

Abstract
The species in family Planctomycetaceae are ideal groups for investigating the origin of eukaryotes. Their cells are divided by a lipidic intracytoplasmic membrane and they share a number of eukaryote-like molecular characteristics. However, their genomic structures, potential abilities, and evolutionary status are still unknown. In this study, we searched for common protein families and a core genome/pan genome based on 11 sequenced species in family Planctomycetaceae. Then, we constructed phylogenetic tree based on their 832 common protein families. We also annotated the 11 genomes using the Clusters of Orthologous Groups database. Moreover, we predicted and reconstructed their core/pan metabolic pathways using the KEGG (Kyoto Encyclopedia of Genes and Genomes) orthology system. Subsequently, we identified genomic islands (GIs) and structural variations (SVs) among the five complete genomes and we specifically investigated the integration of two Planctomycetaceae plasmids in all 11 genomes. The results indicate that Planctomycetaceae species share diverse genomic variations and unique genomic characteristics, as well as have huge potential for human applications.
Introduction
The Planctomycetaceae family is distributed in both soil and water. Previous studies have estimated that 50% of the nitrogen molecules in the atmosphere are generated by anammox planctomycetes [1]. Planctomyces bekefii was the first member of family Planctomycetaceae, reported in 1924 by Gimesi, who originally established genus Planctomyces Gimesi 1924 to accommodate this peculiar aquatic “fungi” [2]. After nearly 50 years, Hirsch reported a freshwater bacterium in 1972 and named it “Blastocaulis sphaerica,” which was later identified as P. bekefii [3]. Further studies provided conclusive evidence that the two organisms are indistinguishable and both are bacteria rather than fungi [4]. The transmission electron micrographs of the negatively stained strains showed that Planctomycetaceae species possess crateriform structures on their cell surface. Some cells have stalks or extensive holdfast materials on one pole. To date, the Planctomycetaceae family includes 11 genera, namely, Aquisphaera [5], Blastopirellula [6], [7], Gemmata [8], Isosphaera [9], Pirellula [10], [11], Planctomyces [12], Rhodopirellula [6], Schlesneria [13], [14], Singulisphaera [14], [15], Telmatocola [16], and Zavarzinella [14], [17], with more than 18 species (http://www.bacterio.cict.fr/p/planctomycetaceae.html). The whole genomes of 11 type strains are available in the National Center for Biotechnology Information (NCBI) databases (ftp://ftp.ncbi.nlm.nih.gov/genomes/).
Planctomycetaceae species exhibit strong tolerance for seawater, acidic peat bogs, hot springs, and low temperatures [13], [15], [18]. Furthermore, members of Planctomycetaceae family possess unusual features, i.e., the absence of peptidoglycans, the synthesis of C30 sterols, the encoding of C1 transfer enzymes, and the presence of clathrin-like membrane coat proteins and anammoxosomes [18]. Their most unique feature is the presence of cell compartmentalization structure called a lipidic intracytoplasmic membrane (ICM) [18], which is unusual in prokaryotic and eukaryotic species. This ICM divides the cell into two parts: the paryphoplasm and the pirellulosome. The pirellulosome contains all the ribosomes, whereas the paryphoplasm is ribosome free. This type of cell compartmentalization occurs in members of Verrucomicrobia. More than seven Planctomycetaceae type species have been reported from different genera, namely, Blastopirellula [6], [7], Gemmata [8], Isosphaera [9], Pirellula [10], [11], Rhodopirellula [6], Schlesneria [13] and Singulisphaera [15]. Only Gemmata obscuriglobus exhibits a double-membrane ICM, whereas other type species have a single membrane. Phylogenetic studies on the conserved regions in the ribosomal RNA sequences and whole proteomes suggest that Planctomycetaceae are closely related with Verrucomicrobia and Chlamydiae, which are all members of the Planctomycetaceae–Verrucomicrobia–Chlamydiae (PVC) superfamily [18]. Jun et al. [19] proposed that Planctomycetaceae should be classified between bacteria and archaea based on their evolutionary relationship. Brochier and Philippe [20] found that Planctomycetales emerged at the base of the bacterial branch, according to the conserved positions in ribosomal RNA.
To date, overviews on the genomic characteristics of planctomycete are insufficient. As the fast development of sequencing technology, the foundations are starting to be laid for comparative genomics to assist in the interpretation of planctomycete cell biology. In this study, we attempted to unveil the relationships among Planctomycetaceae species and to explore their evolutionary status among organisms using whole-genome analysis.
Results and Discussion
Overview of Planctomycetaceae Genomes
We previously sequenced three Planctomycetaceae type strains (Singulisphaera acidiphila DSM 18658T, Schlesneria paludicola DSM 18645T, and Zavarzinella formosa DSM 19928T) [14]. These strains were isolated from acidic wetlands in northern Russia (http://www.dsmz.de/). The whole genomes of type species from six other genera are available in NCBI (Table 1). More than two type strains of the genus Planctomyces were also sequenced. We performed a preliminarily exploration of the resistance genes in 11 Planctomycetaceae genomes (Supplementary Fig. S1) [21].
Table 1. Genome survey of 11 strains in family Planctomycetaceae.
| Species | Accession number(GenBank) | Genome size(Mb) | GC content(%) | Genecount | Proteincount |
| Planctomyces limnophilus DSM 3776T | NC_014148 | 5.46 | 53.70 | 4,373 | 4,258 |
| Isosphaera pallida DSM 9630T | NC_014962 | 5.53 | 62.40 | 3,823 | 3,722 |
| Planctomyces brasiliensis DSM 5305T | NC_015174 | 6.01 | 56.40 | 4,865 | 4,750 |
| Pirellula staleyi DSM 6068T | NC_013720 | 6.20 | 57.50 | 4,825 | 4,717 |
| Blastopirellula marina DSM 3645T | AANZ00000000 | 6.65 | 57.00 | 6,079 | 6,025 |
| Rhodopirellula baltica DSM 10527T | NC_005027 | 7.15 | 55.40 | 7,404 | 7,325 |
| Planctomyces maris DSM 8797T | NZ_ABCE00000000 | 7.78 | 50.50 | 6,530 | 6,480 |
| Schlesneria paludicola DSM 18465T | AHZR00000000 | 8.70 | 56.00 | 8,626 | 8,626 |
| Gemmata obscuriglobus DSM 5831T | ABGO00000000 | 9.16 | 67.20 | 8,080 | 7,989 |
| Singulisphaera acidiphila DSM 18658T | AHZQ00000000 | 9.73 | 63.01 | 8,972 | 8,972 |
| Zavarzinella formosa DSM 19928T | AIAB00000000 | 10.09 | 59.60 | 10,112 | 10,112 |
The Planctomycetaceae genomes are over 5 Mb long and possess more than 3,700 coding genes (CDSs), which are relatively larger than those of most bacteria. The genome size of Z. formosa is 10.09 Mb with 10,112 CDSs, similar to the ∼12 Mb genome of Saccharomyces cerevisiae [22], but with twice the number of CDSs. Except for a small subset of individuals, the number of CDSs is roughly proportional to the genome size (Table 1). Deviations were inevitable because 8 of the 11 genomes were obtained from NCBI and they may contain gaps and sequencing errors. Furthermore, the C-value paradox should be considered. From fig. 1, in the 11 genomes, the length of genes are mostly less than 1,000 bp. Remarkably, in the range of [100–500) bp, the drastic increase of genes in the larger genomes was observed. G. obscuriglobus had the highest GC content, ranging from 50.50% to 67.20% in the 11 Planctomycetaceae genomes (Table 1). Planctomycetaceae has more than 30,000 pan-genes and 832 core gene clusters with 9,928 genes. Jogler et al. [23] used the 8 genomes excluding the 3 genomes sequenced by us, and found that the planctomycetal core genome led to 114 predicted protein clusters containing 2,908 proteins from all eight analyzed planctomycetes after the in silico subtraction of E. coli and B. subtilis genomes with all-against-all BLASTP approach (with stringent filters: coverage higher than 60% and below the E value cutoff of 1e −5 were taken into account). Proteome-based analysis of the predicted protein families in the 11 genomes (Fig. 2A and Supplementary Table S1) showed that Z. formosa and G. obscuriglobus share 3,344 common protein families. Furthermore, P. maris and S. paludicola share 2,821 protein families, P. maris and P. brasiliensis share 2,684 protein families. Z. formosa possesses the highest number of unique families with 1,092 proteins in 355 protein families, which is followed by G. obscuriglobus with 745 proteins in 252 families and Rhodopirellula baltica with 356 proteins in 216 families. In addition, numerous orphan proteins excluded from the final clusters and paralogs found in the predicted proteomes are responsible for the large size of Planctomycetaceae genomes. To confirm their relationships, we analyzed the tetranucleotide frequencies and the average nucleotide identity (ANI) [24], [25] of the 11 genomes (Supplementary Figs. S2A and S2B).
Figure 1. Gene length distribution in the 11 Planctomycetaceae genomes.

The values above each bar (in percent) were calculated as the number of genes that are of specified length divided by the number of total genes in each Planctomycetaceae genome.
Figure 2. Gene family clusters and core genome/pan genome analyses based on whole-genome sequences.

(A) The histogram reveals the number of unique gene families in each of the 11 Planctomycetaceae strains. The color bar on the right shows the number of common gene families between crossed strains. (B) The box plot at the left indicates the number of core genes that correspond with the strain genomes. The box plot at the right indicates the pan gene numbers that correspond with the strain genomes. The box in the figures indicates half the distribution of values in the random sampling; the bold line in box indicates the median.
Evolutionary Research on Planctomycetaceae
Fuerst et al. [26] reconstructed evolutionary models of the NE to illustrate the resemblance between Planctomycetales and fungi in terms of their compartmentalization. Jun et al. [27], [28] used an alignment-free method based on whole proteome FFPs (or monomers) to compare the FFPs of 900 proteomes from 26 phyla. The results revealed that Planctomycetaceae was found between bacteria and archaea in the phylogenetic trees. Moreover, the universal primers of bacterial 16S rRNA genes may not be effective for polymerase chain reaction amplification of the corresponding planctomycete genes [29]. That is, certain species in Planctomycetales may have been overlooked by the metagenomic analyses. Furthermore, the 5S rRNAs of Planctomycetales were shorter (109 nt to 111 nt) than the “minimal” length found in other prokaryotes (118 nt) and uniquely lacked an insertion at position 66. Brochier and Philippe [20] reanalyzed the bacterial phylogeny based on rRNA sequences using the “slow–fast” method [30], which is more reliable for identifying the most slowly evolving positions that are less affected by artifacts. Their study found that the Planctomycetales family might have emerged at the base of the bacteria branch.
A core-gene tree was created based on the concatenated alignments of the core genes and only the conserved regions of the orthologous core protein sequences were used to construct this genome-wide tree. We used protein sequences rather than nucleic acid sequences to minimize the possible influence of base composition and codon preference biases on phylogenic construction. The reasonable branching of the phylogenetic trees was consistent with the different physiologic and biochemical features of Planctomycetaceae species (Fig. 3). For instance, both R. baltica and Blastopirellula marina live in marine environments with high NaCl concentrations. In addition, Pirellula staleyi, R. baltica, and B. marina are strain of genera from the “Pirellula group,” which are closely similar in morphology and physiology despite having less than 90% similarity of their 16S rRNA gene sequences [6]. Sequence alignment of the 16S rRNA genes confirmed that Planctomycetaceae is present in environmental correlation groups [31]. Therefore, we selected three closely related species that belonged to environmental correlation groups as outgroups to determine the evolutionary status of Planctomycetaceae. Results indicate that the predicted proteomes accurately represent the phylogenetic relationships among members of Planctomycetaceae.
Figure 3. Phylogenetic tree constructed using proteomes predicted from 11 genomes of Planctomycetaceae.

The rooted tree generated in MEGA 4.0 with the concatenated alignments of the conserved regions of the orthologous core protein sequences, topology of which illustrates the relationships among Planctomycetaceae species and related taxa. Bootstrap values (in percent) of 1000 simulations are indicated at all branches. Bar represents 0.05 amino acid substitutions per site. Coraliomargarita akajimensis, Parachlamydia acanthamoebae, and Phycisphaera mikurensis were selected as outgroups [31].
Gene Function Annotation and Metabolic Pathways Reconstruction
We annotated the gene function of the 11 Planctomycetaceae genomes with COG databases and predicted their metabolic pathways using the KEGG database. To our surprise, only 29.28% of the total genes in the 11 genomes have been characterized, numerous genes remain unclear (Fig. 4A). Jogler et al. [23] reported that more than 55% of the predicted proteins are of unknown function in each genome on average in Planctomycetaceae. In the 11 sequenced species, only R. baltica (originally classified as Pirellula sp. str. 1) [32] has been described in detail in the literature. P. staleyi and P. limnophilus genomes were described in summary form only [10], [12]. The function could be predicted for only 32% of the ORFs in R. baltica; although around 54% of ORFs with a predicted function were in P. staleyi and P. limnophilus, hypothetical proteins are a feature of planctomycetes, and this remains a challenge for understanding the molecular cell biology of these bacteria [18]. The same case occurred in their core genome. We obtained 832 core families, only 255 of which are characterized in the COG database. For the pan genome, genes related with general function is dominant except G. obscuriglobus, genetic information of which is the most prominent. But the situation is different in their core genome, in which the energy production and conversion is dominant, and then general function. Overall, Genes involving general function, amino acids transport and metabolism, replication/recombination and repair, signal transduction mechanisms and translation/ribosomal structure/biogenesis governed in either pan genome or core genome of Planctomycetaceae (Fig. 4B). Given that most scientists pay more attention to the unique cell biological characteristics, few focus on the whole metabolic pathways of Planctomycetaceae family thus far. Such as, identification protein domains likely to be involved in morphogenesis, cell Division and signal transduction in Planctomycetes by comparative genomics [23]; the endocytosis process in G. obscuriglobus with eukaryotic membrane coat (MC)-like-protein-encoding genes [33]; the unusual peptidoglycan (PG)-free cell wall and ICM structure [18] and so on. Actually, the whole metabolic pathways analysis can help us explain the whole biological features well and provide more information to our research. Since R. baltica is the first planctomycete to be genome completely sequenced and one of the best-studied planctomycetes on the proteomic level, we mainly focused on it for the illustration of the subsequent metabolic pathways research. Glockner et al. [32] partly predicted the metabolic pathways of R. baltica in bioinformatical ways, for instance, C1 metabilism and Entner-Doudoroff pathway. Hieu et al. [34] experimentally identified 1,267 nonredundant proteins (accounting for 17.3% of the total putative protein-coding ORFs) from R. baltica proteome ananlysis, and reconstructed a rough depiction of house-keeping metabolic routes of carbohydrate, amino acid, nucleic acid and fatty acid metabolism pathways of R. baltica from 668 functionally assigned proteins.
Figure 4. Gene annotation.

(A) The inner ring: number of genes in the genomes; The outer ring: characterized/uncharacterized genes. (B) Gene function classification based on COG annotation. Heatmap at the left shows the functional prediction of 11 Planctomycetaceae genome. The core genome consists of the nonredundant core protein families. Heatmap at the right shows the functional prediction of the core genome. The color bar shows gene numbers. (C) ICM structure of four Planctomycetaceae species. These pictures shows the the ICM structure of four Planctomycetaceae species, namely S. acidiphila, R. baltica, Z. formosa, and S. paludicola (left to right), which were observed under transmission electron microscopy.
In this study, the 11 Planctomycetaceae genomes contained 231 predicted metabolic pathways and 15,061 enzymatic reactions, among which 1,280 are in P. limnophilus, 1,264 are in I. pallid, 1,353 are in P. brasiliensis, 1,340 are in P. staleyi, 1,305 are in B. marina, 1,391 are in R. baltica, 1,371 are in P. maris, 1,445 are in S. paludicola, 1,374 are in G. obscuriglobus, 1,565 are in S. acidiphila, and 1,373 are in Z. formosa (Supplementary Table S2). Although the function of some genes in the metabolic pathways may be replaced and cannot be identified during evolution, core metabolic pathways can provide relatively accurate genetic information inherited from their ancestor. In all, 123 metabolic pathways in KEGG pathway database responded to all the 11 genomes of Planctomycetaceae. Their ancestors should have a comprehensive metabolism based on these pathways (Supplementary Fig. S4). Glycolysis/gluconeogenesis, the citrate cycle (TCA cycle), the pentose phosphate pathway, and oxidative phosphorylation are the metabolic junctions of energy generation and intermediate compounds, which was supported in the former proteomic research of R. baltica [34], [35]. Gade et al. [35] found that R. baltica cells could be adapted to growth with eight different carbohydrates: glucose, ribose, N-acetylglucosamine, xylose, maltose, lactose, melibiose, and raffinose, respectively. We found these species also have the potential of using fructose, galactose, starch, sucrose, and mannose. Moreover, they can generate energy through sulfur and nitrogen metabolism. Some photosynthesis- and carbon fixation–related genes were also found. Furthermore, almost all 20 amino acid metabolic pathways were detected, which agreed with the R. baltica’s proteomic study [34]. Remarkably, nearly all the genes for the synthesis of the seven amino acids essential to humans were found. We infer that these species are likely able to synthesize these amino acids, namely, valine, leucine, phenylalanine, tyrosine, lysine, tryptophan, and isoleucine. Kerger et al. [36] reported the presence of hydroxyl fatty acids in planctomycetes and deduced that they originate from lipopolysaccharides (LPS). Schlesner et al. [6] concluded that the absence of measurable amounts of hydroxyl fatty acids indicates the absence of significant amounts of lipopolysaccharides in the cell wall even though the organisms are Gram negative. In our study, some of the peptidoglycan biosynthesis genes and most of the Escherichia coli lipopolysaccharide biosynthesis genes were detected in all 11 genomes (Supplementary Fig. S3).
Most unexpectedly, the 11 species can synthesize unsaturated fatty acids and complex lipids such as steroids [37]. In particular, Gemmata spp. can synthesize C30 sterols such as lanosterol, which are speculated to regulate membrane fluidity in the characteristic internal membranes of planctomycetes [18]. Although R. baltica is not capable of utilizing methanol, methylamine, or methylsulfonate [20], we detected biosynthetic genes for zeatin, steroids, steroid hormones, carotenoids, ubiquinones, and other terpenoid quinones in all 11 genomes. In addition, we found an intact mevalonate pathway conserved among the 11 species that produces the precursor of the terpenoid backbone, which indicates that Planctomycetaceae species are capable of synthesizing complex compounds. Schlesner et al. [6] found that the absorption spectrum of a methanol extract peaks at 495 nm and forms two shoulders at 460 and 520 nm, which are similar to that of carotene. However, few homologous genes related to steroid and carotene biosynthesis were found, which illustrates that the conserved domains of functional genes have been replaced during evolution or that these species use new biosynthetic pathways.
Leary et al. [38] cloned the rpoN gene from P. limnophilus that encodes alternative sigma factor σ54, which is involved in diverse metabolic functions such as nitrogen fixation, hydrogen metabolism, and degradation of aromatic compounds, and they demonstrated complementation in a Salmonella typhimurium mutant. Analysis of the core metabolic pathways of the 11 species revealed that all of them shared metabolic pathways for degrading diverse toxic compounds, including ethylbenzene, aminobenzoate, naphthalene, chloroalkane, polycyclic aromatic hydrocarbon, toluene, bisphenol, benzoate, chlorocyclohexane, and chlorobenzene (Supplementary Fig. S3). This finding indicates that these species may have potential applications in environmental management. We marked the metabolic pathways for degrading aromatic compounds with bold rectangle in supplementary fig. S3. On the other hand, these species possess genes for synthesizing streptomycin and other antibiotics, such as tetracycline, ansamycins, vancomycin, novobiocin, butirosin, and neomycin. Furthermore, we also found biosynthetic genes for alkaloids, which are mostly toxic to humans.
Despite the ABC transporter system has been found in R. baltica [34], [35], F-type ATPases and ATP-binding cassette transporters were detected in all the 11 species (Supplementary Fig. S3). After analyzing the core metabolic pathways, we found a high level of two-component systems, which verify that these species possess complex regulatory systems for adapting to their physiological and functional complexity. Actually, Planctomycetaceae species have an ICM structure [18], which is unusual in prokaryotic and eukaryotic species (Fig. 4C). However, its composition and function remain unclear. We inferred that two-component systems widely regulate the life cycle by phosphorylation/dephosphorylation based on a phosphotransferase system (PTS) in the 11 species.
Structural Variations (SVs) and Genomic Islands (GIs) in the Five Complete Planctomycetaceae Genomes
Many factors are responsible for the genome expansion or shrink, including homologous recombination, horizontal gene transfer (HGT), convergent/divergent evolution and specialization [39], [40]. Glockner et al. [32] found that multicopy genes make up for about 25.4% of the R. baltica genome sequence, which is less than the 30% reported for Bacillus subtilis [41] and the 29% calculated for Escherichia coli K-12. Therefore extensive gene duplication is not the reason for the large genome size of R. baltica. Gade et al. [35] cultivated R. baltica cells with 8 different carbohydrates respectively and profiled with 2-DE for changes in protein patterns. They found that most genes not organized in clusters are functionally related and coordinately regulated, which is in agreement with the previous in silico observation that functionally related genes are often not clustered in operon-like structures in the 7.145 Mb genome of R. baltica [32]. This contrasts the genetic organization known from standard bacteria such as E. coli or B. subtilis, both having considerably smaller genome sizes than R. baltica. Apparently, the number of genes organized in clusters decreases with increasing genome size [42]. In addition, a large genome with an expanded genetic capability might be a prerequisite for environmental adaptability, as already discussed for the genome of Pseudomonas aeruginosa [43]. In this case, it is necessary to investigate the genome arrangement and genomic islands in the 11 genomes. We found that numerous unique segments were inserted between homologous blocks, which indicate that homologous recombination and HGT facilitate the divergent evolution of these genomes (Fig. 5). On the other hand, we selected the 5 completed genomes of P. limnophilus, P. brasiliensis, P. staleyi, I. pallida, and R. baltica to predict accurately the GIs and SVs. GI prediction was performed by integrating the softwares SIGI-HMM, IslandPath-DIMOB and IslandPick (Fig. 6) [44]. In a recent study, the SIGI-HMM method was shown to have the highest precision and overall accuracy among six tested sequence composition GI prediction methods [45]. Based on the prediction results from SIGI-HMM, the proportion of GIs was less than 5.0% of the five genomes and most of the GIs originated from Actinobacteria, and then, from Bacillus, Nitrospira, Bacteroides, Chloroflexi, Gammaproteobacteria in the five completed genomes. Up to 355 genes were in the R. baltica GIs, 65 were in the P. limnophilus GIs, 161 were in the P. staleyi GIs, 234 were in the P. brasiliensis GIs, and 58 were in the I. pallid GIs. However, most of them remain uncharacterized (Supplementary Table S3). We found LPS biosynthesis–related proteins in the GIs of P. limnophilus and R. baltica, streptomycin biosynthetic proteins and its operon regulatory protein in the GIs of R. baltica, CRISPR-associated proteins in I. pallid, cytochrome P450 genes in P. brasiliensis and P. staleyi, bifunctional DNA primase/polymerase in P. brasiliensis and I. pallid, sigma-70 family RNA polymerase sigma factor in P. limnophilus, and WD-40 repeat-containing protein in P. staleyi.
Figure 5. Collinearity of the five completed Planctomycetaceae genomes.
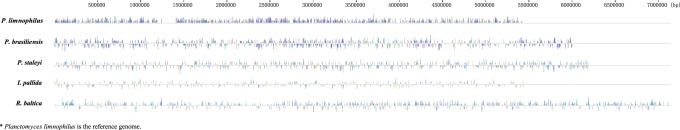
P. limnophilus was set as the reference genome. Different colored lines in each genome indicate its similarity profile. The height of the similarity profile corresponds to the average level of conservation in that region of the genome sequence and was calculated to be inversely proportional to the average alignment column entropy over a region of the alignment. Parts of the similarity plot which are colored mauve are conserved among all the five genomes. The other colors shows the similarity of segments not in all the five genomes, but they are too small to notice in a whole-genome view. The similarity profile above the centerline of the aligned region is in the forward orientation relative to the first genome sequence, and profiles below the centerline indicate regions that align in the inverse orientation. Areas that are completely white were not aligned and probably contain sequence elements specific to a particular genome. The Mauve software was used for this analysis [55]. The seed (minimum match size) we used was 15 mer.
Figure 6. Overview of genomic islands in the five completed genomes.

GI prediction was performed by integrating the following software: SIGI-HMM, IslandPath-DIMOB and IslandPick. The prediction was implemented in Web server IslandViewer (http://pathogenomics.sfu.ca/islandviewer/query.php) [44].
To further explore the HGT, we examined the plasmid integration in all 11 Planctomycetaceae genomes (Fig. 7). Only two plasmids were reported in Planctomycetaceae, from P. limnophilus and I. pallid, and they are available in NCBI. In addition, we selected the closely related Phycisphaera mikurensis and the low-grade fungus Saccharomyces cerevisiae as controls. Strikingly, the I. pallid plasmid genes exhibited high identity with those in the genomes of P. mikurensis, S. paludicola, G. obscuriglobus, S. acidiphila, and Z. formosa, whereas P. limnophilus plasmid genes exhibited high identity with those in the genomes of S. paludicola, G. obscuriglobus, S. acidiphila, and Z. formosa. However, the plasmid genes of I. pallid and P. limnophilus were not detected in the S. cerevisiae genome. Larger genome sizes seemed to be correlated with higher coverage and with homology of plasmid genes. The results indicate that the plasmid integration in the 11 genomes may also lead to larger genome sizes; moreover, it reflects that the potential shuttle ability of Planctomycetaceae plasmids among their close relatives.
Figure 7. Plasmid integration in the 11 genomes.

PL and IS represents the plasmid reported in P. limnophilus and I. pallid respectively. The outermost ring shows the two plasmids’ genes which divided by bold lines. The colors in each genome ring accord with the identity illustrated in the legend. The 11 Planctomycetaceae genomes were arranged according to their genome sizes, the outer the larger. Phycisphaera mikurensis and Saccharomyces cerevisiae were used as a control.
Conclusions
Worldwide interest in the unique cellular biological features and structure in Planctomycetaceae has increased considerably, especially in evolutionary research. Exploring the potential genetic information in this special groups based on comparative genomic ways is essential for conducting the later researches. In this context, we mainly researched the genomic features and metabolic pathways with 11 type strains from different genus of family Planctomycetaceae. The genome-scale information we obtained combined with the already developed physiologic, genetic, and metabolic approaches will increase our knowledge about family Planctomycetaceae and may help us establish strategies for developing them for human applications. In this way, we could learn much about their relationships and potential biological systems of these species. Acquiring of these results were made possible by the use as reference to subsequent study. Further, we might have broader horizon to discover new features in these species responding to their attractive ICM structure, which is significant in evolution from prokaryotes to eukaryotes.
Materials and Methods
Source of Genomes
The genomes of eight species from family Planctomycetaceae were downloaded from GenBank (Table 1), namely, P. limnophilus, I. pallida, P. brasiliensis, P. staleyi, B. marina, R. baltica, P. maris, G. obscuriglobus. The genomes of Schlesneria paludicola, Singulisphaera acidiphila, and Zavarzinella formosa were sequenced in our previously study [14].
Ortholog Retrieval
Orthologs were determined using OrthoMCL [46]. This program first performs an all-against-all BLASTp in BLAST 2.2.25. OrthoMCL then converts the reciprocal BLAST p-values to a normalized similarity matrix that is analyzed using the Markov Cluster algorithm (MCL). The MCL analysis yielded numerous clusters, with each cluster containing a set of orthologs and/or recent paralogs. The BLAST e-value cutoff was ≤10−5; the other parameters were set to their default values.
Core/Pan Genome Analysis
The pan genome of Planctomycetaceae was constructed by selecting one genome as the origin and then gradually adding the other genomes individually. The final pan-genome was obtained by discarding redundant clusters. A set of identity matrices generated through BLAST analyses of all genomes and then selecting the nonredundant clusters conserved in all matrices as the core genome. The parameters used in core genome/pan genome analyses were as follows: e-value ≤10−5, identity ≥50%, and coverage ≥50%.
Calculation of ANI (ANIb) and Tetranucleotide Frequencies
ANI values were calculated as described by Goris et al. [47] (Fig. S2A). The tetranucleotide frequencies and correlation coefficients were calculated based on the algorithm described by Teeling et al. [48] (Fig. S2B).
Construction of Phylogenetic Trees Based on the Predicted Whole Proteomes from the Genomes
Several steps were involved in the phylogenetic tree construction based on the 11 predicted proteomes of Planctomycetaceae. First, performing all-against-all BLASTp using the 11 Planctomycetaceae predicted proteomes with BLAST 2.2.21 and setting appropriate identity and coverage cutoffs to extract their core genome; Then, implementing multiple sequence alignment in MUSCLE 3.8.31 [49] with the core genome; Subsequently, generating tree files in TREEBEST 1.9.2 using the neighbor-joining method; Finally, constructing phylogenetic trees in MEGA 4.0 [50] with the bootstrap value of 1000. A core-genome tree was created based on the concatenated alignments of the core proteins and only the conserved regions of the orthologous core protein sequences were used to construct this genome-wide rooted tree, up to 97 core proteins were obtained using a BLAST query, with e-value ≤10−5, identity ≥30%, and coverage ≥60%. Coraliomargarita akajimensis, Parachlamydia acanthamoebae, and Phycisphaera mikurensis were selected as outgroups according to the 16S rRNA classification [23], [31].
Annotation
Functions of genes were assigned according to the best match of the alignments using BLASTp (E-value ≤10−5) searching against the SwissProt and Uniprot databases (Release 15.10) [51]. The motifs and domains of proteins were determined using InterProScan (Version 4.5) [52] against protein databases. Furthermore, all proteins were aligned against KEGG (Release 48.2) [53] proteins. If the best hit of the proteins in any of these processes was “function unknown,” “putative,” the second best hits were used to assign function until no more hits met the alignment criteria.
Metabolic Reconstruction
We reconstructed the metabolic pathways of the 11 species using the web server Ipath (http://pathways.embl.de/) [54] with their assigned K numbers in KEGG Orthology system.
Collinear Analysis
Collinearity analysis of the five completed genomes was implemented in Mauve [55].
Genomic Island Search
Genomic islands were searched using the web server IslandViewer (http://pathogenomics.sfu.ca/islandviewer/query.php) [44], which integrates the three mainstream methods: SIGI-HMM, which focuses on codon usage; IslandPath-DIMOB, which focuses on dinucleotide and mobile elements; and IslandPick, which focuses on comparative genomic GI prediction. A plasmid integration survey of the 11 genomes was carried out with software BRIG [56], which run local BLAST-2.2.27+ and generated the figure in java.
Supporting Information
Resistance genes in the 11 Planctomycetaceae genomes.
(TIF)
Calculation of ANI (ANIb) and tetranucleotide frequencies.
(TIF)
Metabolic analysis of the 11 Planctomycetaceae species. Color of the heatmap box means gene number, which indicate in the color bar at the right of the heatmap; the colored box in legend at the left shows the same meaning with the colored text in the heatmap.
(PDF)
Reconstruction of the metabolic pathways of the 11 Planctomycetaceae genomes. Reconstruction of the metabolic pathways was implemented in the KEGG orthology system. The common metabolic pathways of the 11 Planctomycetaceae genomes are in green color, and their dispensable metabolic pathways are in red.
(TIF)
Protein families among the 11 Planctomycetaceae genomes.
(XLSX)
Gene distibution in the metabolic pathways of the 11 Planctomycetaceae genomes.
(XLSX)
Genes in the GIs of the five completed Planctomycetaceae genomes.
(XLSX)
Acknowledgments
We are very grateful to Professor Tianshen Tao from Wuhan University for his advice regarding this project. We thank our BGI colleagues for their skilled technical assistance and Professor Zhiping Zhang from Wuhan Institute of Virology, Chinese Academy of Science for his technical support.
Funding Statement
This work was supported by Shenzhen Key Laboratory of Bioenergy (grant CXB201005240001A), Shenzhen Key Laboratory of Environmental Microbial Genomics and Application(grant CXB201108250097A) and by grants from the State Key Development Program for Basic Research of China (2009CB522600), the National Natural Science Foundation of China (Contract 30930001). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Jetten MS (2008) The microbial nitrogen cycle. Environ Microbiol 10: 2903–2909. [DOI] [PubMed] [Google Scholar]
- 2. Lango Z (2005) “Who has first observed planctomyces” (or data to the history of Planctomyces bekefii). Acta Microbiol Immunol Hung 52: 73–84. [DOI] [PubMed] [Google Scholar]
- 3. Henrici AT, Johnson DE (1935) Studies of Freshwater Bacteria: II. Stalked Bacteria, a New Order of Schizomycetes. J Bacteriol 30: 61–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schmidt JM, Starr MP (1979) Corniculate cell surface protrusions in morphotype II of the Blastocaulis-Planctomyces group of budding and appendaged bacteria. Curr Microbiol 3: 187–190. [Google Scholar]
- 5. Bondoso J, Albuquerque L, Nobre MF, Lobo-da-Cunha A, da Costa MS, et al. (2011) Aquisphaera giovannonii gen. nov., sp. nov., a planctomycete isolated from a freshwater aquarium. Int J Syst Evol Microbiol 61: 2844–2850. [DOI] [PubMed] [Google Scholar]
- 6. Schlesner H, Rensmann C, Tindall BJ, Gade D, Rabus R, et al. (2004) Taxonomic heterogeneity within the Planctomycetales as derived by DNA-DNA hybridization, description of Rhodopirellula baltica gen. nov., sp. nov., transfer of Pirellula marina to the genus Blastopirellula gen. nov. as Blastopirellula marina comb. nov. and emended description of the genus Pirellula. Int J Syst Evol Microbiol 54: 1567–1580. [DOI] [PubMed] [Google Scholar]
- 7.Lee HW, Roh SW, Shin NR, Lee J, Whon TW, et al.. (2012) Blastopirellula cremea sp. nov. isolated from an ark clam in Gangjin Bay, South Korea. Int J Syst Evol Microbiol. [DOI] [PubMed]
- 8.Chavan A, Shastri AR, Ross-Russell RI (2012) Branchio-oto-renal syndrome with obstructive sleep apnoea. BMJ Case Rep 2012. [DOI] [PMC free article] [PubMed]
- 9. Giovannoni SJ, Schabtach E, Castenholz RW (1987) lsosphaera pallida, gen. and comb. nov., a gliding, budding eubacterium from hot springs. Microbiology 147: 276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clum A, Tindall BJ, Sikorski J, Ivanova N, Mavrommatis K, et al. (2009) Complete genome sequence of Pirellula staleyi type strain (ATCC 27377). Stand Genomic Sci 1: 308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clum A, Tindall BJ, Sikorski J, Ivanova N, Mavromatis K, et al. (2010) Erratum to: Complete genome sequence of Pirellula staleyi type strain (ATCC 27377). Stand Genomic Sci 2: 228–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Labutti K, Sikorski J, Schneider S, Nolan M, Lucas S, et al. (2010) Complete genome sequence of Planctomyces limnophilus type strain (Mu 290). Stand Genomic Sci 3: 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kulichevskaya IS, Ivanova AO, Belova SE, Baulina OI, Bodelier PL, et al. (2007) Schlesneria paludicola gen. nov., sp. nov., the first acidophilic member of the order Planctomycetales, from Sphagnum-dominated boreal wetlands. Int J Syst Evol Microbiol 57: 2680–2687. [DOI] [PubMed] [Google Scholar]
- 14. Guo M, Han X, Jin T, Zhou L, Yang J, et al. (2012) Genome sequences of three species in the family Planctomycetaceae. J Bacteriol 194: 3740–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kulichevskaya IS, Ivanova AO, Baulina OI, Bodelier PL, Damste JS, et al. (2008) Singulisphaera acidiphila gen. nov., sp. nov., a non-filamentous, Isosphaera-like planctomycete from acidic northern wetlands. Int J Syst Evol Microbiol 58: 1186–1193. [DOI] [PubMed] [Google Scholar]
- 16. Kulichevskaya IS, Serkebaeva YM, Kim Y, Rijpstra WI, Damste JS, et al. (2012) Telmatocola sphagniphila gen. nov., sp. nov., a novel dendriform planctomycete from northern wetlands. Front Microbiol 3: 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nardi E, Palermo A, Cusimano P, Mule G, Cerasola G (2011) Young woman with branchio-oto-renal syndrome and a novel mutation in the EYA-1 gene. Clin Nephrol 76: 330–333. [DOI] [PubMed] [Google Scholar]
- 18. Fuerst JA, Sagulenko E (2011) Beyond the bacterium: planctomycetes challenge our concepts of microbial structure and function. Nat Rev Microbiol 9: 403–413. [DOI] [PubMed] [Google Scholar]
- 19. Jun SR, Sims GE, Wu GA, Kim SH (2010) Whole-proteome phylogeny of prokaryotes by feature frequency profiles: An alignment-free method with optimal feature resolution. Proc Natl Acad Sci U S A 107: 133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brochier C, Philippe H (2002) Phylogeny: a non-hyperthermophilic ancestor for bacteria. Nature 417: 244. [DOI] [PubMed] [Google Scholar]
- 21. Wecker P, Klockow C, Ellrott A, Quast C, Langhammer P, et al. (2009) Transcriptional response of the model planctomycete Rhodopirellula baltica SH1(T) to changing environmental conditions. BMC Genomics 10: 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Akao T, Yashiro I, Hosoyama A, Kitagaki H, Horikawa H, et al. (2011) Whole-genome sequencing of sake yeast Saccharomyces cerevisiae Kyokai no. 7. DNA Res 18: 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jogler C, Waldmann J, Huang X, Jogler M, Glockner FO, et al. (2012) Identification of proteins likely to be involved in morphogenesis, cell division, and signal transduction in Planctomycetes by comparative genomics. J Bacteriol 194: 6419–6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Richter M, Rossello-Mora R (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A 106: 19126–19131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Konstantinidis KT, Tiedje JM (2005) Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A 102: 2567–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fuerst JA, Sagulenko E (2012) Keys to eukaryality: planctomycetes and ancestral evolution of cellular complexity. Front Microbiol 3: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. LaCasse EC, Lochnan HA, Walker P, Lefebvre YA (1993) Identification of binding proteins for nuclear localization signals of the glucocorticoid and thyroid hormone receptors. Endocrinology 133: 2760. [DOI] [PubMed] [Google Scholar]
- 28. Sims GE, Jun SR, Wu GA, Kim SH (2009) Alignment-free genome comparison with feature frequency profiles (FFP) and optimal resolutions. Proc Natl Acad Sci U S A 106: 2677–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vergin KL, Urbach E, Stein JL, DeLong EF, Lanoil BD, et al. (1998) Screening of a fosmid library of marine environmental genomic DNA fragments reveals four clones related to members of the order Planctomycetales. Appl Environ Microbiol 64: 3075–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brinkmann H, Philippe H (1999) Archaea sister group of Bacteria? Indications from tree reconstruction artifacts in ancient phylogenies. Mol Biol Evol 16: 817–825. [DOI] [PubMed] [Google Scholar]
- 31. Yarza P, Richter M, Peplies J, Euzeby J, Amann R, et al. (2008) The All-Species Living Tree project: a 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst Appl Microbiol 31: 241–250. [DOI] [PubMed] [Google Scholar]
- 32. Glockner FO, Kube M, Bauer M, Teeling H, Lombardot T, et al. (2003) Complete genome sequence of the marine planctomycete Pirellula sp. strain 1. Proc Natl Acad Sci U S A 100: 8298–8303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lonhienne TG, Sagulenko E, Webb RI, Lee KC, Franke J, et al. (2010) Endocytosis-like protein uptake in the bacterium Gemmata obscuriglobus. Proc Natl Acad Sci U S A 107: 12883–12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hieu CX, Voigt B, Albrecht D, Becher D, Lombardot T, et al. (2008) Detailed proteome analysis of growing cells of the planctomycete Rhodopirellula baltica SH1T. Proteomics 8: 1608–1623. [DOI] [PubMed] [Google Scholar]
- 35. Gade D, Gobom J, Rabus R (2005) Proteomic analysis of carbohydrate catabolism and regulation in the marine bacterium Rhodopirellula baltica. Proteomics 5: 3672–3683. [DOI] [PubMed] [Google Scholar]
- 36. Kerger BD, Mancuso CA, Nichols PD, White DC, Langworthy T, et al. (1988) The budding bacteria, Pirellula and Planctomyces,with atypical 16S rRNA and absence of peptidoglycan, show eubacterial phospholipids and uniquely high portions of long chain beta-hydroxy fatty acids in the lipopolysaccharide lipid A. Arch Microbiol. 149: 255–260. [Google Scholar]
- 37. Sinninghe Damste JS, Rijpstra WI, Geenevasen JA, Strous M, Jetten MS (2005) Structural identification of ladderane and other membrane lipids of planctomycetes capable of anaerobic ammonium oxidation (anammox). FEBS J 272: 4270–4283. [DOI] [PubMed] [Google Scholar]
- 38. Leary BA, Ward-Rainey N, Hoover TR (1998) Cloning and characterization of Planctomyces limnophilus rpoN: complementation of a Salmonella typhimurium rpoN mutant strain. Gene 221: 151–157. [DOI] [PubMed] [Google Scholar]
- 39. Cornell MJ, Alam I, Soanes DM, Wong HM, Hedeler C, et al. (2007) Comparative genome analysis across a kingdom of eukaryotic organisms: specialization and diversification in the fungi. Genome Res 17: 1809–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kleiner M, Petersen JM, Dubilier N (2012) Convergent and divergent evolution of metabolism in sulfur-oxidizing symbionts and the role of horizontal gene transfer. Curr Opin Microbiol 15: 621–631. [DOI] [PubMed] [Google Scholar]
- 41. Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, et al. (1997) The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390: 249–256. [DOI] [PubMed] [Google Scholar]
- 42. Ermolaeva MD, White O, Salzberg SL (2001) Prediction of operons in microbial genomes. Nucleic Acids Res 29: 1216–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, et al. (2000) Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406: 959–964. [DOI] [PubMed] [Google Scholar]
- 44. Dhillon BK, Chiu TA, Laird MR, Langille MG, Brinkman FS (2013) IslandViewer update: improved genomic island discovery and visualization. Nucleic Acids Res 41: W129–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Langille MG, Hsiao WW, Brinkman FS (2008) Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinformatics 9: 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li L, Stoeckert CJ Jr, Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13: 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, et al. (2007) DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57: 81–91. [DOI] [PubMed] [Google Scholar]
- 48. Teeling H, Meyerdierks A, Bauer M, Amann R, Glockner FO (2004) Application of tetranucleotide frequencies for the assignment of genomic fragments. Environ Microbiol 6: 938–947. [DOI] [PubMed] [Google Scholar]
- 49. Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599. [DOI] [PubMed] [Google Scholar]
- 51. Gade D, Schlesner H, Glockner FO, Amann R, Pfeiffer S, et al. (2004) Identification of planctomycetes with order-, genus-, and strain-specific 16S rRNA-targeted probes. Microb Ecol 47: 243–251. [DOI] [PubMed] [Google Scholar]
- 52. Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, et al. (2005) InterProScan: protein domains identifier. Nucleic Acids Res 33: W116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Letunic I, Yamada T, Kanehisa M, Bork P (2008) iPath: interactive exploration of biochemical pathways and networks. Trends Biochem Sci 33: 101–103. [DOI] [PubMed] [Google Scholar]
- 55. Darling AC, Mau B, Blattner FR, Perna NT (2004) Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14: 1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA (2011) BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12: 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Resistance genes in the 11 Planctomycetaceae genomes.
(TIF)
Calculation of ANI (ANIb) and tetranucleotide frequencies.
(TIF)
Metabolic analysis of the 11 Planctomycetaceae species. Color of the heatmap box means gene number, which indicate in the color bar at the right of the heatmap; the colored box in legend at the left shows the same meaning with the colored text in the heatmap.
(PDF)
Reconstruction of the metabolic pathways of the 11 Planctomycetaceae genomes. Reconstruction of the metabolic pathways was implemented in the KEGG orthology system. The common metabolic pathways of the 11 Planctomycetaceae genomes are in green color, and their dispensable metabolic pathways are in red.
(TIF)
Protein families among the 11 Planctomycetaceae genomes.
(XLSX)
Gene distibution in the metabolic pathways of the 11 Planctomycetaceae genomes.
(XLSX)
Genes in the GIs of the five completed Planctomycetaceae genomes.
(XLSX)