


Abstract
HELQ is a superfamily 2 DNA helicase found in archaea and metazoans. It has been implicated in processing stalled replication forks and in repairing DNA double-strand breaks and inter-strand crosslinks. Though previous studies have suggested the possibility that HELQ is involved in the Fanconi anemia (FA) pathway, a dominant mechanism for inter-strand crosslink repair in vertebrates, this connection remains elusive. Here, we investigated this question in mice using the Helqgt and Fancc− strains. Compared with Fancc−/− mice lacking FANCC, a component of the FA core complex, Helqgt/gt mice exhibited a mild of form of FA-like phenotypes including hypogonadism and cellular sensitivity to the crosslinker mitomycin C. However, unlike Fancc−/− primary fibroblasts, Helqgt/gt cells had intact FANCD2 mono-ubiquitination and focus formation. Notably, for all traits examined, Helq was non-epistatic with Fancc, as Helqgt/gt;Fancc−/− double mutants displayed significantly worsened phenotypes than either single mutant. Importantly, this was most noticeable for the suppression of spontaneous chromosome instability such as micronuclei and 53BP1 nuclear bodies, known consequences of persistently stalled replication forks. These findings suggest that mammalian HELQ contributes to genome stability in unchallenged conditions through a mechanism distinct from the function of FANCC.
INTRODUCTION
Maintaining genome stability is critical for cells, given that the genome is under constant attack by numerous exogenous and endogenous agents (1). It is inevitable that cells will have to proceed with genome duplication using damaged template DNA, which can perturb the normal progression of replication forks. To cope with this problem, organisms have developed multiple mechanisms to allow the rescue of stalled replication forks (2). Such mechanisms include firing of dormant origins, translesion synthesis with error-prone DNA polymerases and homology-directed fork recovery (3–5). Evidence suggests that the latter two mechanisms are in part coordinated by the concerted work of proteins that are mutated in Fanconi anemia (FA) (6), a rare polygenic human genetic disorder (7). FA patients exhibit genome instability, congenital abnormalities, bone marrow failure, hypogonadism and a heightened predisposition to cancer (7,8). FA is uniquely characterized by its cellular hypersensitivity to agents that induce DNA inter-strand crosslinks (ICLs) (7,9), although it is unclear how ICL repair is functionally linked with stalled fork recovery in unchallenged conditions.
The DNA helicase HELQ was first discovered in the human and mouse genomes through its homology to MUS308 (10), a DNA repair enzyme required for ICL resistance in Drosophila melanogaster (11,12). Although it is unlikely that its vertebrate ortholog POLQ plays a major role in ICL repair (13–15), together they make up a unique family of DNA polymerases that possess a helicase domain in the N-terminus in addition to a C-terminal polymerase domain (16–18). Unlike its paralog POLQ, HELQ lacks a polymerase domain, and several lines of evidence indicate that HELQ performs a distinct function from POLQ. HELQ is an ortholog of the Drosophila mus301 gene (19), which is allelic to the female-sterile mutation spindle-C (spn-C) (20). Mutations in spn-C result in the failed repair of meiotic double-strand breaks (DSB) and activation of the meiotic checkpoint (20), which was not observed in mus308 mutants. In line with this observation, it was also reported that the Caenorhabditis elegans ortholog helq-1 plays a role in meiotic DSB repair by promoting postsynaptic RAD-51 filament disassembly (21). These findings suggest that HELQ has a role in meiotic DSB repair through homologous recombination (HR) in these species. In humans, HELQ is expressed in the testes, ovaries, heart and skeletal muscle (22). However, its function is largely unknown.
Biochemically, human HELQ exhibits ATP-dependent 3′–5′ DNA helicase activity in vitro (10,23). A recent study demonstrated that human HELQ preferentially unwinds the parental strands of forked structures with a nascent lagging strand, and that this activity is stimulated by replication protein A (RPA) (23). These findings suggest that HELQ is likely to participate in the recovery of stalled or collapsed replication forks. Several studies have suggested that this role of HELQ is closely linked with the FA pathway. A genetic study in C. elegans demonstrated that helq-1 is required for ICL repair and is epistatic to fcd-2 (24), an ortholog of FANCD2 whose product is mono-ubiquitinated by the FA core complex as a key step in this pathway (25). However, C. elegans contains only a few FA proteins and lacks multiple members comprising the FA core complex (26). HELQ may belong to a primitive FA pathway in C. elegans, but its evolution seems to have taken a complex path. Paradoxically, disruption of Helq in chicken DT40 cells, which contain all of the FA proteins, did not confer hypersensitivity to ICL inducing agents (14). In human cells, HELQ depletion confers hypersensitivity to the crosslinker mitomycin C (MMC) and HR deficiency, the latter reported to be epistatic to FANCD2 (27). Consistent with this observation, exogenously expressed GFP-tagged HELQ co-localizes with RAD51 foci as well as FANCD2 foci after treatment with the topoisomerase I inhibitor camptothecin (CPT) (23). There is little information about the link between HELQ and the FA pathway in mammals, particularly in the absence of exogenous DNA damage.
To decipher the enigmatic connection between HELQ and the FA pathway, we have generated Helq deficient mice using a gene-trap allele named Helqgt for phenotypic comparisons to mice deficient for Fancc, encoding FANCC, a component of the FA core complex (28) in the same genetic background. For all traits examined including hypogonadism and MMC sensitivity, we found that loss of Helq results in phenotypes considerably milder than Fancc deficiency. Moreover, our data show that combined loss of Helq and Fancc leads to further severe phenotypes than single mutants, presenting no evidence for epistasis. Importantly, the strongest inter-dependence for Helq and Fancc was observed for the suppression of spontaneous genome instability derived from replication fork failures rather than MMC resistance. These findings collectively suggest that HELQ contributes to genome stability in unperturbed conditions in a manner that is distinct from the function of FANCC.
MATERIALS AND METHODS
Mouse strains and mouse embryonic fibroblasts
All experiments were performed using mice from a C57BL/6J background and were approved by the Institutional Animal Care and Use Committee. Mouse embryonic fibroblasts (MEFs) were generated from 12.5–14.5 dpc embryos and cultured using standard procedures as described previously (29). All mice were genotyped by PCR. The primers used are available upon request.
Quantitative reverse-transcription-PCR
RNA was isolated from either cultured MEFs or testes tissue using the PureLink RNA Mini Kit (Ambion, Life Technologies) and the RNeasy Kit (QIAGEN). cDNA was then synthesized using the Superscript VILO cDNA Synthesis Kit (Invitrogen, Life Technologies). q-PCR analysis was performed on the LightCycler 480 (Roche) using primer pairs specific for exons 1–2, exons 11–12 and the chimeric mutant transcript spanning between exon 11 and the inserted vector. Expression was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Western blotting and immunofluorescence microscopy
Western blotting and immunofluorescence staining were carried out using standard procedures as described previously (29).
Antibodies
For immunofluorescence and western blotting procedures, we used anti-phospho-histone H3, anti-RPA32, anti-γH2AX, anti-CENP-A, anti-phospho-CHK1 (Cell Signaling; #9706, #2208, #2577, #2048, #2341, respectively), anti-FANCD2 for foci staining, anti-FANCI, anti-53BP1, anti-MCM4 (Abcam; ab2187 or ab108928, ab74332, ab36823, ab4459, respectively), anti-FANCA (Bethyl Laboratories; #A301-980A), anti-CHK1 (Santa Cruz; sc-8408), anti-FANCD2 for western blots (Epitomics; #2986-1) and anti-HELQ (MyBioSource; #MBS120320). For the DNA fiber assay, anti-digoxigenin antibody conjugated with rhodamine from Roche (11207750910) and the streptavidin-AlexaFluor488 from Invitrogen (S-32354) were used.
siRNA transfection in MEFs, HEK 293T and PD331 cells
One million cells were seeded per well in a six-well dish followed by transfection with either 50 (MEFs) or 25 nM (HEK 293T, PD331) of non-targeted control small interfering RNA (siRNA) (#D-001206-13-20, siGENOME Smart pool), HELQ siRNA (#M-015379-01-0005, siGENOME Smart pool) or FANCA siRNA (#M-019283-02-0005, siGENOME Smart pool) from Dharmacon. This was performed using OPTI-MEM and Lipofectamine RNAiMAX (Life technologies) transfection reagents. Twenty-four hours later, a second round of transfection was performed using the same concentration of siRNA, followed by 24 h culture. Cells were then re-plated according to the analysis performed. The PD331 cell lines were obtained from the Oregon Health & Science University Fanconi Anemia Cell Repository (Portland, Oregon).
Metaphase analysis
MEFs were treated with 600 nM MMC for 2 h and allowed 22 h to recover before harvest. For experiments using the HEK 293T cell line, cells were treated with 300 nM MMC for 24 h before harvest. In all experiments, cells were treated with colcemid for 1–2 h before harvest. Following hypotonic treatment, cells were fixed with fixative (3:1 methanol: acetic acid in volume) and dropped on slides in a humidified environment to optimize spreading. Slides were mounted in 1× 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI) anti-fade solution the following day and blinded for analysis. Only metaphases with 38–41 chromosomes (MEFs) or 64–72 chromosomes (HEK 293T cells) were included in the analysis. Chromosome aberrations including radials, gaps/breaks, fragments and ring chromosomes were scored for all experiments. HEK 293T cells were cultured using the same procedures used for MEFs.
DNA fiber assay
All techniques and methods of analysis used were performed as described previously (29,30). Briefly, replication forks were sequentially labeled with deoxyuridine triphosphates (dUTPs) conjugated with digoxigenin (digoxigenin-dUTPs) for 20 min and with biotin-dUTPs for 30 min. Labeled cells were dropped onto slides, fixed and dipped into lysis buffer for the release and extension of DNA fibers. Incorporated dUTPs were visualized by anti-digoxigenin rhodamine conjugate (Roche, Branford, CT) and streptavidin, -Alexa Fluor 488 (Invitrogen, Carlsbad, CA).
Colony formation assay
Five hundred cells were plated in 6 cm dishes along with the corresponding doses of MMC. For HEK 293T cells, the bottoms of the dishes were coated with poly-L-lysine beforehand to aid in cell adhesion. Colonies were stained using crystal violet after a period of 1 week (HEK 293T) or 2 weeks (PD331 and PD331+FANCC) and counted.
MTT assay
The Vybrant MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) Cell Proliferation Assay Kit (Life Technologies) was used. Briefly, 5 × 104 (experiments with MEFs) or 1 × 104 (experiments with PD331 cell lines) cells of each genotype were plated per well in a 96-well plate. The next day, cells were either treated with the corresponding drug or left untreated for 5 days before the assay was performed according to the manufacturer’s instructions. A570 was used to measure relative cell proliferation, whereas A670 was used as a reference for background absorbance.
Cytokinesis-block micronucleus assay and G1 phase cell analyses
The cytokinesis-block micronucleus (MN) assay was performed as described previously (29,31), except that cells were treated with cytochalasin B (0.72 µg/ml) for 4–5 h. The same procedure was used for all analyses of G1 cells.
Measuring HR events using the fluorescent yellow direct repeat transgenic locus system
Wild-type (WT) and Helqgt/gt MEFs carrying the fluorescent yellow direct repeat (FYDR) transgenic locus (32) in the hemizygous state were generated as described earlier in the text. Cells at passage 2 were plated, grown for 3 days and then re-plated into three separate dishes. The corresponding drug treatments (untreated, MMC, CPT) were then administered the following day and washed out after 24 h. After a 48 h recovery period, cells were analyzed by flow cytometry using the FL1-H and FL2-H channels of the FACSCalibur (BD Biosciences).
RESULTS
Helq deficient mice carry a gene-trap allele that generates HELQΔ-β-Geo
To generate a mouse Helq mutant, we searched the BayGenomics database for mutant mouse embryonic stem (ES) cell clones (33,34). Gene-trap vectors are designed to have a splice acceptor site upstream of a reporter gene, typically β-Geo, a fusion gene of the β-galactosidase and neomycin resistance genes. We found that the ES cell clone RRF112 has an insertion of the gene-trap vector pGT0Lxf in the intron between exons 11 and 12 of the Helq locus (Supplementary Figure S1A). This gene-trap allele was named Helqgt. The Helqgt allele is expected to create a chimeric transcript containing exons 1–11 of Helq and β-Geo (Figure 1A), producing a truncated HELQ protein that is fused with β-Geo at its C-terminal end (Figure 1B). This mutant protein still retains its helicase domain but lacks the three C-terminal domains with highly conserved motifs thorough archaea to metazoans (35,36). It has been demonstrated that these domains are required for normal helicase activity in other species (36–38). To generate mice that carry the Helqgt allele, we microinjected RRF112 ES cells into blastocysts from an inbred strain of C57BL/6J (B6) females using a standard method. High-percentage chimera males were mated with B6 inbred females to produce carriers of the Helqgt allele, which were identified by genomic PCR using primer pairs that amplify the boundary sequences of the insertion site (Supplementary Figure S1B). Helqgt heterozygous (Helqgt/+) carriers appear normal in every aspect and are indistinguishable from WT mice (data not shown). A congenic line of Helqgt has been established by backcrossing Helqgt/+ mice to inbred B6 mice at least 10 generations. This B6 congenic Helqgt line was used for the following studies unless otherwise indicated.
Figure 1.
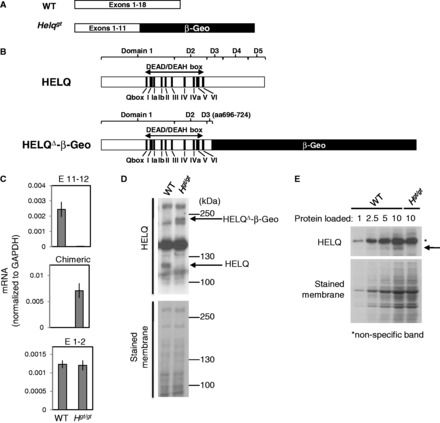
Characterization of the Helqgt allele. Diagrams (drawn to scale) depicting the transcripts (A) and resulting peptides (B) for the WT Helq and Helqgt alleles. The HELQ protein is split into five domains, the first two of which contain the well conserved motifs of the DEAD/DEAH helicase box. The Helqgt allele has most of domain 3 and all of domains 4 and 5 replaced by β-Geo. (C) qRT-PCR analysis using total RNA extracted from WT or Helqgt/gt (Hgt/gt) MEFs is shown. Data for the WT transcript containing exons 11 and 12 (top), the chimeric transcript containing exon 11 and the gene-trap vector sequence (middle) and transcript containing exons 1 and 2 upstream of the insertion site (bottom) are shown. Helqgt/gt MEFs have less than 1/100th of the levels of the WT Helq mRNA compared with WT cells (see top). Experiments were duplicated using RNA samples from different MEF lines to confirm reproducibility. A representative qRT-PCR data set is shown. (D) Western blotting shows no detectable levels of WT HELQ protein in Helqgt/gt MEFs. A band corresponding to the HELQΔ-β-Geo fusion peptide appears at the predicted molecular weight of ∼230 kDa only in the lysate from Helqgt/gt cells. (E) Loading differing ratios of protein reveals that the amount of WT HELQ protein (indicated by arrow) is extremely low in Helqgt/gt MEFs. Asterisk indicates non-specific bands. A stained membrane was used as a loading control for (D) and (E).
WT Helq mRNA and protein are virtually undetectable in Helqgt homozygous cells
Helqgt/+ mice were timed-mated to generate MEFs. We extracted total RNA from WT and Helqgt homozygous (Helqgt/gt) MEFs for reverse-transcription (RT)-PCR and verified the presence of the chimeric message in Helqgt/gt MEFs and the WT mRNA in WT MEFs (Figure 1A) by sequencing the RT-PCR products (Supplementary Figure S1C). Furthermore, quantitative (q-)RT-PCR on RNA from WT and Helqgt/gt cells revealed that the WT transcript containing exons 11–12 was nearly absent in Helqgt/gt cells (<1% of WT) that predominantly express the chimeric transcript containing exon 11 and β-Geo sequences (Figure 1C). As we found no difference between WT and Helqgt/gt cells for the levels of transcript containing exons 1–2 far upstream of the insertion site, the presence of the gene-trap vector likely has no effect on Helq expression. Next, we performed western blots on whole-cell extracts from WT and Helqgt/gt MEFs (Figure 1D). Consistent with the q-RT-PCR results, WT HELQ (∼120 kD) was undetectable in Helqgt/gt cells. Instead, they predominantly express the mutant HELQ protein (HELQΔ-β-Geo). A semi-quantitative analysis indicated that Helqgt/gt cells express WT HELQ protein <10% of the levels seen in WT cells, if any at all (Figure 1E). These data suggest that the splice acceptor site at the Helqgt allele is efficient, leading to little expression of normal full-length HELQ in Helqgt/gt cells.
Helq deficiency causes a mild form of hypogonadism, which is not epistatic to Fancc
As previous studies suggested an intriguing connection of HELQ to the FA pathway (23,24,27), we generated Helqgt/gt mice along with WT control mice to examine hypogonadism, one of the most consistent phenotypes seen in the FA mouse models (39). We found that Helqgt/gt males have significantly smaller testes (P < 0.005, t-test), ∼62% of WT males by weight at 6 weeks of age (0.165 ± 0.01 g and 0.102 ± 0.01 g for the average WT and Helqgt/gt testes weights, respectively, Figure 2A). Histological analysis revealed that ∼10–20% seminiferous tubules in Helqgt/gt males are atrophied and devoid of spermatocytes and spermatogonia at this stage (Figure 2B). This mosaic pattern of normal and empty seminiferous tubules is similar to what has been seen in a number of FA mouse models (40–48). For comparison, we also generated mice homozygous for a Fancc allele (Fancc−) (28) in the same background. As reported earlier (28,49,50), Fancc−/− males had extremely small testes weighing an average of only 0.027 ± 0.001 g (only 16% of WT by weight, P < 0.0001, t-test) with >90% of seminiferous tubules exhibiting atrophy or hypotrophy (Figure 2A and B). When compared with Helqgt/gt males, Fancc−/− testes were only 26% of Helqgt/gt testes by weight (P < 0.0001, t-test). These data suggest that the hypogonadism observed in Helqgt/gt testes is not as severe as in Fancc−/− testes. To test for an epistatic relationship between Helq and Fancc for this trait, we generated mice doubly homozygous for Helqgt and Fancc−. Testes from Helqgt/gt;Fancc−/− males were even smaller (average of 0.021 ± 0.002 g) than those from Fancc−/− males (P < 0.05, t-test), having all seminiferous tubules completely devoid of spermatogonia and spermatocytes. Although Fancc−/− and Helqgt/gt;Fancc−/− mice are significantly smaller in size than WT and Helqgt/gt mice, these observations still hold true after taking this into consideration (Supplementary Figures S2A and B). Collectively, these findings indicate that mutations in Helq and Fancc are not epistatic to each other in causing hypogonadism.
Figure 2.
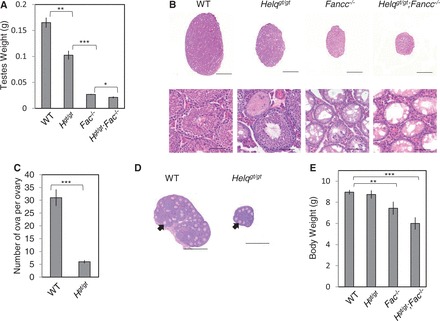
Helqgt/gt mice exhibit a hypogonadism phenotype reminiscent of mouse models of FA. (A) Helqgt/gt, Fancc−/− and Helqgt/gt;Fancc−/− males show significantly reduced testes weights at 6 weeks of age. At least five males were observed per genotype. (B) Histological analysis by hematoxylin and eosin staining shows a mosaic pattern of normal and empty seminiferous tubules in Helqgt/gt, Fancc−/− and Helqgt/gt;Fancc−/− mice, with the respective phenotypes becoming increasingly worse. Scale bars are 1500 µm for the whole testis sections (top) and 75 µm for the enlarged images (bottom). (C) Helqgt/gt females exhibit smaller ovaries with a reduced number of follicles at 3 weeks of age. Six ovaries from three WT females and 10 ovaries from five Helqgt/gt females were observed. (D) Hematoxylin and eosin staining of ovaries from WT and Helqgt/gt females. Example ova-containing follicles are indicated by arrows. Scale bars are 500 µm. (E) Helqgt/gt mice display normal body weights at weaning age, unlike Fancc−/− and Helqgt/gt;Fancc−/− mice, which are significantly smaller. In (A), (C) and (E), error bars represent the standard error of the means (SEMs) and significance was determined by t-test. Statistical significance at P < 0.05, P < 0.01, and P < 0.001 are indicated as *, ** and ***, respectively. WT, Hgt/gt, Fac−/− and Hgt/gt;Fac−/− refer to WT, Helqgt/gt, Fancc−/− and Helqgt/gt;Fancc−/−, respectively.
Female-specific sub-fertility in Helqgt/gt mice is consistent with germ cell hypoplasia during embryogenesis
It has been reported that hypogonadism in FA mouse models is attributed to severely compromised proliferation of primordial germ cells (40,41,45,51). This leads to sterility in a significant fraction of Fancc−/− mice (28,49). However, as hypogonadism in Helqgt/gt males is modest, they are fertile, producing litters at sizes comparable with Helqgt/+ males (Supplementary Table S1). Younger Helqgt/gt males have a greatly increased fraction (>50%) of seminiferous tubules exhibiting atrophy or hypotrophy (Supplementary Figure S2C). However, as they get older, the number of such tubules decreases. This is most likely because surviving germ cells in Helqgt/gt males can repopulate tubules as spermatogonial stem cells and support fertility as previously seen in FA mouse models (42). Therefore, much like the FA genes, Helq is required for normal proliferation of germ cells during embryogenesis but has no effect on spermatogonial stem cells at later stages. Different from males, Helqgt/gt females were more severely affected with hypogonadism (see Figure 2C and D). The number of ova per ovary was reduced to 6 ± 0.61 in Helqgt/gt females compared with 31 ± 3.2 in WT females (P < 0.0001, t-test). When initially tested in a 129/B6 mixed background, four of seven Helqgt/gt females were sterile. Fertile Helqgt/gt females tended to have small litters, with an average litter size of only 3.5 (n = 6). This female-specific sub-fertility is consistent with germ cell hypoplasia during embryogenesis, as it is believed that the total oocyte pool is determined at this stage. Taken together, although Helqgt/gt mice exhibit hypogonadsim that is phenotypically similar to FA mouse models, its underlying mechanism is distinct, given the non-epistatic relationship between Helq and Fancc.
Helqgt/gt mice are born in the expected Mendelian ratio, showing no growth retardation
As Helqgt/gt males are fertile, we performed crosses between Helqgt/gt;Fancc+/− males and Helqgt/+;Fancc+/− females to efficiently generate Helqgt/gt;Fancc−/− mice in the B6 background. A total of 105 mice were genotyped at 3 weeks of age (Supplementary Table S2). Although Helqgt/gt mice were found at the expected ratio, the number of Fancc−/− mice in this background was reduced to ∼65% of the expected number (17 versus 26.25, P < 0.05, χ2-test) as described previously (52). Only seven Helqgt/gt;Fancc−/− mice were observed at this age, but this number was not statistically different from 13.125, the expected number (P > 0.05, χ2-test). This relatively small number of Helqgt/gt;Fancc−/− mice is most likely attributed to the sub-lethality of Fancc−/− mice in this background. As shown in Figure 2E, we also found that Fancc−/− mice are significantly smaller in size (average weight of 7.4 g ± 0.61) compared with WT (8.7 ± 0.15 g) and Helqgt/gt mice (8.8 ± 0.39 g). Helqgt/gt;Fancc−/− mice were even smaller than Fancc−/− mice, weighing 6.0 g ± 0.71 on average, though this difference did not reach statistical significance likely due to the small number of mice examined. Overall, Helqgt/gt mice are healthy, exhibiting phenotypes milder than Fancc−/− mice. It was reported that Fancc−/− mice in the B6 background are essentially tumor-free (53). Similarly, a small-scale aging study with 11 Helqgt/gt mice (5 males and 6 females) in this background showed no significant increase in spontaneous tumor incidence up to 21 months of age. This was not surprising, given the milder phenotypes of Helqgt/gt mice compared with Fancc−/− mice.
Mono-ubiquitination and focus formation of FANCD2 are intact in Helqgt/gt cells
Our data so far did not support epistasis between Helq and Fancc. Therefore, we next tested the role of Helq in FANCD2 focus formation, a signature of FA pathway activation (25). For this purpose, we used primary WT and Helqgt/gt MEFs, as Fancc−/− and Helqgt/gt;Fancc−/− cells, both lacking a functional FA core complex, do not form FANCD2 foci (25). As shown in Figure 3A and B, no significant difference was observed in the percentage of cells positive for FANCD2 foci between WT and Helqgt/gt cells even after treatment with the crosslinker MMC. The same was true after treatment with aphidicolin (APH), a replication inhibitor and robust inducer of FANCD2 and FANCI foci (54) (Figure 3C and D). Focus formation of FANCD2/FANCI at prophase, markers of unresolved replication intermediates (55), were also unchanged between these two genotypes in response to MMC or APH (Supplementary Figure S3A and B). Agreeing with these data, mono-ubiquitination of FANCD2 was also robustly induced in Helqgt/gt cells following treatment with either MMC or APH (Figures 3E and F). In comparison, no mono-ubiquitinated FANCD2 could be detected in Fancc−/− and Helqgt/gt;Fancc−/− cells. These data collectively suggest that Helq is not required for FANCD2 mono-ubiquitination or focus formation.
Figure 3.
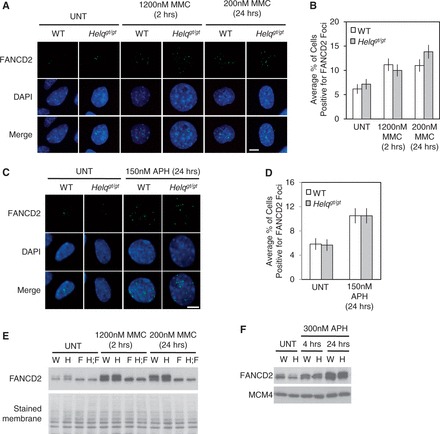
FANCD2 mono-ubiquitination and focus formation remain intact in Helqgt/gt cells. Shown are representative images of FANCD2 foci (green) in response to MMC (A) or APH (C) in WT and Helqgt/gt cells. Cells were harvested immediately following treatment except for the 2 h treatment of 1200 nM MMC, in which cells were given 22 h of recovery time before harvest. Nuclei were stained with DAPI (blue). Scale bar is 10 µm. The average percentages of cells positive for FANCD2 foci in response to MMC or APH are shown in (B) and (D), respectively. (E) Western blotting shows that Helqgt/gt cells exhibit normal FANCD2 mono-ubiquitination in response to MMC. The defect observed in Fancc−/− and Helqgt/gt;Fancc−/− cells is shown for a better comparison. (F) Helqgt/gt cells do not display any defect in FANCD2 mono-ubiquitination in response to APH. Cells were treated with 300 nM APH for either 4 or 24 h before harvest. Error bars (B, D) represent the binomial error for the combined data set. A stained membrane and MCM4 were used as loading controls in (E) and (F), respectively. W, H, F and H;F in (E, F) refer to WT, Helqgt/gt, Fancc−/− and Helqgt/gt;Fancc−/− cells, respectively. UNT refers to untreated cells.
HELQ plays a minor role in MMC resistance in a manner non-epistatic with FANCC
Previous studies reported that HELQ is required for ICL resistance in worms and humans (24,27) but not in chicken DT40 cells (14). Therefore, we tested Helqgt/gt cells for MMC hypersensitivity, another hallmark of FA cells (6). For this line of experiments, we used primary MEFs with the following four genotypes, WT, Helqgt/gt, Fancc−/− and Helqgt/gt;Fancc−/−. First, we examined MMC-induced chromosome aberrations. Representative metaphase spreads for each genotype after MMC treatment are shown in Figure 4A. Although we scored 120 metaphases per experimental group, the total number of MMC-induced chromosome aberrations was not statistically different between Helqgt/gt and WT cells (Figure 4B and C). However, Helqgt/gt cells did exhibit a slight but significant increase in radials (9.2 ± 2.6% as opposed to 5.0 ± 2.0% in WT), a type of complex chromosome aberration that occurs frequently in MMC-treated FA cells (Figure 4A and D). Consistent with their hypersensitivity to MMC, the majority of Fancc−/− cells exhibited chromosome aberrations (72.5 ± 4.09%, Figure 4B) with a drastic increase in radials (36.7 ± 4.42%, Figure 4D). Intriguingly, Helqgt/gt;Fancc−/− cells showed statistically higher levels of chromosome aberrations including radials (84.5 ± 3.35% and 46.7 ± 4.57% in Figure 4B and D, respectively) compared with Fancc−/− cells. These data suggest that Helqgt/gt cells are not as extremely sensitive to MMC as Fancc−/− cells. However, as Helqgt/gt;Fancc−/− cells showed a significantly higher number of MMC-induced chromosome aberrations than Fancc−/− cells, HELQ contributes to MMC resistance through a mechanism that is distinct from the function of FANCC. Given the mild sensitivity of Helqgt/gt cells to MMC, this mechanism is likely to be a secondary alternative to the FA pathway. Finally, we tested the effect of MMC on the proliferation of these cells using a MTT assay. In agreement with the metaphase analysis, a 5 days culture in multiple low doses of MMC significantly reduced the proliferation of Fancc−/− and Helqgt/gt;Fancc−/−, but not Helqgt/gt, cells (Figure 4E). Although Helqgt/gt;Fancc−/− cells appeared to display an even greater reduction in proliferation than Fancc−/− cells, this did not reach statistical significance. Together, these data are consistent with the idea of HELQ performing a minor role in MMC resistance.
Figure 4.
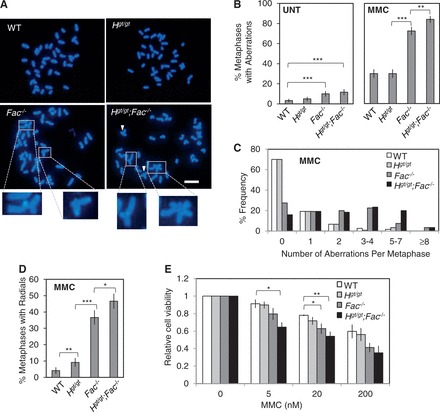
Helqgt/gt cells display modest sensitivity to MMC. (A) Shown are representative images of DAPI-stained metaphase spreads from all genotypes following MMC treatment (600 nM MMC for 2 h followed by 22 h recovery before harvest). White arrowheads indicate chromosome aberrations. Enlarged images of radial structures from the Fancc−/− and Helqgt/gt;Fancc−/− samples are shown at bottom. Scale bar is 10 µm. (B) Shown are the average percentages of metaphases positive for chromosomal aberrations in the untreated (left) or MMC-treated (right) conditions. For each experimental group, 120 metaphases were scored. (C) A histogram displaying the number of aberrations per metaphase for each genotype after MMC treatment is shown. (D) The average percentages of metaphases positive for radial structures after MMC treatment are shown. (E) The MTT assay reveals that Helqgt/gt cells display little, if any MMC sensitivity compared with Fancc−/− or Helqgt/gt;Fancc−/− cells. Cells were treated with the indicated doses of MMC for 5 days before analysis. Error bars show either the binomial error of the combined data set (B, D) or the SEMs for at least three independent experiments (E). Significance was determined by either χ2-test (B, D) or t-test (E). Statistical significance at P < 0.05, P < 0.01 and P < 0.001 is indicated as *, ** and ***, respectively. WT, Hgt/gt, Fac−/− and Hgt/gt;Fac−/− refer to WT, Helqgt/gt, Fancc−/− and Helqgt/gt;Fancc−/− respectively.
HELQ depletion leads to only mild MMC sensitivity compared with FANCA-depleted/FANCC-deficient human cells
Although Helqgt/gt cells apparently lack normal full-length HELQ, they do express the mutant HELQΔ-β-Geo protein. Therefore, we wondered whether the mild MMC sensitivity of Helqgt/gt cells truly reflects a consequence of HELQ deficiency. To test this possibility, we depleted HELQ and/or FANCA, another FA core complex member (56), in human HEK 293T cells via small interfering RNAs (siRNA, see Supplementary Figure S4A). At two different doses, FANCA depletion, but not HELQ depletion, led to MMC hypersensitivity as measured by colony formation assay (Supplementary Figure S4B). Similar results were obtained for MMC-induced chromosome aberrations (Supplementary Figures S4C and D). To further confirm these results, we also performed siRNA-mediated depletion of HELQ in PD331 cells (Supplementary Figure S4E), a human cell line deficient for FANCC, or their complemented counterparts (PD331 + FANCC). In a colony formation assay, HELQ-depleted PD331 + FANCC cells exhibited modestly decreased survival following MMC treatment (at 300 nM) compared with control siRNA-treated counterparts, though this was still mild compared with that of the PD331 cells (Supplementary Figure S4F). Only when measured by MTT assay (5 days culture, 15–45 nM doses) could we observe a clear non-epistatic relationship between HELQ and FANCC (Supplementary Figure S4G). Together, these data collectively support the idea that HELQ plays a minor, backup role in MMC resistance in mammalian cells that is most likely non-epistatic to FANCC.
A loss of HELQ and/or FANCC alters the distribution of replication fork speed in unperturbed S phase, increasing persistent stalled forks
Several studies have suggested that HELQ and its orthologs have a role in the recovery of stalled or collapsed replication forks (23,38,57). As we noticed a modest but significant increase in spontaneous chromosome aberrations in Fancc−/− and Helqgt/gt;Fancc−/− cells (Figure 4B), we examined replication fork speed in WT, Helqgt/gt, Fancc−/− and Helqgt/gt;Fancc−/− cells in unperturbed S phase using the DNA fiber technique (Supplementary Figure S5A) (29). Although the mean fork speeds were not different among the four genotypes (Figure 5A), the distributions of fork speed values were significantly different (P < 0.001, Kolmogorov–Smirnov test, Supplementary Figure S5B). Categorizing fork speeds into three ranges (slow, mid and fast), we found that compared with WT cells, Helqgt/gt cells exhibited a slight increase of forks in mid-range speed and a decrease in slow forks (Figure 5B). Fancc−/− cells had a higher frequency of faster forks with a decrease of mid-speed forks. Increases in mid-speed and faster forks in these cells might have contributed to faster median fork speeds in these cells (Figure 5A). Helqgt/gt;Fancc−/− cells displayed increases in both faster and slower fork speeds, indicating that their fork movement is greatly altered from WT cells even in unchallenged S phase. Although counterintuitive, an increase in faster forks may suggest an increase in fork stalling events, as we previously reported (29). This is due to the number of fork termination events. Faster forks that terminate within the duration of the assay likely escape detection. Thus, an increase in fork stalling leads to a decreased number of fork termination events, thereby leaving a greater number of faster forks visible for measurement. To address whether this was the case, we next looked at RPA32 foci, markers of stalled replication forks (Figure 5C) (58,59). Compared with WT cells (7.06 ± 0.61%), increased percentages of Helqgt/gt and Fancc−/− cells (9.20 ± 0.61% and 12.5 ± 0.88%, respectively) were positive for RPA32 foci (Figure 5D), with an even further increase observed in Helqgt/gt;Fancc−/− cells (15.6 ± 0.98%). These differences were all significant to one another (P < 0.001, χ2-test). The number of RPA32 foci co-localizing with γH2AX foci, a marker of DSBs (60), was also significantly increased in Helqgt/gt and Fancc−/− cells (5.46 ± 0.48% and 7.44 ± 0.70%, respectively) compared with WT cells (4.08 ± 0.47%). However, unlike RPA32 foci, there was no significant increase in RPA/γH2AX co-localizing foci in Helqgt/gt;Fancc−/− cells (8.02 ± 0.73%) compared with Fancc−/− cells. These data present a new line of evidence that Helq and Fancc are not epistatic to each other in the recovery of stalled/collapsed replication forks even in unchallenged S phase.
Figure 5.
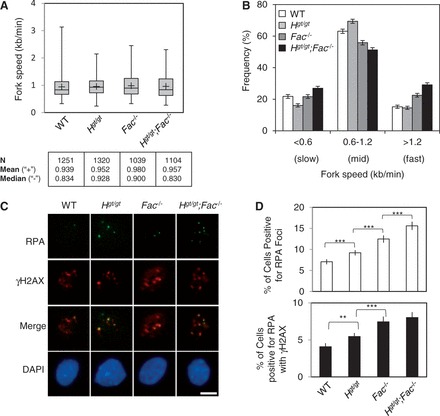
A combined loss of Helq and Fancc greatly alters the distribution of replication fork speeds and leads to increased levels of RPA/γH2AX foci. (A) Box plots show the range of fork speed values for the four genotypes. The line through the middle of the shaded box represents the median, whereas the ‘+’ sign shows the location of the mean (values shown at bottom along with the number of tracts, N, analyzed). (B) Separating fork speed values into slow, mid and fast forks reveals statistically significant differences among the ratios of the four genotypes (P < 0.001, χ2-test). Error bars show the binomial error. (C) Shown are representative images of cells from all four genotypes co-stained for RPA (green) and γH2AX (red). Nuclei were stained with DAPI (blue). For RPA foci analysis, cells were pre-extracted before fixation using a 0.5% Triton X-100 solution. Scale bar is 10 µm. (D) The average percentages of cells positive for RPA foci (top) or RPA/γH2AX co-localization events (bottom) are shown. Error bars show the binomial error for the combined data set. Statistical significance (determined by χ2-test) at P < 0.01 and P < 0.001 are indicated as ** and ***, respectively. WT, Hgt/gt, Fac−/− and Hgt/gt;Fac−/− refer to WT, Helqgt/gt, Fancc−/− and Helqgt/gt;Fancc−/−, respectively.
Helq and Fancc are independently required to prevent the formation of spontaneous MN
Persistent stalled forks can lead to the formation of micronuclei (MN) if unresolved before M phase entry (29,61). Therefore, we measured spontaneous MN levels using the cytokinesis-block MN assay (Figure 6A), a standard assay for this purpose (31). Compared with WT cells (4.33 ± 0.48%, see Figure 6B), significantly increased numbers of Helqgt/gt (9.56 ± 0.98%) and Fancc−/− (14.0 ± 1.16%) cells contained spontaneous MN (P < 0.001, χ2-test for both). Helqgt/gt;Fancc−/− cells exhibited a >6-fold increase (26.1 ± 1.47%) compared with WT cells, and this number was also statistically higher than that of Fancc−/− cells (P < 0.001, χ2-test). These data indicate that Helq and Fancc function in a non-epistatic manner to prevent spontaneous MN likely through contributing to the recovery of stalled forks. We exploited this modest but significant increase in spontaneous MN in Helqgt/gt cells to validate that this allele accurately reflects the consequences of loss of HELQ function. We performed siRNA-mediated knockdown of Helq (or Helqgt) transcripts in WT and Helqgt/gt MEFs, respectively (Supplementary Figures S6A and B). This resulted in significantly increased spontaneous MN levels in WT cells (P < 0.001, χ2-test) but not Helqgt/gt cells (Supplementary Figure S6C), suggesting that the HELQΔ-β-Geo mutant protein is likely devoid of any activity. Unresolved replication intermediates can cause the formation of MN through two mechanisms; non-disjunction of sister chromatids and chromosome/chromatid breaks. These two mechanisms can be distinguished by staining MN for the centromeric marker CENP-A (62) (Supplementary Figure S6D). We found that both types of MN were increased in these mutant cells with similar ratios (Supplementary Figure S6E). We also measured MMC-induced MN in these cells. Because of the relatively higher levels of spontaneous MN in mutant cells, we subtracted spontaneous MN frequency values from those in the MMC treatment to obtained differences (numbers in the white bars in Figure 6B). These were then compared to evaluate the effect of MMC on MN formation. Such values were similar between WT and Helqgt/gt cells (7.44 and 7.22, respectively), whereas Fancc−/− cells showed a 2-fold larger value (14.1) at the same dose used for the metaphase analysis (Figure 4). These findings are consistent with the idea that the FA pathway is a major pathway for MMC resistance for which HELQ performs only a minor role. Helqgt/gt;Fancc−/− cells showed a value (15.0) that was not much higher than that of Fancc−/− cells. We think this may be due to the extreme sensitivity of Helqgt/gt;Fancc−/− cells to MMC. As the majority of them have multiple abnormal metaphase chromosomes (Figure 4C), a fraction of them may not complete M phase to form MN in the subsequent G1 phase, making this value smaller. Unlike spontaneous conditions, MMC increased exclusively CENP-A- MN (Supplementary Figure S6E). We also examined CPT-induced MN (Supplementary Figure S6F). Both Helqgt/gt and Fancc−/− cells showed significantly increased levels of CPT-induced MN (14.2 and 14.2, respectively) compared with WT cells (9.93). The largest increase was observed in Helqgt/gt;Fancc−/− cells (16.2). However, we were unable to determine epistasis for this, due to the limited sensitivity of the MN assay.
Figure 6.
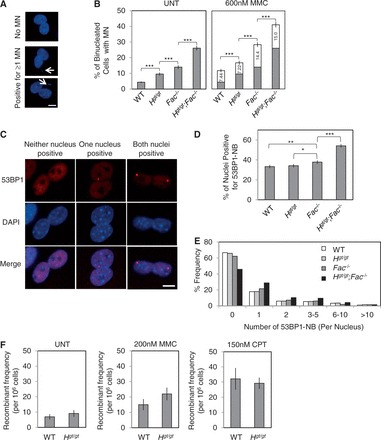
Helq suppresses multiple forms of spontaneous genome instability in a manner that is not epistatic with Fancc, but Helqgt/gt cells show levels of recombination comparable with WT. (A) Shown are representative images of binucleated cells with or without micronuclei (MN, indicated by white arrows). Nuclei and MN were stained with DAPI (blue). Scale bar is 10 µm. (B) Shown are the average percentages of binucleated cells positive for spontaneous MN (left) or MMC-induced MN (right). For the latter, the levels for the untreated condition are shown in gray with numbers in the white box showing the increase after MMC treatment. At least three independent experiments were performed and >900 cells were observed per experimental group. (C) Simultaneous disruption of Helq and Fancc results in a significant increase in 53BP1-NB. Shown are representative images of binucleated cells with or without nuclei positive for 53BP1-NB (red). Nuclei were stained with DAPI (blue). Scale bar is 10 µm. (D) Shown are the average percentages of nuclei positive for 53BP1-NB. (E) A histogram detailing the number of 53BP1-NB per nucleus is shown. Four independent experiments were performed and >750 cells were observed per experimental group. (F) No significant difference in the number of HR events was detected between WT and Helqgt/gt MEFs as measured by the FYDR transgenic locus system. Shown are the average numbers of eYFP+ recombinants per 106 cells analyzed. Scale bars in (B, D) show the binomial error of the combined data sets, wherease those in (F) show the SEMs for data obtained from at least seven different embryos per genotype. Statistical significance was determined by either χ2-test (B, D) or t-test (F). P < 0.05, P < 0.01 and P < 0.001 are indicated as*, ** and ***, respectively. WT, Hgt/gt, Fac−/− and Hgt/gt;Fac−/− refer to WT, Helqgt/gt, Fancc−/− and Helqgt/gt;Fancc−/−, respectively.
Helq and Fancc are not epistatic to suppress the formation of 53BP1 nuclear bodies
Recently, it was shown that unresolved replication intermediates can rupture during passage through M phase, leading to the formation of what are known as 53BP1 nuclear bodies (53BP1-NB) in G1 phase cells (63,64). Interestingly, such bodies are often exquisitely symmetrical in terms of their appearance within the daughter nuclei (Figure 6C). To score 53BP1-NB, G1 phase cells are typically identified as cyclin A negative (63,64). However, owing to the lack of a cyclin A antibody that works well for mouse cells, we used the cytokinesis-blocking reagent, cytochalasin B, to identify G1 phase daughter nuclei as those contained within binucleated cells (Figure 6C). Although there was no significant difference in the percentage of 53BP1-NB positive nuclei between WT and Helqgt/gt cells (33.3 ± 1.67% and 34.2 ± 1.68%, respectively), the number of nuclei containing 53BP1-NB was statistically higher in Fancc−/− cells (37.8 ± 1.71%, P < 0.01, χ2 test) (Figure 6D). Much like spontaneous MN formation (Figure 6B), Helqgt/gt;Fancc−/− cells showed a drastic increase in 53BP1-NB containing nuclei (54.1 ± 1.77%) compared with Fancc−/− cells (P < 0.001, χ2 test) with more nuclei containing multiple 53BP1-NB (Figure 6E). These data are consistent with the non-epistatic relationship between Helq and Fancc in preventing genome instability derived from persistent stalled forks.
Helqgt/gt cells display recombinant frequencies at the FYDR locus that are comparable with WT
Given its involvement in meiotic DSB repair in flies and worms (20,21), we postulated that HELQ may function downstream of the FA pathway in HR, similar to other proteins such as BRCA2 and PALB2 (65,66). Supporting this idea, it has been reported that depletion of HELQ in human cells lowers HR efficiency (27). To test this, we used the FYDR transgenic locus system (32) to measure the levels of spontaneous, MMC-induced and CPT-induced HR events in Helqgt/gt MEFs (Supplementary Figures S7A and B). Under all conditions tested, Helqgt/gt cells showed levels of recombination that were comparable with WT (Figure 6F and Supplementary Figure S7C), suggesting the possibility that HELQ is not a major player in HR.
DISCUSSION
In this study, we have investigated a possible involvement of HELQ in the FA pathway using the Helqgt strain as a model. We showed that primary Helqgt/gt MEFs are capable of FANCD2 mono-ubiquitination and focus formation (Figure 3). By phenotypic comparison to Fancc−/− mice/cells in the same genetic background, we found that Helqgt/gt mice/cells exhibit a mild form of FA-like phenotypes such as hypogonadism (Figure 2) and MMC sensitivity (Figure 4). Importantly, double mutants for Helqgt and Fancc− had more severe phenotypes than single mutants (Table 1). These findings are in stark contrast to the complete epistasis reported for Fanca/Fancc and Fanca/Fancg in mice (50,67). Collectively, our data show that Helq and Fancc are not epistatic to one another for any trait tested.
Table 1.
Summary of phenotypes following Helq and/or Fancc disruption
| Trait | WT | Helqgt/gt | Fancc−/− | Helqgt/gt; Fancc−/− |
|---|---|---|---|---|
| Sub-lethality | − | − | + | + |
| Growth retardation | − | − | + | ++? |
| Tumor | − | − | −(52) | N.D. |
| Hypogonadism | − | + | ++ | +++ |
| MMC sensitivity | − | +/− | ++ | +++ |
| MN formation | − | + | ++ | +++ |
| 53BP1-NB formation | − | − | + | ++ |
| HR (measured by FYDR) | − | − | N.D. | N.D. |
‘−’ indicates no significant change from WT, whereas ‘+’, ‘++’ and ‘+++’ refer to progressively more severe phenotypes. ‘+/−’ refers to a small but significant effect. N.D. means not determined.
Although our data strongly indicate that HELQ and FANCC function in parallel, it remains possible that HELQ could function in HR as a downstream step in the FA pathway. If this is the case, then the non-epistatic relationship between Helq and Fancc implicates that the FA core complex and HR machinery have a complicated non-linear relationship as seen previously for fancc and brca2 in chicken DT40 cells (68). However, using the FYDR transgenic locus system, we found that HELQ’s role in HR is likely to be non-essential or minor (Figure 6F). This is further supported by the fact that (i) Helqgt/gt mice are fully viable as opposed to early embryonic lethality seen for disruption of major HR genes (69), (ii) loss of HELQ results in only modest sensitivity to MMC (Figure 4), and (iii) Helqgt/gt males are fertile, showing no apparent meiosis defects. Alternatively, it may be that the role of HELQ in HR is only visible when a major player in HR is compromised. Thus, the precise role of HELQ in HR requires further investigation.
Our data show that loss of Helq in mice results in phenotypes considerably milder compared with those seen in Fancc−/− mice (Table 1). This observation was not limited to Helqgt homozygosity, as HELQ depletion in human HEK 293T cells did not cause a statistically higher level of MMC-induced chromosome aberrations or affect cellular survival as measured by colony formation assay (Supplementary Figure S4A–D). Furthermore, HELQ-depleted PD331 + FANCC cells exhibited only modestly reduced cellular survival at 300 nM MMC (colony formation assay) (Supplementary Figure S4F). Although our data show a consistent trend towards HELQ and FANCA/FANCC being non-epistatic in human cells, the minor role of HELQ in MMC resistance may have prevented it from being manifested as statistically significant, except in the MTT assay (Supplementary Figure S4G).
It is noteworthy that HELQ orthologs do not exist in bacteria or yeast but archaea have HELQ-like helicases (HELQa) (70). Atomic structures of HELQa from three species revealed the presence of five structural domains in HELQ, which are also conserved in HELQ in metazoans (36,38,71). Mutagenesis studies have demonstrated that normal helicase activity requires the three C-terminal domains (36,72), which are missing from HELQΔ-β-Geo in Helqgt/gt mice (Figure 1B). Therefore, it is likely that HELQΔ-β-Geo is also devoid of helicase activity. Furthermore, the presence of β-Geo is also likely to jeopardize its enzymatic activity. In line with this, siRNA-mediated knockdown of the Helqgt transcript in Helqgt/gt cells did not lead to increased levels of spontaneous MN (Supplementary Figure S6C). Furthermore, given the normal phenotypes of Helqgt/+ mice, we do not think that this allele confers any dominant-negative effect. Future studies using a null allele may give definitive answers for these issues.
Although there are definitely certain differences, HELQa and mammalian HELQ share similar biochemical properties with preference for structures resembling stalled replication forks, suggesting their role in stalled fork recovery (23,57,73). Consistent with this idea, loss of HELQ caused an increase in stalled forks even in unchallenged conditions, and this role of Helq was not epistatic to Fancc (Figure 5D). The non-epistatic relationship for Helq and Fancc is much clearer for the formation of spontaneous MN and 53BP1-NB (Figure 6), which are derived from persistent stalled forks, rather than MMC-induced chromosome aberrations (Figure 4). Therefore, we propose that the major role of HELQ is the rescue of stalled forks in normal S phase. Given that HELQ and the FA core complex function in parallel, elucidating such a role of HELQ may provide clues to decipher the function of the FA pathway in physiological conditions beyond ICL repair. Furthermore, as HELQ remains functional in FA mutant cells, it could potentially be exploited to provide a therapeutic benefit against cancers with FA pathway disruption.
Recent genome-wide association studies have identified single nucleotide polymorphisms at loci within or near HELQ that are associated with increased risks for several different cancers including upper aerodigestive tract cancers and head and neck cancers (74–77). Our study was unable to detect an increased incidence of spontaneous tumors in Helqgt/gt mice. This could be due to several different factors, such as genetic background and species difference. The majority of FA mouse models do not show a strong cancer phenotype, despite the FA pathway’s tumor suppressive role in humans (39). Therefore, it may be necessary to test the role of Helq in tumor suppression in a sensitized background.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
A postdoctoral fellowship from the Japan Society for the promotion of Science (to T.K.); March of Dimes [5-FY07-110 to N.S.]; and National Institutes of Health (NIH) [R01CA148806 to N.S.]. Funding for open access charge: University of Minnesota.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr John Schimenti’s Laboratory for initial support for this study, Dr Markus Grompe for providing them with the Fancc− strain, Dr Bevin Engelward for providing the FYDR strain, Dr Alexandra Sobeck for her critical reading of this manuscript and Drs Hung-Ji Tsai and Matt Anderson for their assistance with qRT-PCR experiments.
REFERENCES
- 1.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell. Biol. 2010;11:208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 3.Blow JJ, Ge XQ. A model for DNA replication showing how dormant origins safeguard against replication fork failure. EMBO Rep. 2009;10:406–412. doi: 10.1038/embor.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell. Biol. 2010;11:683–687. doi: 10.1038/nrm2974. [DOI] [PubMed] [Google Scholar]
- 5.Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell. Biol. 2012;13:141–152. doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim H, D'Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–1408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crossan GP, Patel KJ. The Fanconi anaemia pathway orchestrates incisions at sites of crosslinked DNA. J. Pathol. 2012;226:326–337. doi: 10.1002/path.3002. [DOI] [PubMed] [Google Scholar]
- 9.Kee Y, D'Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010;24:1680–1694. doi: 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marini F, Wood RD. A human DNA helicase homologous to the DNA cross-link sensitivity protein Mus308. J. Biol. Chem. 2002;277:8716–8723. doi: 10.1074/jbc.M110271200. [DOI] [PubMed] [Google Scholar]
- 11.Boyd JB, Sakaguchi K, Harris PV. mus308 mutants of Drosophila exhibit hypersensitivity to DNA cross-linking agents and are defective in a deoxyribonuclease. Genetics. 1990;125:813–819. doi: 10.1093/genetics/125.4.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leonhardt EA, Henderson DS, Rinehart JE, Boyd JB. Characterization of the mus308 gene in Drosophila melanogaster. Genetics. 1993;133:87–96. doi: 10.1093/genetics/133.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shima N, Munroe RJ, Schimenti JC. The mouse genomic instability mutation chaos1 is an allele of Polq that exhibits genetic interaction with Atm. Mol. Cell. Biol. 2004;24:10381–10389. doi: 10.1128/MCB.24.23.10381-10389.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshimura M, Kohzaki M, Nakamura J, Asagoshi K, Sonoda E, Hou E, Prasad R, Wilson SH, Tano K, Yasui A, et al. Vertebrate POLQ and POLbeta cooperate in base excision repair of oxidative DNA damage. Mol. Cell. 2006;24:115–125. doi: 10.1016/j.molcel.2006.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yousefzadeh MJ, Wood RD. DNA polymerase POLQ and cellular defense against DNA damage. DNA Repair (Amst.) 2013;12:1–9. doi: 10.1016/j.dnarep.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris PV, Mazina OM, Leonhardt EA, Case RB, Boyd JB, Burtis KC. Molecular cloning of Drosophila mus308, a gene involved in DNA cross-link repair with homology to prokaryotic DNA polymerase I genes. Mol. Cell. Biol. 1996;16:5764–5771. doi: 10.1128/mcb.16.10.5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seki M, Marini F, Wood RD. POLQ (Pol theta), a DNA polymerase and DNA-dependent ATPase in human cells. Nucleic Acids Res. 2003;31:6117–6126. doi: 10.1093/nar/gkg814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shima N, Hartford SA, Duffy T, Wilson LA, Schimenti KJ, Schimenti JC. Phenotype-based identification of mouse chromosome instability mutants. Genetics. 2003;163:1031–1040. doi: 10.1093/genetics/163.3.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laurencon A, Orme CM, Peters HK, Boulton CL, Vladar EK, Langley SA, Bakis EP, Harris DT, Harris NJ, Wayson SM, et al. A large-scale screen for mutagen-sensitive loci in Drosophila. Genetics. 2004;167:217–231. doi: 10.1534/genetics.167.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCaffrey R, St Johnston D, Gonzalez-Reyes A. Drosophila mus301/spindle-C encodes a helicase with an essential role in double-strand DNA break repair and meiotic progression. Genetics. 2006;174:1273–1285. doi: 10.1534/genetics.106.058289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ward JD, Muzzini DM, Petalcorin MI, Martinez-Perez E, Martin JS, Plevani P, Cassata G, Marini F, Boulton SJ. Overlapping mechanisms promote postsynaptic RAD-51 filament disassembly during meiotic double-strand break repair. Mol. Cell. 2010;37:259–272. doi: 10.1016/j.molcel.2009.12.026. [DOI] [PubMed] [Google Scholar]
- 22.Marini F, Kim N, Schuffert A, Wood RD. POLN, a nuclear PolA family DNA polymerase homologous to the DNA cross-link sensitivity protein Mus308. J. Biol. Chem. 2003;278:32014–32019. doi: 10.1074/jbc.M305646200. [DOI] [PubMed] [Google Scholar]
- 23.Tafel AA, Wu L, McHugh PJ. Human HEL308 localizes to damaged replication forks and unwinds lagging strand structures. J. Biol. Chem. 2011;286:15832–15840. doi: 10.1074/jbc.M111.228189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muzzini DM, Plevani P, Boulton SJ, Cassata G, Marini F. Caenorhabditis elegans POLQ-1 and HEL-308 function in two distinct DNA interstrand cross-link repair pathways. DNA Repair (Amst.) 2008;7:941–950. doi: 10.1016/j.dnarep.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 26.Youds JL, Barber LJ, Boulton SJ. C. elegans: A model of Fanconi anemia and ICL repair. Mutat. Res. 2009;668:103–116. doi: 10.1016/j.mrfmmm.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 27.Moldovan GL, Madhavan MV, Mirchandani KD, McCaffrey RM, Vinciguerra P, D'Andrea AD. DNA polymerase POLN participates in cross-link repair and homologous recombination. Mol. Cell. Biol. 2010;30:1088–1096. doi: 10.1128/MCB.01124-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitney MA, Royle G, Low MJ, Kelly MA, Axthelm MK, Reifsteck C, Olson S, Braun RE, Heinrich MC, Rathbun RK, et al. Germ cell defects and hematopoietic hypersensitivity to gamma-interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood. 1996;88:49–58. [PubMed] [Google Scholar]
- 29.Kawabata T, Luebben SW, Yamaguchi S, Ilves I, Matise I, Buske T, Botchan MR, Shima N. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol. Cell. 2011;41:543–553. doi: 10.1016/j.molcel.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugimura K, Takebayashi S, Ogata S, Taguchi H, Okumura K. Non-denaturing fluorescence in situ hybridization to find replication origins in a specific genome region on the DNA fiber. Biosci. Biotechnol. Biochem. 2007;71:627–632. doi: 10.1271/bbb.60662. [DOI] [PubMed] [Google Scholar]
- 31.Fenech M. Cytokinesis-block micronucleus cytome assay. Nat. Protoc. 2007;2:1084–1104. doi: 10.1038/nprot.2007.77. [DOI] [PubMed] [Google Scholar]
- 32.Hendricks CA, Almeida KH, Stitt MS, Jonnalagadda VS, Rugo RE, Kerrison GF, Engelward BP. Spontaneous mitotic homologous recombination at an enhanced yellow fluorescent protein (EYFP) cDNA direct repeat in transgenic mice. Proc. Natl Acad. Sci. USA. 2003;100:6325–6330. doi: 10.1073/pnas.1232231100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stryke D, Kawamoto M, Huang CC, Johns SJ, King LA, Harper CA, Meng EC, Lee RE, Yee A, L'Italien L, et al. BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 2003;31:278–281. doi: 10.1093/nar/gkg064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stanford WL, Cohn JB, Cordes SP. Gene-trap mutagenesis: past, present and beyond. Nat. Rev. Genet. 2001;2:756–768. doi: 10.1038/35093548. [DOI] [PubMed] [Google Scholar]
- 35.Fujikane R, Komori K, Shinagawa H, Ishino Y. Identification of a novel helicase activity unwinding branched DNAs from the hyperthermophilic archaeon, Pyrococcus furiosus. J. Biol. Chem. 2005;280:12351–12358. doi: 10.1074/jbc.M413417200. [DOI] [PubMed] [Google Scholar]
- 36.Buttner K, Nehring S, Hopfner KP. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat. Struct. Mol. Biol. 2007;14:647–652. doi: 10.1038/nsmb1246. [DOI] [PubMed] [Google Scholar]
- 37.Woodman IL, Briggs GS, Bolt EL. Archaeal Hel308 domain V couples DNA binding to ATP hydrolysis and positions DNA for unwinding over the helicase ratchet. J. Mol. Biol. 2007;374:1139–1144. doi: 10.1016/j.jmb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 38.Richards JD, Johnson KA, Liu H, McRobbie AM, McMahon S, Oke M, Carter L, Naismith JH, White MF. Structure of the DNA repair helicase hel308 reveals DNA binding and autoinhibitory domains. J. Biol. Chem. 2008;283:5118–5126. doi: 10.1074/jbc.M707548200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parmar K, D'Andrea A, Niedernhofer LJ. Mouse models of Fanconi anemia. Mutat. Res. 2009;668:133–140. doi: 10.1016/j.mrfmmm.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong JC, Alon N, McKerlie C, Huang JR, Meyn MS, Buchwald M. Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum. Mol. Genet. 2003;12:2063–2076. doi: 10.1093/hmg/ddg219. [DOI] [PubMed] [Google Scholar]
- 41.Agoulnik AI, Lu B, Zhu Q, Truong C, Ty MT, Arango N, Chada KK, Bishop CE. A novel gene, Pog, is necessary for primordial germ cell proliferation in the mouse and underlies the germ cell deficient mutation, gcd. Hum. Mol. Genet. 2002;11:3047–3053. doi: 10.1093/hmg/11.24.3047. [DOI] [PubMed] [Google Scholar]
- 42.Lu B, Bishop CE. Late onset of spermatogenesis and gain of fertility in POG-deficient mice indicate that POG is not necessary for the proliferation of spermatogonia. Biol. Reprod. 2003;69:161–168. doi: 10.1095/biolreprod.102.014654. [DOI] [PubMed] [Google Scholar]
- 43.Yang Y, Kuang Y, Montes De Oca R, Hays T, Moreau L, Lu N, Seed B, D'Andrea AD. Targeted disruption of the murine Fanconi anemia gene, Fancg/Xrcc9. Blood. 2001;98:3435–3440. doi: 10.1182/blood.v98.12.3435. [DOI] [PubMed] [Google Scholar]
- 44.Crossan GP, van der Weyden L, Rosado IV, Langevin F, Gaillard PH, McIntyre RE, Gallagher F, Kettunen MI, Lewis DY, Brindle K, et al. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat. Genet. 2011;43:147–152. doi: 10.1038/ng.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holloway JK, Mohan S, Balmus G, Sun X, Modzelewski A, Borst PL, Freire R, Weiss RS, Cohen PE. Mammalian BTBD12 (SLX4) protects against genomic instability during mammalian spermatogenesis. PLoS Genet. 2011;7:e1002094. doi: 10.1371/journal.pgen.1002094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bakker ST, van de Vrugt HJ, Rooimans MA, Oostra AB, Steltenpool J, Delzenne-Goette E, van der Wal A, van der Valk M, Joenje H, te Riele H, et al. Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum. Mol. Genet. 2009;18:3484–3495. doi: 10.1093/hmg/ddp297. [DOI] [PubMed] [Google Scholar]
- 47.Bakker ST, van de Vrugt HJ, Visser JA, Delzenne-Goette E, van der Wal A, Berns MA, van de Ven M, Oostra AB, de Vries S, Kramer P, et al. Fancf-deficient mice are prone to develop ovarian tumours. J. Pathol. 2011;226:28–39. doi: 10.1002/path.2992. [DOI] [PubMed] [Google Scholar]
- 48.Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, Grompe M. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev. 2003;17:2021–2035. doi: 10.1101/gad.1103403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen M, Tomkins DJ, Auerbach W, McKerlie C, Youssoufian H, Liu L, Gan O, Carreau M, Auerbach A, Groves T, et al. Inactivation of Fac in mice produces inducible chromosomal instability and reduced fertility reminiscent of Fanconi anaemia. Nat. Genet. 1996;12:448–451. doi: 10.1038/ng0496-448. [DOI] [PubMed] [Google Scholar]
- 50.Noll M, Battaile KP, Bateman R, Lax TP, Rathbun K, Reifsteck C, Bagby G, Finegold M, Olson S, Grompe M. Fanconi anemia group A and C double-mutant mice: functional evidence for a multi-protein Fanconi anemia complex. Exp. Hematol. 2002;30:679–688. doi: 10.1016/s0301-472x(02)00838-x. [DOI] [PubMed] [Google Scholar]
- 51.Nadler JJ, Braun RE. Fanconi anemia complementation group C is required for proliferation of murine primordial germ cells. Genesis. 2000;27:117–123. doi: 10.1002/1526-968x(200007)27:3<117::aid-gene40>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 52.Carreau M. Not-so-novel phenotypes in the Fanconi anemia group D2 mouse model. Blood. 2004;103:2430. doi: 10.1182/blood-2003-11-3946. [DOI] [PubMed] [Google Scholar]
- 53.Freie B, Li X, Ciccone SL, Nawa K, Cooper S, Vogelweid C, Schantz L, Haneline LS, Orazi A, Broxmeyer HE, et al. Fanconi anemia type C and p53 cooperate in apoptosis and tumorigenesis. Blood. 2003;102:4146–4152. doi: 10.1182/blood-2003-03-0971. [DOI] [PubMed] [Google Scholar]
- 54.Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum. Mol. Genet. 2005;14:693–701. doi: 10.1093/hmg/ddi065. [DOI] [PubMed] [Google Scholar]
- 55.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell. Biol. 2009;11:753–760. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 56.Foe JR, Rooimans MA, Bosnoyan-Collins L, Alon N, Wijker M, Parker L, Lightfoot J, Carreau M, Callen DF, Savoia A, et al. Expression cloning of a cDNA for the major Fanconi anaemia gene, FAA. Nat. Genet. 1996;14:488. doi: 10.1038/ng1296-488. [DOI] [PubMed] [Google Scholar]
- 57.Guy CP, Bolt EL. Archaeal Hel308 helicase targets replication forks in vivo and in vitro and unwinds lagging strands. Nucleic Acids Res. 2005;33:3678–3690. doi: 10.1093/nar/gki685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 59.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 61.Naim V, Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell. Biol. 2009;11:761–768. doi: 10.1038/ncb1883. [DOI] [PubMed] [Google Scholar]
- 62.Howman EV, Fowler KJ, Newson AJ, Redward S, MacDonald AC, Kalitsis P, Choo KH. Early disruption of centromeric chromatin organization in centromere protein A (Cenpa) null mice. Proc. Natl Acad. Sci. USA. 2000;97:1148–1153. doi: 10.1073/pnas.97.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell. Biol. 2011;13:243–253. doi: 10.1038/ncb2201. [DOI] [PubMed] [Google Scholar]
- 64.Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011;193:97–108. doi: 10.1083/jcb.201011083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 66.Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 2007;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 67.van de Vrugt HJ, Koomen M, Bakker S, Berns MA, Cheng NC, van der Valk MA, de Vries Y, Rooimans MA, Oostra AB, Hoatlin ME, et al. Evidence for complete epistasis of null mutations in murine Fanconi anemia genes Fanca and Fancg. DNA Repair (Amst.) 2011;10:1252–1261. doi: 10.1016/j.dnarep.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 68.Kitao H, Yamamoto K, Matsushita N, Ohzeki M, Ishiai M, Takata M. Functional interplay between BRCA2/FancD1 and FancC in DNA repair. J. Biol. Chem. 2006;281:21312–21320. doi: 10.1074/jbc.M603290200. [DOI] [PubMed] [Google Scholar]
- 69.Friedberg EC, Meira LB. Database of mouse strains carrying targeted mutations in genes affecting biological responses to DNA damage Version 7. DNA Repair (Amst.) 2006;5:189–209. doi: 10.1016/j.dnarep.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 70.Woodman IL, Bolt EL. Molecular biology of Hel308 helicase in archaea. Biochem. Soc. Trans. 2009;37:74–78. doi: 10.1042/BST0370074. [DOI] [PubMed] [Google Scholar]
- 71.Oyama T, Oka H, Mayanagi K, Shirai T, Matoba K, Fujikane R, Ishino Y, Morikawa K. Atomic structures and functional implications of the archaeal RecQ-like helicase Hjm. BMC Struct. Biol. 2009;9:2. doi: 10.1186/1472-6807-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang C, Tian B, Li S, Ao X, Dalgaard K, Gokce S, Liang Y, She Q. Genetic manipulation in Sulfolobus islandicus and functional analysis of DNA repair genes. Biochem. Soc. Trans. 2013;41:405–410. doi: 10.1042/BST20120285. [DOI] [PubMed] [Google Scholar]
- 73.Fujikane R, Shinagawa H, Ishino Y. The archaeal Hjm helicase has recQ-like functions, and may be involved in repair of stalled replication fork. Genes Cells. 2006;11:99–110. doi: 10.1111/j.1365-2443.2006.00925.x. [DOI] [PubMed] [Google Scholar]
- 74.Li WQ, Hu N, Hyland PL, Gao Y, Wang ZM, Yu K, Su H, Wang CY, Wang LM, Chanock SJ, et al. Genetic variants in DNA repair pathway genes and risk of esophageal squamous cell carcinoma and gastric adenocarcinoma in a Chinese population. Carcinogenesis. 2013;34:1536–1542. doi: 10.1093/carcin/bgt094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao Y, He Y, Xu J, Xu L, Du J, Zhu C, Gu H, Ma H, Hu Z, Jin G, et al. Genetic variants at 4q21, 4q23 and 12q24 are associated with esophageal squamous cell carcinoma risk in a Chinese population. Hum. Genet. 2013;132:649–656. doi: 10.1007/s00439-013-1276-5. [DOI] [PubMed] [Google Scholar]
- 76.Liang C, Marsit CJ, Houseman EA, Butler R, Nelson HH, McClean MD, Kelsey KT. Gene-environment interactions of novel variants associated with head and neck cancer. Head Neck. 2012;34:1111–1118. doi: 10.1002/hed.21867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McKay JD, Truong T, Gaborieau V, Chabrier A, Chuang SC, Byrnes G, Zaridze D, Shangina O, Szeszenia-Dabrowska N, Lissowska J, et al. A genome-wide association study of upper aerodigestive tract cancers conducted within the INHANCE consortium. PLoS Genet. 2011;7:e1001333. doi: 10.1371/journal.pgen.1001333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.