

Cell migration is an essential process in which physical and chemical cues are provided by the extracellular matrix (ECM) (Lauffenburger and Horwitz, 1996; Petrie et al., 2009). Contact guidance from topographical features (Doyle et al., 2009; Teixeira et al., 2003) or three-dimensional (3D) microenvironments (Dallon et al., 1999; Dickinson et al., 1994; Guido and Tranquillo, 1993; Provenzano et al., 2008a) is the phenomena by which the ECM provides directional cues that influence cell orientation and migration direction. In 3D collagen matrices, contact guidance from collagen fibers regulates cell migration via anisotropy in the local microenvironment (Dickinson et al., 1994; Provenzano et al., 2008a), with cells migrating preferentially along aligned collagen fibers.
In this unit, basic methods for engineering 3D collagen matrices to generate contact guidance cues are presented. The matrices closely resemble architectures observed in vivo (Provenzano et al., 2006). Utilizing in vivo-mimicking ECM conditions facilitates the acquisition of physiologically relevant information, while permitting manipulation of the system within a manageable experimental window. Therefore, to achieve this, two basic methods for generating 3D migration/invasion assays with defined architecture are described. The first system, described in Basic Protocol 1 makes use of placing cell-seeded collagen gels (CSCGs) or tissue explants into collagen matrices in a defined manner to create collagen architectures via cell contraction-mediated collagen reorganization. The second basic protocol (Basic Protocol 2) describes methods to align collagen matrices during matrix polymerization using a magnetic field. The aligned matrices may then be oriented and combined to develop 3D migration assays in which the cells face different contact guidance cues.
NOTE: All solutions and equipment used during the protocols must be sterile for use with cell culture. Likewise, all procedures should be performed in a sterile tissue culture hood with proper aseptic technique used throughout the protocols.
NOTE: Collagen matrix polymerization times should be optimized for each individuals’ laboratory conditions. Moreover, parameters may need to be adjusted to accommodate different cell types, experimental designs and collagen matrix concentration.
BASIC PROTOCOL 1: PREPARATION OF A 3D MIGRATION ASSAY USING NESTED CELL-SEEDED COLLAGEN GELS
Cell-seeded collagen gels (CSCGs) placed into an encasing matrix will contact and reorganize the matrix to generate regions of collagen alignment and regions of non-alignment that provide contact guidance cues for motile cells. In this assay, cells are seeded into collagen matrices to make plugs that are then inserted into an encasing collagen matrix. Over time, cells reorganize the matrix and invade into the newly organized encasing matrix.
Materials
Sub-confluent epithelial or fibroblast cells on a standard tissue culture dish/flask
Culture media of choice for the chosen cell type
Trypsin/EDTA solution (Cellgro)
Rat tail collagen (BDBiosciences)
0.1 M HEPES buffer in 2X PBS
96-well ultra-low adhesion culture plate (Corning)
6-well culture dish(s) (Corning ultra-low attachment plates work very well, but other more standard plates may be substituted as needed).
P1000 pipettes with the tips cut off
27 gauge syringe needle to help detach CSCGs
Detach and count cells
-
1
Aspirate the cell culture medium from the culture dish containing sub-confluent cells. Rinse the cells with PBS or serum-free culture media to remove residual serum. Aspirate buffer/media from the dish.
-
2
Add 2–3 ml of trypsin/EDTA solution to the cell dish and incubate for ~3–5 minutes (or until sufficient cell detachment is achieved) at room temperature. Gentle agitation of the dish may be necessary to detach cells.
Note that the concentration of the trypsin/EDTA solution may depend on the chosen cell line and the volume of trypsin/EDTA that is necessary will depend on the size of the culture dish. In general, the lowest concentration and volume of trypsin/EDTA solution that results in sufficient cell detachment within 10 minutes should be used. Furthermore, any cell culture reagent that adequately detaches the cells of interest may be substituted for trypsin/EDTA solution. -
3
After the cells detach from dish, transfer the cell-containing trypsin/EDTA solution to a 15 ml screw cap conical tube and add 2 volumes of culture media for every one volume of trypsin/EDTA solution to the tube. Centrifuge the tube to pellet the cells.
-
4
Discard the supernatant and resuspend the cells in 1–2 ml of culture media. Count the cells.
Prepare Cell Seeded Collagen Gels (CSCGs)
-
5
Determine the volume of cell-containing media that contains at least 150,000, but no more than 500,000 cells.
The number of cells per CSCG may be adjusted for cell type and experimental design, but in general fewer than 150,000 cells does not adequately fill the CSCG with cells, while cell numbers greater than 500,000 cells/CSCG are commonly too cell dense. -
6
Calculate the volume of collagen mixture needed for the experiment.
The amount of collagen added will depend on the desired final concentration of the collagen matrix and the initial concentration (in mg/ml) of the collagen solution. The volume of neutralizing HEPES buffer solution will be equal to the volume of collagen solution. Add regular culture medium to achieve the final volume. In our hands, 1–2 mg/ml collagen matrices have worked very well for MDA-MB-231 and T47D breast carcinoma cells. Therefore, as an example, to make a 2 mg/ml collagen matrix from a 10 mg/ml initial stock of collagen, add 200 μl of collagen solution, 200 μl of HEPES buffer solution, and 600 μl of culture medium for every ml of matrix solution needed. The 600 μl of culture medium includes the volume of culture medium needed to add the correct number of cells. For each CSCG, 150–250 μl (150 μl CSCGs work very well but larger volumes may used to accommodate more cells, i.e. 250 μl for 500,00 cells) of the collagen/HEPES/media mixture is added to a well within the 96-well plate.Since the collagen solution is very viscous, pipette very carefully, allowing adequate time for the collagen solution to enter and exit the pipette. After releasing the collagen solution from the pipette allow the remaining volume to pool in at the bottom of the pipette tip and then clear the pipette again. In addition, make extra solution to ensure that enough CSCGs can be poured for the experiment. -
7
In a new conical tube add the calculated volume of collagen solution to an equal volume of 100 mM HEPES buffer (in 2X PBS). Add culture medium and culture medium containing cells to achieve the correct final volume and cell number per CSCG.
All solutions should be kept on ice until the mixture is pipetted to the culture plate. -
8
Pipette 150–250 μl of collagen mixture (containing cells as described in step 6) to an individual well in the 96-well culture plate.
-
9
Allow the collagen matrix to polymerize for 20 minutes at room temperature, then at least 2 but not more than 16 hours in a cell culture incubator at 37°C (Figure 1a).
Figure 1. Key steps in Basic Protocol 1.

(a) Polymerized CSCGs in a 96-well plate. (b) Detach CSCGs from the culture dish wall. (c) P1000 pipette with the tip cut off. (d) Transfer the CSCG from the culture well with care taken not to pull the CSCG into the pipette. (e) Place the CSCG into a culture plate. (f) Float the CSCG in culture media with care not to allow multiple CSCGs to contact one another. (g–j) Place CSCGs into the encasing matrix.
Implant CSCGs into the encasing matrix
-
10
Gently detach the CSCG from the sides of the culture plate well using a fine needle, such as 28 gauge syringe needle. Insert the needle at the side of the CSCG, touching the well wall, and while maintaining contact with the wall, move the needle along the perimeter of the CSCG (Figure 1b). Gently shake and tap the plate to help detach the CSCGs from the culture well surfaces.
-
11
Using a P1000 pipette with the tip cut off to increase the opening diameter (Figure 1c), gently pull the CSCG up out of the culture well. The CSCG should never enter the pipette. The CSCG should remain suspended below the entrance to the pipette tip (Figure 1d).
-
12
Suspend the CSCGs in culture medium for 20 minutes to allow the matrix to contract. Allow ample room in the culture plate for the CSCGs to float without coming into contact with one another (Figure 1e-f).
-
13
During step 12 make an additional collagen mixture for the encasing matrix. For each well of a 6-well tissue culture plate, 1.5 ml of 2 mg/ml collagen mixture is needed.
-
14
Add 1.5 ml of the collagen mixture to a well of the 6-well plate. Allow the collagen matrix to polymerize for ~10–15 minutes at room temperature.
This step helps ensure that the CSCG will not directly rest on the bottom of the culture dish and attach to the bottom surface. By placing the CSCG into a matrix that is just starting to polymerize, and is therefore less fluid, the CSCG becomes suspended in the encasing matrix. -
15
Place the CSCGs into the encasing matrix, making sure that the CSCGs does not sink to the bottom of the culture dish. The CSCGs are placed 7 mm apart (from edge to edge) along the maximal diameter midline of the well (Figure 1g–j). A custom reference grid may be placed under the well to help facilitate placement.
-
16
Allow the collagen matrix to polymerize undisturbed at room temperature for at least 20 minutes. After the matrix has sufficiently polymerized to allow movement of the plate without disturbing the CSCG placement, move the plate to the cell culture incubator at 37°C.
-
17
Sixteen to twenty four hours after matrix construction, detach the encasing matrix and float the matrix in cell culture medium.
Detach the matrix from the culture dish by gently inserting a small pipette tip on the side of the matrix (while touching the well wall with the pipette tip). Gently move the tip around the perimeter of the matrix while maintaining contact with the well wall. Place the pipette tip at the side of the well and gently pipette 2 ml of media into the well, under the matrix. Gently rotate the plate to detach the matrices and float them in the media. Extreme care must be taken during this step so that the CSCGs do not tear away from the encasing matrix. -
18
Incubate the nested CSCGs for one to several days changing the media every 3 days or for more even stimulation change 25% of the media daily.
We find that collagen alignment can be observed within the first 24 hours, but that collagen alignment is maintained and enhanced over 6 to 10 days. -
19
Analyze 3D cell migration/invasion with optical imaging (see Support Protocols)
Note: A single plug nested assay can be employed as well (Grinnell et al., 2006; Miron-Mendoza et al., 2008; Provenzano et al., 2006), but does not provide the consistent contact guidance cues as the alignment is more distributed in random locations around the plug. However, this assay still provides a very good 3D migration assay, particularly for cancer biology studies where the CSCG can simulate an epithelial mass where carcinoma cells invade into the collagenous stroma. Likewise, more than two CSCGs can be employed to create more complex architectures.
BASIC PROTOCOL 2: PREPARATION OF ALIGNED COLLAGEN MATRICES TO CONSTRUCT A 3D MIGRATION ASSAY WITH DEFINED CONTACT GUIDANCE CUES
One limitation of the nested-plug assay, above, is that it does not readily allow separation of signaling events that lead to an aligned matrix, from those necessary to migrate along a matrix that is already aligned. Here we describe an approach to create a pre-aligned collagen matrix (as presented in Provenzano et al (2008) and adapted from Guo and Kaufman (Guo and Kaufman, 2007), which provides cells with contact guidance cues from perpendicular, parallel, and/or unaligned matrices. This approach allows one to address the question of which molecular events are specifically necessary for recognizing the topography of the matrix, and mediating contact-guided 3D cell migration.
Materials
Sub-confluent epithelial or fibroblast cells on a standard tissue culture dish/flask
Culture media of choice for the chosen cell type
Trypsin/EDTA solution (Cellgro)
Rat tail collagen (BDBiosciences)
0.1 M HEPES buffer in 2X PBS
1.5 μm diameter streptavidin coated iron oxide beads (Bangs Labs: BM551)
Cylindrical magnet (~2G; McMaster: 5862K32)
Nunc Multidish 4-well rectangular
Detach and count cells
-
1
Detach and count cells as described in steps 1–4 of Basic Protocol 1.
Prepare rectangular CSCGs
-
2
In a new conical tube add the appropriate volume of collagen solution (as described in Basic Protocol 1) needed to make 2.5 milliliters (for every two samples) of a mixture that will generate matrices with a final collagen matrix concentration of 2.0 mg/ml. Add an equal volume of 100 mM HEPES buffer (in 2X PBS). Add culture medium and culture medium containing cells to achieve the correct final volume and cell number per CSCG.
At least 2 million cells per 2.5 ml matrix should be included. This number may vary by cell type and experimental question, but in general a smaller number of cells will not sufficiently fill the CSCG and the cell distribution at the CSCG-aligned matrix interface will be uneven. -
3
Pipette the collagen mixture into a rectangular mold (~1 × 3 inches) or culture dish (Nunc 4-well rectangular multidishes work very well), and allow the matrix to polymerize for at least 2 hours. During this time, move forward with step number 4.
While herein rectangular CSCGs are described and have worked well, in general, any CSCG geometry that is needed to satisfy the final assay architecture may be substituted. Moreover, the size of the assay can be scaled as needed. However, the matrix volume should be optimized for each condition.
Generating aligned collagen matrices
-
4
In a new conical tube add the appropriate volume of collagen solution (as described in Basic Protocol 1) needed to make 2.5 milliliters of a mixture that will generate matrices with a final collagen matrix concentration of 2.0 mg/ml. Add an equal volume of 100 mM HEPES buffer (in 2X PBS). Add culture medium and then streptavidin coated iron oxide beads (1.5 μm diameter; Bangs Labs) to achieve the correct final volume with a bead concentration of 0.1 mg/ml.
-
5
Pipette the collagen mixture (containing magnetic beads) into a rectangular dish with the same geometry as the dish used in step 3.
-
6
Place the cylindrical magnet underneath the culture well with the poles of the magnet along the long axis of the gel mold, in the direction of alignment, to align the collagen matrix during collagen fibrillogenesis at room temperature for 20 minutes.
The culture plate well should be slowly translated over the magnet continuously, along the direction of alignment, during the first 10 minutes (with each pass taking ~15 seconds) of the polymerization period in a single direction. If multiple matrices are being generated, an array of magnets can be employed. In this case, four magnets can be separated by plastic so that a magnet is below each well (in the case of a Nunc 4-well plate). Note that care must be taken when handling the magnets as they will strongly attract or repel (depending on their orientation to one another) and can cause injury. -
7
Place the culture dish, with the magnets remaining underneath the wells, in a cell culture incubator at 37°C for an additional 60 minutes.
Implant Aligned matrices CSCGs into the encasing matrix
-
8
Remove the CSCG(s) and aligned matrices from the incubator. Using a small spatula or cell scraper, carefully lift each CSCG from the culture plate and float the matrix in culture media for 20 minutes.
-
9
During step 8 make an additional collagen mixture for the encasing matrix.
The volume of the encasing matrix will depend on the choice of final geometry. -
10
Cut a CSCG and an aligned matrix into two equal pieces and then pipette a thin, evenly distributed, volume of encasing matrix onto the culture plate. Immediately place the CSCG at the center of the dish and place the aligned matrices parallel and perpendicular to the CSCG. Gently pipette the encasing matrix over CSCG-aligned matrix pieces to achieve the configuration shown in Figure 2.
The aligned matrices should be pressed directly against the CSCG with no space between the matrices at the CSCG/aligned matrix interface. Care should be taken so that the thin volume of collagen mixture at the bottom of the plate is not pushed between the CSCG and the aligned matrices. Likewise, when pipetting the encasing matrix, care should be taken not to flow an excess amount of collagen into the interface. Note that the CSCGs and aligned collagen matrix can be carefully cut and shaped to achieve the desired architecture as shown in Figure 2. However, care should be taken to prevent an uneven surface from placed against the CSCG. -
11
Culture for one to several days.
-
12
Analyze 3D cell migration/invasion with optical imaging (see Support Protocols)
Figure 2.
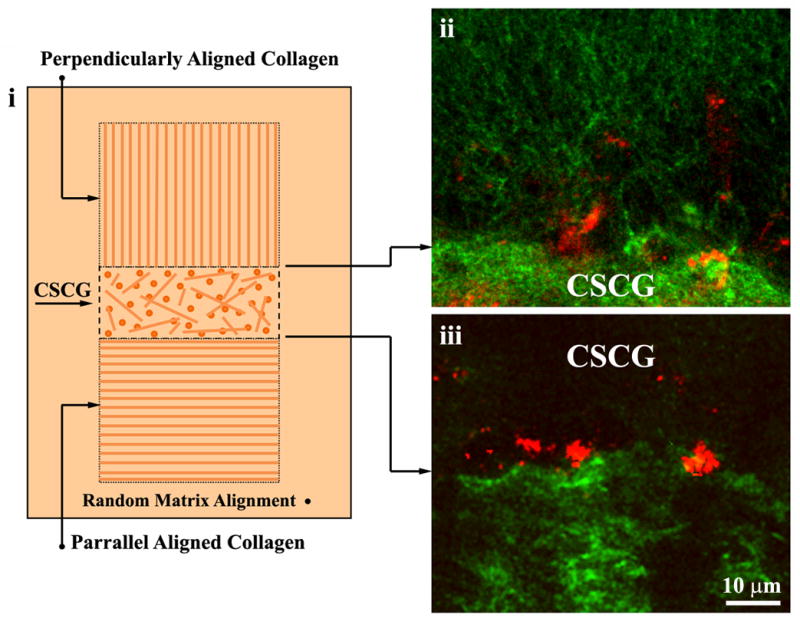
(i) Diagram illustrating 3D Migration/Invasion assay described in Basic Protocol 2. In the assay, collagen is magnetically aligned and aligned collagen matrices are connected to the CSCG so that the collagen alignment is either perpendicular (ii) or parallel (iii) to the CSCG boundary. Collagen is shown in green and cells in red. Reproduced from Provenzano et al., 2008a with permission.
SUPPORT PROTOCOL 1: IMAGING COLLAGEN MATRIX ALIGNMENT AND CELL MIGRATION WITH MULTIPHOTON EXCITATION AND SECOND HARMONIC GENERATION MICROSCOPY
Note: A number of multiphoton laser-scanning microscopy (MPLSM) systems are now commercially available that can be used to image collagen and the cells in the assays described above. Custom systems are also commonplace and the reader is encouraged to examine the references included in this chapter for additional details.
Note: The magnetic beads used in Basic Protocol 2 scatter light and can overload the microscope detector if care is not taken. Begin imaging with a low power or detector gain until it is determined how the beads will influence the microscope setup.
Multiphoton laser-scanning microscopy, MPLSM (Denk et al., 1990), is an optical sectioning technique where excitation is restricted to the plane of focus because the probability of two or more photons simultaneously exciting a fluorophore outside the focal volume is extremely low, significantly reducing phototobleaching by not exciting the out-of-focus-plane tissue volume (for detailed reviews of MPLSM and Second Harmonic Generation (SHG) see refs.(Diaspro and Sheppard, 2002; Helmchen and Denk, 2005; Mohler et al., 2003; Provenzano et al., 2009; Zipfel et al., 2003) and detailed protocols for live cell imaging and SHG, including equipment considerations, are provided in Units 19.7 and 4.15, respectively). With MPLSM, the excitation wavelengths are long, typically 650–1050 nm excitation for 2- and 3-photon excitation, which has practical advantages for being able to excite a wide range of fluorophores (both endogenous and engineered) while facilitating deeper penetration into tissues, since the longer wavelengths are less prone to scatter. Additionally, MPLSM is advantageous due to its ability to produce second harmonic generation signals useful for imaging fibrillar collagen. As such, MPLSM has emerged as a popular and powerful tool for analyzing cells and the extracellular matrix under live culture conditions.
To image the collagen matrix of the 3D migration assays described in Basic Protocols 1 and 2, we image the live cell constructs with MPLSM (as described in (Provenzano et al., 2006; Provenzano et al., 2008a; Provenzano et al., 2008b) at an 890 nm excitation wavelength to simultaneously generate SHG signals from fibrillar collagen and endogenous fluorescence from the metabolite FAD (Provenzano et al., 2008a). Since SHG signal is found at exactly half of the excitation wavelength, SHG signals and endogenous fluorescence are separated with a 445 nm narrow band-pass filter and a 464 nm long pass filter (TFI Technologies, Greenfield, MA), respectively. Collagen fiber orientation is examined at regions of the CSCG-encasing or –aligned matrix interface and as the level of the single cell using a Nikon CFI Plan Apo 60X water-immersion lens (N.A. =1.2). The number of cells invading into the collagen gel is collected within three focal plains (center ± 30 μm) in regions of interest. Alternatively, full z-stacks can be created in regions of interest.
Alternative approaches for imaging 3D cell migration in collagen matrices exist. For instance, confocal reflectance microscopy has been used to successfully image the collagen matrix (i.e. (Hegerfeldt et al., 2002)). In combination with confocal reflectance or MPLSM, live cells can be imaged while expressing fluorescent proteins (i.e. (Hegerfeldt et al., 2002; Provenzano et al., in press)). Furthermore, the cells in 3D constructs can be imaged with transmitted or phase-contrast light microscopy, and in samples fixed with paraformaldehyde the distribution and number of cells that have migrated out of the CSCG can be analyzed following staining for cellular components, such as the nucleus (Grinnell et al., 2006; Miron-Mendoza et al., 2008), while MPSLM can be used to image cells and collagen in fixed samples as well (Gehler et al., 2009). Note, that when studying cell migration in the context of 3D collagen matrix architecture, as is the case in both Basic Protocols, a method to image the collagen matrix in regions of interest for cell migration must be included as changes in the organization of collagen strongly influence cell behavior.
COMMENTARY
Background Information
In three-dimensional microenvironments in vivo, cell migration plays a fundamental role in numerous physiologic and pathological processes, including tissue morphogenesis and cancer metastasis (Condeelis and Segall, 2003; Friedl and Gilmour, 2009). In vivo, alignment of collagen fibers has been shown to facilitate gland development (Ingman et al., 2006) and carcinoma cell invasion (Provenzano et al., 2006), while endothelial tube formation in 3D constructs in vitro also shows a dependence on collagen matrix alignment (Lee et al., 2009). Therefore, in order to recapitulate matrix architecture associated with invasion into the collagenous stroma in intact tumors (Provenzano et al., 2006), we engineered 3D collagen matrix assays for studying contact guidance in vitro (Provenzano et al., 2008a). The first assay makes use of the cells themselves to generate contact guidance cues through cell contraction-mediated matrix reorganization, while the second assay pre-aligns the collagen matrix to facilitate separation of signals associated with 3D migration and motility events to reorganize the collagenous ECM.
Live cell imaging with MPLSM has emerged as a powerful tool for studying cell migration in 3D microenvironments not only due to it ability to image endogenous fluorescence in live unlabeled cells, but also due to its ability to simultaneously image the collagen matrix. The use of SHG to image collagen fibers in 3D volumes provides novel insight into how the stromal ECM influences cell behavior. Furthermore, it also provides a means to understand how the cell influences matrix architecture under both normal and pathological conditions. While other techniques are valuable and can often be used in conjunction with MPLSM (i.e. imaging live cells with transmitted light, fixing samples after imaging to stain for specific features, etc.), MPLSM is unique in its ability to noninvasively image deep into 3D collagen environments and concurrently image cells and collagen with high resolution and good viability.
Critical Parameters and Troubleshooting
The ability of cells to contract a collagen matrix and cause the realignment (as covered in Protocol 1) is dependent in part on the stiffness of the matrix, and in part on the ability of the cells to generate a contractile force. We have found that a 2 mg/ml collagen gel works well for many cell types including MDA-MB-231 cells, NMuMG cells, mouse embryonic fibroblasts, and mammary tumor explants. Dramatically increasing the collagen concentration of the encasing matrix may diminish the extent of the collagen rearrangement, and consequently may alter 3D cell migration. However, stiffer matrices may be useful for highly contractile cells, or when one wants to determine if changes in signaling events have altered cellular contractility (as in (Gehler et al., 2009)). In addition, the density of cells in the CSCG can influence the time course of collagen reorganization and cell migration. Our interests have revolved around simulating carcinoma cells invading into the collagenous stroma. As such, we have utilized a high cell density within the CSCGs, however, lower cell densities may be used if desired. The optimal cell density for a specific experimental condition should be optimized by each investigator. Finally, if the CSCG is detaching from the encasing matrix lengthening the time of CSCG contraction before implantation may reduce pulling away from the encasing matrix. Alternatively, using low-adhesion culture plates can help facilitate detachment of the encasing matrix with the CSCG from the culture plate reducing the need to agitate the plate.. Lastly, increasing the collagen density of the CSCG relative to the encasing matrix can help reduce detachment.
Anticipated Results
Cells migrate preferentially through collagen-aligned perpendicular to the CSCG. In addition, cells migrate efficiently through aligned collagen matrices in the direction of alignment. With the assay described in Basic Protocol 1, the cells exert contractile forces on the matrix between the CSCGs, aligning the collagen matrix (collagen fibers oriented ~90º relative to the CSCG boundary). In regions 90º from the CSCG-CSCG axis (i.e. the “top” and “bottom” of the CSCG) collagen is oriented parallel to the CSCG boundary, likely due to collagen being pushed during growth expansion of the CSCG. Moreover, due to force balance resistance to contraction, in regions in the “back” (i.e. 180º from the interface nearest to the other CSCG) collagen is also reorganized perpendicular to the CSCG, providing a valuable control to discount the possibility that cell migration from one explant to another in regions of alignment is being driven by contact guidance and not being driven exclusively by chemoattraction between the two CSCGs. Likewise, in pre-aligned matrices (Basic Protocol 2), cells are expected to migrate preferentially into collagen matrix with alignment perpendicular to the CSCG.
Time Considerations
After cells are detached from the culture plate for seeding into CSCGs, the steps should be performed decisively so that the cells do not remain in suspension for prolonged periods of time. In addition, care must be taken to ensure that the collagen matrices are adequately polymerized before transferring out of the culture so that matrix integrity is maintained. Likewise, in step 14 of Basic Protocol 1, the time should be optimized so that the matrix is neither under or over polymerized, so that the CSCG can easily flow into the matrix, but does not immediately drop to the bottom of the culture dish. Overall, once optimized the entire assembly for procedure for protocol 1 can be performed in 1–2 hours, depending on the number of samples being constructed. Basic Protocol 2 takes ~2.5 hours to assemble, with additional time required as the number of samples increases.
Migration into the aligned collagen regions in both protocols begins within 24 hours and continues for days. The exact time will depend on the cell lines used as well as the dimensions of the assay and the matrix conditions. However, following migration over 14 days is not uncommon. It is worth noting, however, that in prealigned matrices, cells that migrate into the matrix can remodel the matrix and in some regions reduce alignment while promoting alignment in other regions. As such, time points for these experiments are typically shorter (~12–72 hours) to limit the influence of cell-mediated matrix remodeling. Longer times may be used, but changes in matrix architecture should be noted as they may influence the migration pattern.
Literature Cited
- Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nat Rev Cancer. 2003;3:921–30. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- Dallon JC, Sherratt JA, Maini PK. Mathematical modelling of extracellular matrix dynamics using discrete cells: fiber orientation and tissue regeneration. J Theor Biol. 1999;199:449–71. doi: 10.1006/jtbi.1999.0971. [DOI] [PubMed] [Google Scholar]
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–6. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Diaspro A, Sheppard CJR. Two-Photon Excitation Fluorescence Microscopy. In: Diaspro A, editor. Confocal and Two-Photon Microscopy: Foundations, Applications, and Advances. Wiley-Liss, Inc; New York: 2002. pp. 39–73. [Google Scholar]
- Dickinson RB, Guido S, Tranquillo RT. Biased cell migration of fibroblasts exhibiting contact guidance in oriented collagen gels. Ann Biomed Eng. 1994;22:342–56. doi: 10.1007/BF02368241. [DOI] [PubMed] [Google Scholar]
- Doyle AD, Wang FW, Matsumoto K, Yamada KM. One-dimensional topography underlies three-dimensional fibrillar cell migration. J Cell Biol. 2009;184:481–90. doi: 10.1083/jcb.200810041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10:445–57. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- Gehler S, Baldassarre M, Lad Y, Leight JL, Wozniak MA, Riching KM, Eliceiri KW, Weaver VM, Calderwood DA, Keely PJ. Filamin A-beta1 integrin complex tunes epithelial cell response to matrix tension. Mol Biol Cell. 2009;20:3224–38. doi: 10.1091/mbc.E08-12-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinnell F, Rocha LB, Iucu C, Rhee S, Jiang H. Nested collagen matrices: a new model to study migration of human fibroblast populations in three dimensions. Exp Cell Res. 2006;312:86–94. doi: 10.1016/j.yexcr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Guido S, Tranquillo RT. A methodology for the systematic and quantitative study of cell contact guidance in oriented collagen gels. Correlation of fibroblast orientation and gel birefringence. J Cell Sci. 1993;105(Pt 2):317–31. doi: 10.1242/jcs.105.2.317. [DOI] [PubMed] [Google Scholar]
- Guo C, Kaufman LJ. Flow and magnetic field induced collagen alignment. Biomaterials. 2007;28:1105–1114. doi: 10.1016/j.biomaterials.2006.10.010. The original work from which the protocol for aligning collagen with magnetic beads was adapted. [DOI] [PubMed] [Google Scholar]
- Hegerfeldt Y, Tusch M, Brocker EB, Friedl P. Collective cell movement in primary melanoma explants: plasticity of cell-cell interaction, beta1-integrin function, and migration strategies. Cancer Res. 2002;62:2125–30. [PubMed] [Google Scholar]
- Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods. 2005;2:932–40. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- Ingman WV, Wyckoff J, Gouon-Evans V, Condeelis J, Pollard JW. Macrophages promote collagen fibrillogenesis around terminal end buds of the developing mammary gland. Dev Dyn. 2006;235:3222–9. doi: 10.1002/dvdy.20972. [DOI] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–69. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Lee PF, Yeh AT, Bayless KJ. Nonlinear optical microscopy reveals invading endothelial cells anisotropically alter three-dimensional collagen matrices. Exp Cell Res. 2009;315:396–410. doi: 10.1016/j.yexcr.2008.10.040. [DOI] [PubMed] [Google Scholar]
- Miron-Mendoza M, Seemann J, Grinnell F. Collagen Fibril Flow and Tissue Translocation Coupled to Fibroblast Migration in 3D Collagen Matrices. Mol Biol Cell. 2008;19:2051–8. doi: 10.1091/mbc.E07-09-0930. This study describes the use of nested matrices to study fibroblast migration in collagen matrices. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler W, Millard AC, Campagnola PJ. Second harmonic generation imaging of endogenous structural proteins. Methods. 2003;29:97–109. doi: 10.1016/s1046-2023(02)00292-x. [DOI] [PubMed] [Google Scholar]
- Petrie RJ, Doyle AD, Yamada KM. Random versus directionally persistent cell migration. Nat Rev Mol Cell Biol. 2009;10:538–49. doi: 10.1038/nrm2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Eliceiri KW, Keely PJ. Multiphoton microscopy and fluorescence lifetime imaging microscopy (FLIM) to monitor metastasis and the tumor microenvironment. Clin Exp Metastasis. 2009;26:357–70. doi: 10.1007/s10585-008-9204-0. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling, and gene expression through a FAK-ERK linkage. Oncogene. doi: 10.1038/onc.2009.299. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Trier SM, Keely PJ. Contact guidance mediated three-dimensional cell migration is regulated by Rho/ROCK-dependent matrix reorganization. Biophys J. 2008a;95:5374–84. doi: 10.1529/biophysj.108.133116. The source of procedures described in this unit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Rueden CT, Trier SM, Yan L, Ponik SM, Inman DR, Keely PJ, Eliceiri KW. Nonlinear optical imaging and spectral-lifetime computational analysis of endogenous and exogenous fluorophores in breast cancer. J Biomed Opt. 2008b;13:031220. doi: 10.1117/1.2940365. [DOI] [PubMed] [Google Scholar]
- Teixeira AI, Abrams GA, Bertics PJ, Murphy CJ, Nealey PF. Epithelial contact guidance on well-defined micro- and nanostructured substrates. J Cell Sci. 2003;116:1881–92. doi: 10.1242/jcs.00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel WR, Williams RM, Webb WW. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol. 2003;21:1369–77. doi: 10.1038/nbt899. [DOI] [PubMed] [Google Scholar]