug marker generates gene fusions. Creating point mutations or changes of a few bases can be accomplished with extremely high efficiency using Red Beta-mediated oligonucleotide recombination and screening for recombinants by a polymerase chain reaction (PCR) with primers specific for the mutation (8). However, a two-step process of selection and counter-selection is required when low efficiency events are expected, i.e. to construct seamless deletions or fusions in which large segments of DNA are removed or inserted without leaving a mark. First, a counter-selection cassette is inserted, and subsequently removed to generate the appropriate modification. Both steps use recombineering. Many such two-step systems exist. Here we describe the tetA-sacB cassette, a particularly useful two-step system, which provides unique advantages over other counter-selection methods.
The tetA gene product, located in the cytoplasmic membrane, prevents cellular accumulation of tetracycline, conferring resistance (TcR). TetA also causes the cell to become sensitive to lipophilic chelators, like fusaric acid, and to have increased sensitivity to kanamycin and osmotic shock (9). Thus, the TetA protein has both positive and negative selection effects on cell growth (10,11). The sacB gene product converts sucrose to levan, which accumulates in the periplasm and is toxic to E. coli (12,13). Combining both of these genes into a tetA-sacB cassette provides TcR as the selection for insertion, and each gene product exerts independent toxic effects that allow removal by counter-selection. We have developed a new medium that selects against both tetA and sacB. An additional benefit of this rich Tet/SacB medium is that incubation times are reduced relative to minimal media selections. This tetA-sacB cassette also has the advantage of not imposing any restraints on strain genotype, unlike most counter-selectable markers that require special mutant alleles such as rpsL (StrR), galK, thyA or tolC to be present in the genome (See the respective references 14–17).
MATERIALS AND METHODS
Bacterial strains and plasmids
Bacterial strains are described in Table 1. Standard genetic methods including recombineering and P1 transduction (20–22) were used for strain construction. Three sequenced strains MG1655, W3110 and DH10B (23–25) have been used to test the efficiencies of counter-selection. The pSIM5 and pSIM18 plasmids, which provide Red recombination functions and confer either chloramphenicol or hygromycin resistance, respectively, have been transformed into these strains and their derivatives for recombineering purposes (26).
Table 1.
Bacterial strains
| Strain | Genotype | Reference |
|---|---|---|
| CC4231 | W3110 ΔlacU169 gal490 pglΔ8 [λcI857 Δ(cro-bioA)] araD<>cat-sacB-amp | This work |
| DH10B | mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara-leu)7697 galU galK rpsL nupG | (18) |
| DY330 | W3110 ΔlacU169 gal490 pglΔ8 [λcI857 Δ(cro-bioA)] | (2) |
| LT1594 | MG1655 ΔlacZ<>hyg [pSIM5] | This work |
| LT1610 | MG1655 ΔlacZ<>hyg sfi-lacZ | This work |
| LT1619 | MG1655 ΔlacZ<>hyg sfiA<>tetA-sacB [pSIM5] | This work |
| MG1655 | (19) | |
| T-SACK | W3110 araD<>tetA-sacB-amp fliC<>cat argG::Tn5 | This work |
| W3110 | IN(rrnD-rrnE)1 rph-1 | (19) |
| XTL298 | CC4231 araD<>tetA-sacB-amp | This work |
| XTL425 | DH10B sfiA<>tetA-sacB [pSIM18] | This work |
| XTL426 | DH10B galM<tetA-sacB>gpmA [pSIM18] | This work |
| XTL621 | W3110 sfiA<>tetA-sacB | This work |
| XTL622 | W3110 galM<tetA-sacB>gpmA | This work |
| XTL623 | MG1655 sfiA<>tetA-sacB | This work |
| XTL624 | MG1655 galM<tetA-sacB>gpmA | This work |
| XTL629 | W3110 sfiA<>tetA-sacB [pSIM18] | This work |
| XTL630 | W3110 galM<tetA-sacB>gpmA [pSIM18] | This work |
| XTL631 | MG1655 sfiA<>tetA-sacB [pSIM18] | This work |
| XTL632 | MG1655 galM<tetA-sacB>gpmA [pSIM18] | This work |
| XTL634 | W3110 araD<>tetA-sacB-amp | This work |
| XTL635 | MG1655 sfi<>luc | This work |
| XTL671 | DH10B galM<tetA-sacB>gpmA | This work |
Materials
DNA oligos are listed in Table 2 and were purchased from Integrated DNA Technologies and used in the unpurified, salt-free form. The sequences of DNA primers to amplify tetA, cat, amp, cat-sacB and tet-sacB cassettes are described (see FAQs in the http://redrecombineering.ncifcrf.gov/ website). The High Fidelity Platinum® Taq DNA polymerase from Invitrogen was used for PCRs. Sequencing was done by SAIC-Frederick, Inc. and results were analyzed with Sequencher version 4.8.
Table 2.
Oligonucleotides for PCR amplification and oligo recombinationa
| galM / tet | AAATCACCAGCAAACACCGACATATTTGCAACTCAATATTCACAACAACCTCCTAATTTTTGTTGACACTCTATC |
| gpmA / sacB | CAACAGCAATGCTTACGCATAACCATAGCGAAAATAGTGGCGCAGTGTAAATCAAAGGGAAAACTGTCCATATGC |
| sfiA / tetA | AACTCACAGGGGCTGGATTGATTatgTACACTTCAGGCTATGCACATCGTTCCTAATTTTTGTTGACACTCTATC |
| sfiA / sacB | CATTGGCTGGGCGACAAAAAAAGTTCCAGGATTAATCCTAAATTTACttaATCAAAGGGAAAACTGTCCATATGC |
| Oligo A | GATTGATTatgTACACTTCAGGCTATGCACATCGT//taaGTAAATTTAGGATTAATCCTGGAACTTTTTTT |
| Oligo B | ACCGACATATTTGCAACTCAATATTCACAACAACC//TTACACTGCGCCACTATTTTCGCTATGGTTATGCG |
| galM / parS-1 | AAATCACCAGCAAACACCGACATATTTGCAACTCAATATTCACAACAACCCGATAAAAAGCCGAAGCCTTAAA |
| gpmA / parS-2 | CAACAGCAATGCTTACGCATAACCATAGCGAAAATAGTGGCGCAGTGTAATTGTTGACTTTCTCGTGACAAGC |
| sfiA / luc atg | ACTGGATGTACTGTACATCCATACAGTAACTCACAGGGGCTGGATTGATTatgGAAGACGCCAAAAACATAAAG |
| sfiA / luc stop | AAGCATTGGCTGGGCGACAAAAAAAGTTCCAGGATTAATCCTAAATTTACttaCACGGCGATCTTTCCGCCCTTCT |
| sfiA / lacZ atg | AACTCACAGGGGCTGGATTGATTatgTACACTTCAGGCTATGCACATCGTGATTCACTGGCCGTCGTTTTAC |
| sfiA / lacZ stop | CATTGGCTGGGCGACAAAAAAAGTTCCAGGATTAATCCTAAATTTACttaTTTTTGACACCAGACCAACTGG |
aUnderlined sequence is the primer for PCR amplification. The lowercase sequences represent the start and stop codons within the sequence. The // represents the center point in the 70 base Oligos A and B with flanking homologies to delete tetA-sacB. Oligo A and B correspond to the lagging strand during DNA replication of the bacterial chromosome.
Standard cultures were grown with LB broth or agar (1.5%) containing per liter: 10 g tryptone, 5 g yeast extract and 5 g NaCl. Counter-selection against sacB-carrying strains is done on L sucrose agar (1.5%) containing per liter 10 g tryptone, 5 g yeast extract and 60 g sucrose (no NaCl). The sucrose (60% solution) is autoclaved separately and added to the molten agar after its sterilization. Tetracycline (12.5 μg/ml), hygromycin (200 μg/ml), chloramphenicol (10 μg/ml) and ampicillin (30 μg/ml) are used as indicated. Bochner/Maloy agar was prepared as described previously (11).
Tet/SacB counter-selection medium
The Tet/SacB counter-selection agar that was developed here contains, per liter, 15 g of Difco agar, 4 g of tryptone, 4 g of yeast extract, 8 g of NaCl, 8 g of NaH2PO4·H2O, 0.11 g ZnCl2, 24 mg fusaric acid and 60 g sucrose. The agar, tryptone and yeast extract were autoclaved in a 400-ml volume with water. The NaCl and NaH2PO4·H2O were mixed and autoclaved in a 400-ml volume with water. Sucrose at 60 g in 100 ml was autoclaved. The molten agar mix, salt mix and sucrose were combined together after autoclaving. The fusaric acid (Sigma) was stored at 48 mg/ml in ethanol at −20° in a lightproof container. The ZnCl2 was stored in water at 25 mM after filter sterilizing; note that higher concentrations precipitate. Fusaric acid (0.5 ml) and ZnCl2 (32 ml) were added to the molten agar mixture after cooling to 55°. The final volume was brought to 1 liter with sterile H2O. Petri plates (100 mm) were poured so that each contained a 40-ml volume of the final molten agar mixture. The large volume ensures that the plates will remain hydrated over several days of incubation at 42°C. After hardening, the plates were placed in their original plastic bags, wrapped in aluminum foil and stored at 4°. We emphasize the importance of precisely following these procedures as variations can dramatically affect the selection.
P1 Tet/SacB counter-selection agar
When Tet/SacB counter-selection agar is used during a P1 transduction (22), it is modified by adding 5 ml of a sterile 1 M sodium citrate solution to the agar for a final concentration of 5 mM. This is the P1 Tet/SacB counter-selection agar.
Creating the tetA-sacB counter-selection cassette
We have created a new dual counter-selection system, tetA-sacB, starting with strain CC4231, which is DY330 with the cat-sacB cassette inserted to replace araD with an amp cassette located just beyond sacB (Table 1). We note that this intermediate strain can be used to place genes under arabinose control in the chromosome using sucrose counter-selection. To broaden the application and usefulness of this construct, we replaced cat with a second counter-selectable cassette, tetA, as follows. The tetA gene was amplified from a Tn10 transposon by PCR with primers that retain its promoter region but do not amplify the adjacent tetR gene encoding the repressor of tetA. Replacement of cat with tetA leaves sacB with its own promoter downstream of the tetA gene. This generates the tetA-sacB dual cassette with independent promoters for each gene and with tetA constitutively expressed (Figure 1). A TcR derivative, XTL298, contains tetA-sacB in the arabinose operon. It remains sensitive to sucrose and was confirmed by sequence analysis (see http://redrecombineering.ncifcrf.gov/).
Figure 1.
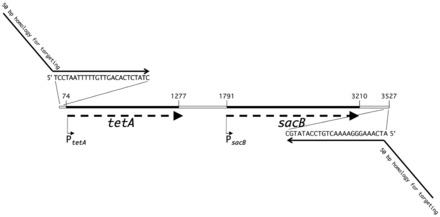
The tetA-sacB cassette. The diagram shows the relative positions of the two genes and their promoters. Dashed arrows indicate the direction of transcription through tetA and sacB coding sequences (black bars). Numbers indicate base pair positions. The sequences of primers used for PCR amplification of the cassette are indicated. These priming sequences are placed at the 3′-ends of chimeric primers that contain 50 nt of homology for targeting the dual cassette to a region of choice. With the exception of the chimeric primers, this diagram is to scale.
Tet/SacB counter-selection and recombineering conditions
Counter-selections for tetA alone (11) and sacB alone (27) have been defined previously; we reproduced both selection conditions with specific modifications to combine them in one agar plate. The protocol for replacing tetA-sacB is similar to our general recombineering protocols (http://redrecombineering.ncifcrf.gov/, 20,21) with minor modifications. Cells are grown overnight at 30° in LB with either chloramphenicol or hygromycin to maintain pSIM5 or pSIM18, respectively. The overnight culture, diluted 80-fold in LB without drug, is grown at 32° to OD600 ∼0.4, induced for 15 min at 42° to express Red, and then made electro-competent. After DNA electroporation, the cells are transferred to 5 ml of LB at 32° and grown for >4 h with aeration before plating on pre-warmed (42°) Tet/SacB counter-selection agar and incubated at 42° for 2 days. Total viable cells were determined on LB agar at 32°. The same cells electroporated without added DNA were the control for spontaneous mutants that survive the double selection.
Insertion of tetA-sacB into host strains DH10B, W3110 and MG1655
The tetA-sacB DNA flanked by appropriate homology arms was amplified from XTL298 cells by colony touch PCR (21) using chimeric primers (Table 2) as illustrated in Figure 1. These tetA-sacB products were inserted into DH10B[pSIM18] either within sfiA or near galM selecting for TcR by using previously described recombineering methods (20,21) to generate strains XTL425 and XTL426, respectively. These genomic regions, sfiA<>tetA-sacB and galM<tetA-sacB>gpmA, were then transferred by P1 transduction (22) from XTL425 and XTL426 into W3110 (generating strains XTL621 and XTL622, respectively) and MG1655 (generating strains XTL623 and XTL624, respectively). The <> designation indicates insertion of a construct made by recombineering within the designated gene, whereas the <tetA-sacB> flanking symbols indicate insertion of the construct in an intergenic region.
General considerations when constructing new tetA-sacB insertions by recombineering
In generating XTL425 and XTL426, 16 independent TcR recombinant colonies of DH10B[pSIM18] were patched to LB tetracycline and L sucrose agar. The patches on L sucrose were compared to each other and to a XTL298 control patch to ensure that the most sucrose sensitive recombinants were chosen; this helps to avoid any PCR-generated mutations that reduce sucrose sensitivity. We have also noticed that in some genomic locations sucrose sensitivity of all recombinants is reduced. Where this occurs, we either insert the construct in the same site but in the opposite orientation and retest, or if necessary and where possible, change to a nearby insertion site. Once several of the most sucrose sensitive recombinants are isolated for a particular location, these are then retested directly on Tet/SacB counter-selection agar and compared with XTL298 to ensure that the most sensitive candidate is used.
Modified P1 transduction method for replacement of tetA-sacB by counter-selection
P1 transductions were carried out as described previously (22) with the modification that recipient cells infected with P1vir were diluted and grown in 5 ml LB broth containing 200 mM sodium citrate for 8 h to ensure cellular segregation of the DNA markers, as well as the TetA and SacB proteins present in the starting cells. This culture was diluted and titered for total cells on LB. The remaining culture was concentrated into 100 μl of M9 salts. The concentrated cells were plated for recombinants on the selective P1 Tet/SacB counter-selection agar.
Creation of T-SACK, a versatile template strain for cassette amplification by PCR
The araD<>tetA-sacB amp region was moved from XTL298 to W3110 by P1 transduction, selecting for ampicillin resistance, creating XTL634. We also transferred the argG::Tn5 transposon carrying the kan (KmR) cassette into this strain by P1. Finally, a cat (CmR) cassette, fliC<>cat was introduced by P1. This new strain, T-SACK, has several drug resistance cassettes [tetA-sacB, amp, cat and kan]; all of which can be used as templates for PCR amplification to generate recombineering reagents by colony touch PCR. This strain is used as a template for colony touch PCR, instead of plasmid DNA. Even small amounts of plasmid DNA, which efficiently transform, show up as drug-resistant colonies, making true recombinants hard to find. Note that a tetA PCR product can be generated and used for λ Red-mediated homologous recombination when appropriate 50 base homology arms are included on the 5′-ends of the tetA primers (28).
RESULTS AND DISCUSSION
The presence of TetA enhances sucrose counter-selection
CC4231 and XTL298 differ by having cat or tetA, respectively, upstream of sacB. We tested whether those functions differentially affect the sensitivity caused by the sacB cassette on L sucrose agar. The two strains were grown to OD600 ∼0.4 and titered directly on L sucrose or LB agar at 32°. In three independent trials, there was a 4-fold increase in sucrose sensitivity for XTL298. In a similar experiment after growth to OD600 ∼0.4, both cell types were made electro-competent, subjected to electroporation without DNA and plated on L sucrose and LB agar. Sucrose resistant colonies appeared with an average frequency of 1.8 × 103 per 108 colonies on LB agar for CC4231 and 4.4 × 102 per 108 colonies on LB agar for XTL298. These data indicate that sucrose counter-selection improves ∼ 4-fold in cells expressing the TetA protein. We suspect that the presence of TetA in the membrane accentuates the toxicity of the periplasmic levan produced from the sucrose. It is also possible that sacB expression is somewhat higher in the tetA-sacB construct.
Plating on Bochner/Maloy and Tet/SacB counter-selection media
We tested the inhibitory effect of tetA-sacB expression when challenged on Bochner/Maloy agar, which is designed to be counter-selective only for tetA expression (11). A log phase culture of XTL671 (DH10B galM<tetA-sacB>gpmA) was titered with a relative average survival of 8.2 × 103 colonies on Bochner/Maloy versus 108 colonies on LB agar, but surviving colonies grew very poorly. When XTL622 and XTL624 (W3110 and MG1655 containing galM<tetA-sacB>gpmA, respectively) were tested on Bochner/Maloy agar, very little counter-selection occurred with ∼4 × 107 survivors per 108 colonies on LB agar. Thus, as described previously, Bochner/Maloy agar is selective for the recA DH10B background but not for healthier genetic backgrounds like W3110 and MG1655 (11). As a first attempt to create a double selection plate, we added 6% sucrose to Bochner/Maloy agar; however, W3110 and MG1655 tetA-sacB derivatives still gave high background growth on this medium (data not shown). We therefore developed a new Tet/SacB counter-selection agar (see ‘Materials and Methods’ section), optimized for coselection against both gene products. It has been noted that the counter-selection growth condition against tetA on fusaric acid media is most efficient at 42° (29), and we have confirmed that our modified system is also most efficient at 42°. Therefore, when removing the tetA-sacB cassette by recombineering methods, strains have been used that express the Red recombination functions from plasmids (26) rather than from the defective prophage, since bacteria with the prophage are unable to grow at 42° (2).
The three parental strains MG1655, W3110 and DH10B all form colonies on the Tet/SacB counter-selection agar at 42° with a final efficiency equal to that on LB agar. However, colonies are only barely visible after 1 day; 2 days are required for full colony formation on Tet/SacB counter-selection agar because these selective conditions reduce cell growth even without tetA-sacB present. We also note that MG1655 grows better than W3110, which grows better than DH10B on LB as well as on Tet/SacB counter-selection agar. We next tested survival of the six MG1655, W3110 and DH10B derivatives containing tetA-sacB at either sfi or galM on the Tet/SacB counter-selection agar (Table 3). Here, when both gene products are present, they exert a synergistic toxicity, causing an extremely low survival of ≤6 × 10−7. We have not found reports of any counter-selection system this restrictive.
Table 3.
Counter-selection used to replace tetA-sacB cassettes by recombineering
| Colonies on Tet/SacB Counter-Selection Agara |
|||||||
|---|---|---|---|---|---|---|---|
| Strainb (Parent) |
sfiA<>tetA-sacB |
Strainb (Parent) |
galM<tetA-sacB>gpmA |
||||
| No DNAc | Oligo Ad | sfiA-luce | No DNAc | Oligo Bd | parSe | ||
| XTL425 (DH10B) | 7 | 21 000 | 1500 | XTL426 (DH10B) | 53 | 50 000 | 61 000 |
| XTL629 (W3110) | 21 | 57 000 | 1600 | XTL630 (W3110) | 23 | 56 000 | 110 000 |
| XTL631 (MG1655) | 26 | 21 000 | 1500 | XTL632 (MG1655) | 42 | 25 000 | 44 000 |
aSurvivors on Tet/SacB Counter-Selection Agar are normalized to 108 cells on LB agar.
bAll strains carry the pSIM18 plasmid for Red recombination.
cControl without added DNA. The average survival of the same six strains on L sucrose was 28-fold higher than on Tet/SacB medium, with a range from 14- to 53-fold. Note that the sucrose counter-selection for cells containing tetA is already 4-fold more selective than for cells lacking tetA, as shown by comparing CC4231, the cat-sacB strain, to XTL298, the tetA-sacB strain.
d70 base oligos used to remove the tetA-sacB cassette by Red-mediated recombination.
ePCR products of either sfiA-luc (∼1.6 kb) or parS (279 bp) to replace the respective tetA-sacB cassette in sfiA or the tetA-sacB cassette between galM and gpmA.
Recombineering with the tetA-sacB dual counter-selection system
Single-stranded DNA recombination with 70 base long oligonucleotides was used to delete the tetA-sacB cassette located at sfiA<>tetA-sacB or galM<tetA-sacB>gpmA from all six derivatives of the host strains (Table 3). Oligo-generated recombinants were orders of magnitude more frequent than spontaneous survivors in the control electroporation without DNA. All colonies tested were recombinants and had lost the tetA and sacB markers.
To test the efficiency of counter-selection with a short (279 bp) PCR product, a dsDNA containing a protein binding site, P1 parS (30), was used to replace the tetA-sacB cassette at galM<tetA-sacB>gpm (Table 3). The frequency of removing tetA-sacB by recombination with this short PCR product was similar to that found using oligo B (Table 3). All 16 survivors, tested by PCR analysis, had lost the tetA-sacB and gained the parS site.
Generation of sfiA-luc and sfiA-lacZ translational gene fusions by replacing sfiA<>tetA-sacB
We sought to test the efficacy of the tetA-sacB counter-selection with large inserts by replacing tetA-sacB with the luciferase gene, luc. DNA damage by agents like mitomycin C induces gene expression of sfiA, an SOS response gene (31). A fusion between the luciferase gene, luc, and sfiA was made in DH10B, MG1655 and W3110 derivatives carrying sfiA<>tetA-sacB [pSIM18]. The luc open reading frame starting with the initiation codon replaces just the open reading frame of sfiA. The sfiA-luc recombinants were generated at a frequency of ∼2 × 103/108 viable cells (Table 3). Those derived from MG1655 were analyzed further. All 16 independent colonies tested were recombinant and carried luc as judged by PCR amplification. When assayed for luciferase, five out of eight recombinants had higher mitomycin C-inducible activity. The remaining three recombinants may have PCR-generated mutations that lowered the luciferase activity. One of the MG1655 sfiA-luc fusions with the highest inducible luciferase activity was saved as XTL635.
We also created a sfiA-lacZ translational gene fusion by Red-mediated recombination that can be used to observe and quantitate SOS induction kinetics. LT1594, the lacZ deletion mutant (ΔlacZ<>hyg) of MG1655 carrying pSIM5 was used. Into this strain, the sfiA<>tetA-sacB counter-selection cassette was introduced by recombineering selecting for TcR. To make the sfiA-lacZ gene fusion, we amplified lacZ by PCR using a primer with the first nine codons of sfiA fused in frame to the 6th codon of lacZ, and a primer containing the last eight codons of lacZ, including the TAA stop codon. These chimeric primers were used to generate a PCR product containing from codon 6 to the stop codon of lacZ complete with flanking homologies. The entire sfiA-lacZ gene fusion was used to replace the sfiA<>tetA-sacB counter-selection cassette by recombineering. LT1619 (MG1655 ΔlacZ<>hyg sfiA<>tetA-sacB [pSIM5]) expressing the Red recombination functions was electroporated with the sfiA-lacZ PCR fragment and following outgrowth recombinant colonies were selected on Tet/SacB counter-selection agar. A low frequency of Tet/SacB resistant colonies (∼1 × 102 /108 viable cells) was found, and this frequency was similar to that of a control electroporation without DNA. The lower level of lacZ recombinants likely reflects the greater difficulty in replacing the ∼3.5 kb tetA-sacB cassette with the similar sized ∼3.2 kb lacZ fragment rather than with the smaller ∼1.6 kb luc fragment. Plating the same recombinant cell mixture on L sucrose gave ∼50-fold higher background than did the Tet/SacB plates, indicating the difficulty in identifying recombinants on L sucrose. Examination of 16 survivors on the Tet/SacB counter-selection agar revealed that five had become TcS and now expressed functional LacZ, as evidenced by colonies that turned red on MacConkey lactose agar but only in the presence of mitomycin C. The other 11 colonies remained Lac− and still carried some form of the tetA-sacB cassette as evidenced by PCR amplification and reduced TcR. All five recombinants expressed similar levels of β-galactosidase, and one was saved as LT1610. It is shown to increase LacZ expression as mitomycin C levels increase (Figure 2).
Figure 2.
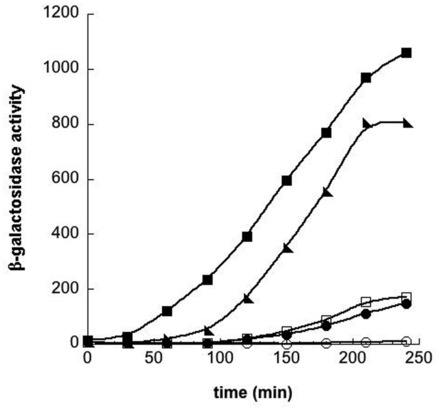
SOS induction of the sfiA-lacZ reporter construct. An overnight culture of LT1610 was diluted 1/500 into LB, distributed among five flasks, placed at 37° in a shaking water bath, and grown to an OD600 of 0.1. Mitomycin C was then added (t = 0) to the following concentrations (μg/ml): open circles, no drug; filled circles, 3 × 10−3; open squares, 1 × 10−2; filled triangles, 0.1; and filled squares, 1.0. Time points were taken every 30 min during growth and β-galactosidase activity in each sample was determined according to Miller (32), and plotted on the y-axis; time of exposure to Mitomycin C is plotted on the x-axis.
P1 transductants can be isolated by Tet/SacB counter-selection
P1 transduction was used to transfer the sfiA-luc fusion construct from the MG1655 derivative XTL635 into the recipient XTL621 (W3110 sfiA<>tetA-sacB). P1 transduction occurs by recombination between the recipient genome and the donor strain DNA, which P1 transfers to the recipient. Since only a few recombinants are generated in such crosses, transduction necessitates that there be a direct selection conferred on the recipient by the recombining donor DNA. Counter-selections are not normally stringent enough to enable selection of rare recombinants generated by P1 transduction. Here, sfiA-luc is the donor DNA, and the only selection is the Tet/SacB counter-selection against the sfiA<>tetA-sacB cassette present in the recipient. A control culture yielded fewer resistant colonies than cultures that had been infected with the P1 donor lysate (Table 4). Examination of the target region by PCR analysis demonstrated that the sfiA-luc fusion had been transferred into W3110 replacing the tetA-sacB cassette in 10 out of 16 resistant colonies tested (Table 4). Our ability to readily identify transductants using the P1 Tet/SacB counter-selection agar demonstrates the efficacy and general usefulness of this system.
Table 4.
Counter-selection used to replace tetA-sacB cassettes by P1 transduction
| Donor Straina | Recipient Strain | P1 lysate (µl)b | CFUc |
|---|---|---|---|
| XTL635 | XTL621 | 0 | 31 |
| MG1655 sfiA<>luc | W3110 sfiA<>tetA-sacB | 1 | 185 |
| 10 | 132 |
aA lysate of P1 vir was prepared by infection of XTL635.
bVolume of the donor P1 lysate used to infect the recipient XTL621.
cThe raw number of colonies resistant to Tet/SacB counter-selection agar when plating 100 µl of concentrated cells where the total number of cells was about 5 × 109. The frequency of P1 transduced recombinants among resistant colonies tested by PCR analysis was 63%. In a second independent transduction (data not shown), the frequency of recombinants was 56%.
CONCLUSIONS
The Tet/SacB dual counter-selection system described here has unique and substantial advantages over other selection/counter-selection systems. The tetA and sacB genes of the counter-selection cassettes each express a protein that is toxic to the bacterial cell growing on the newly developed Tet/SacB counter-selection agar. This toxic combination causes a stronger counter-selection than achieved by either gene product alone. This two-gene cassette can be used without regard to the genotype of the strain in which selection is imposed. Importantly, when combined with a homologous recombination method, like recombineering, this counter-selection system allows facile replacement of the tetA-sacB cassette in generating deletion, substitution or fusion mutants at the targeted region. This system is likely to be useful for genetic manipulations in other bacterial species and modification of genomic clones on bacterial artificial chromosomes.
FUNDING
Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (in part); federal funds from the National Cancer Institute, National Institutes of Health, under contract [HHSN261200800001E] (in part); The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. Funding for open access charge: This research is funded by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Conflict of interest statement. The National Institutes of Health has a patent covering methods used for recombineering. Authors Court, Costantino, Li and Sawitzke are inventors on these patents and employees of NIH.
ACKNOWLEDGEMENTS
We thank Adam Parks for critiquing the article, and David Friedman for critical reading and Tet/SacB counter-selection experiments carried out in his laboratory with his strains. We offer special thanks to Suki Sathyanarayana, Lauren Mora-Smith and Smruti Patel from the Media Laboratory of the Frederick National Laboratory for Cancer Research for preparation of many iterations of the Tet/SacB counter-selection agar.
REFERENCES
- 1.Muyrers JP, Zhang Y, Buchholz F, Stewart AF. RecE/RecT and Redα/Redβ initiate double-stranded break repair by specifically interacting with their respective partners. Genes Dev. 2000;14:1971–1982. [PMC free article] [PubMed] [Google Scholar]
- 2.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl Acad. Sci. USA. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Court DL, Sawitzke JA, Thomason LC. Genetic engineering using homologous recombination. Annu. Rev. Genet. 2002;36:361–388. doi: 10.1146/annurev.genet.36.061102.093104. [DOI] [PubMed] [Google Scholar]
- 4.Copeland NG, Jenkins NA, Court DL. Recombineering: a powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2001;2:769–779. doi: 10.1038/35093556. [DOI] [PubMed] [Google Scholar]
- 5.Murphy KC. Use of bacteriophage λ recombination functions to promote gene replacement in Escherichia coli. J. Bacteriol. 1998;180:2063–2071. doi: 10.1128/jb.180.8.2063-2071.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellis HM, Yu D, DiTizio T, Court DL. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl Acad. Sci. USA. 2001;98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Datta S, Costantino N, Zhou X, Court DL. Identification and analysis of recombineering functions from Gram-negative and Gram-positive bacteria and their phages. Proc. Natl Acad. Sci. USA. 2008;105:1626–1631. doi: 10.1073/pnas.0709089105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sawitzke JA, Costantino N, Li XT, Thomason LC, Bubunenko M, Court C, Court DL. Probing cellular processes with oligo-mediated recombination and using the knowledge gained to optimize recombineering. J. Mol. Biol. 2011;407:45–59. doi: 10.1016/j.jmb.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stavropoulos TA, Strathdee CA. Synergy between tetA and rpsL provides high-stringency positive and negative selection in bacterial artificial chromosome vectors. Genomics. 2001;72:99–104. doi: 10.1006/geno.2000.6481. [DOI] [PubMed] [Google Scholar]
- 10.Bochner BR, Huang HC, Schieven GL, Ames BN. Positive selection for loss of tetracycline resistance. J. Bacteriol. 1980;143:926–933. doi: 10.1128/jb.143.2.926-933.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maloy SR, Nunn WD. Selection for loss of tetracycline resistance by Escherichia coli. J. Bacteriol. 1981;145:1110–1111. doi: 10.1128/jb.145.2.1110-1111.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gay P, Le Coq D, Steinmetz M, Ferrari E, Hoch JA. Cloning structural gene sacB, which codes for exoenzyme levansucrase of Bacillus subtilis: expression of the gene in Escherichia coli. J. Bacteriol. 1983;153:1424–1431. doi: 10.1128/jb.153.3.1424-1431.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinmetz M, Le Coq D, Djemia HB, Gay P. Genetic analysis of sacB, the structural gene of a secreted enzyme, levansucrase of Bacillus subtilis Marburg. MGG. 1983;191:138–144. doi: 10.1007/BF00330901. [DOI] [PubMed] [Google Scholar]
- 14.Russell CB, Dahlquist FW. Exchange of chromosomal and plasmid alleles in Escherichia coli by selection for loss of a dominant antibiotic sensitivity marker. J. Bacteriol. 1989;171:2614–2618. doi: 10.1128/jb.171.5.2614-2618.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong QN, Ng VC, Lin MC, Kung HF, Chan D, Huang JD. Efficient and seamless DNA recombineering using a thymidylate synthase A selection system in Escherichia coli. Nucleic Acids Res. 2005;33:e59. doi: 10.1093/nar/gni059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeVito JA. Recombineering with tolC as a selectable/counter-selectable marker: remodeling the rRNA operons of Escherichia coli. Nucleic Acids Res. 2008;36:e4. doi: 10.1093/nar/gkm1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grant SG, Jessee J, Bloom FR, Hanahan D. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl Acad. Sci. USA. 1990;87:4645–4649. doi: 10.1073/pnas.87.12.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bachmann BJ. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE, editors. Vol. 2. Washington, DC: ASM Press; 1996. pp. 2460–2488. [Google Scholar]
- 20.Sawitzke JA, Thomason LC, Costantino N, Bubunenko M, Datta S, Court DL. Recombineering: in vivo genetic engineering in E. coli, S. enterica, and beyond. Methods Enzymol. 2007;421:171–199. doi: 10.1016/S0076-6879(06)21015-2. [DOI] [PubMed] [Google Scholar]
- 21.Thomason L, Court DL, Bubunenko M, Costantino N, Wilson H, Datta S, Oppenheim A. Recombineering: genetic engineering in bacteria using homologous recombination. Curr. Protoc. Mol. Biol. 2007 doi: 10.1002/0471142727.mb0116s78. Chapter 1, Unit 1 16. [DOI] [PubMed] [Google Scholar]
- 22.Thomason LC, Costantino N, Court DL. E. coli genome manipulation by P1 transduction. Curr. Protoc. Mol. Biol. 2007 doi: 10.1002/0471142727.mb0117s79. Chapter 1, Unit 1 17. [DOI] [PubMed] [Google Scholar]
- 23.Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 24.Hayashi K, Morooka N, Yamamoto Y, Fujita K, Isono K, Choi S, Ohtsubo E, Baba T, Wanner BL, Mori H, et al. Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100049. 2006 0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durfee T, Nelson R, Baldwin S, Plunkett G, 3rd, Burland V, Mau B, Petrosino JF, Qin X, Muzny DM, Ayele M, et al. The complete genome sequence of Escherichia coli DH10B: insights into the biology of a laboratory workhorse. J. Bacteriol. 2008;190:2597–2606. doi: 10.1128/JB.01695-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Datta S, Costantino N, Court DL. A set of recombineering plasmids for gram-negative bacteria. Gene. 2006;379:109–115. doi: 10.1016/j.gene.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 27.Blomfield IC, Vaughn V, Rest RF, Eisenstein BI. Allelic exchange in Escherichia coli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol. Microbiol. 1991;5:1447–1457. doi: 10.1111/j.1365-2958.1991.tb00791.x. [DOI] [PubMed] [Google Scholar]
- 28.Sharan SK, Thomason LC, Kuznetsov SG, Court DL. Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 2009;4:206–223. doi: 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerlach RG, Jackel D, Holzer SU, Hensel M. Rapid oligonucleotide-based recombineering of the chromosome of Salmonella enterica. Appl. Environ. Microbiol. 2009;75:1575–1580. doi: 10.1128/AEM.02509-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nielsen HJ, Youngren B, Hansen FG, Austin S. Dynamics of Escherichia coli chromosome segregation during multifork replication. J. Bacteriol. 2007;189:8660–8666. doi: 10.1128/JB.01212-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Power EG, Phillips I. Induction of the SOS gene (umuC) by 4-quinolone antibacterial drugs. J. Med. Microbiol. 1992;36:78–82. doi: 10.1099/00222615-36-2-78. [DOI] [PubMed] [Google Scholar]
- 32.Miller JH. A Short Course in Bacterial Genetics. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]