

Abstract
DDB2 is a protein playing an essential role in the lesion recognition step of the global genome sub-pathway of nucleotide excision repair (GG-NER) process. Among the proteins involved in the DNA damage response, p21CDKN1A (p21) has been reported to participate in NER, but also to be removed by proteolytic degradation, thanks to its association with PCNA. DDB2 is involved in the CUL4-DDB1 complex mediating p21 degradation; however, the direct interaction between DDB2, p21 and PCNA has been never investigated. Here, we show that DDB2 co-localizes with PCNA and p21 at local UV-induced DNA-damage sites, and these proteins co-immunoprecipitate in the same complex. In addition, we provide evidence that p21 is not able to bind directly DDB2, but, to this end, the presence of PCNA is required. Direct physical association of recombinant DDB2 protein with PCNA is mediated by a conserved PIP-box present in the N-terminal region of DDB2. Mutation of the PIP-box resulted in the loss of protein interaction. Interestingly, the same mutation, or depletion of PCNA by RNA interference, greatly impaired DDB2 degradation induced by UV irradiation. These results indicate that DDB2 is a PCNA-binding protein, and that this association is required for DDB2 proteolytic degradation.
Keywords: DDB2, PCNA, UV damage, nucleotide excision repair, p21CDKN1A
Introduction
Damage DNA binding protein subunit 2 (DDB2) forms, together with DDB1, the complex UV-DDB,1 which has affinity for the major types of lesions introduced in DNA by UV irradiation, such as 6–4 photoproducts (6–4PPs) and cyclobutane pyrimidine dimers (CPDs).2,3 Cells, to remove the UV damage, may use nucleotide excision repair (NER) mechanism that is divided in 2 sub-pathways called transcriptional coupled repair (TC-NER) and global genome repair (GG-NER).4 In GG-NER, DDB1 translocates into the nucleus and binds DDB2 promoting the recognition of photolesions.5 In particular, DDB2, encoded by the XP-E gene, is able to bind UV-damaged DNA and, in the early GG-NER process, it has a crucial role as initial damage recognition factor.6 Moreover, DDB2 and DDB1 associate with Cullin 4A (CUL4A),7 and DDB2 is necessary for DDB1 binding to DNA damage sites, as shown by the crystal structure of the UV-DDB complexed with damaged DNA .8,9 The CUL4A-DDB1-DDB2 (CRL4) complex has an ubiquitin ligase activity targeting histones H3 and H4, whose ubiquitination is important for recruiting the NER factor XPC to UV-damaged chromatin.10,11 Recently, it has been also demonstrated that DDB2 plays a role in the chromatin decondensation to favor the DNA damage repair, independently of the CRL4 complex.12
DDB2 itself is a substrate for CRL4 complex, and its degradation regulates the XPC recruitment to damaged DNA and, consequently, the repair of UV-induced CPDs. These results support the idea that DDB2 degradation is crucial for XPC binding to DNA damage sites during the initial steps of NER.6,13,14
DDB2 regulates the cellular levels of the cell cycle inhibitor p21CDKN1A (p21), an important player in the DNA damage response that was suggested to participate in various DNA repair processes thanks to its interaction with PCNA.14 In particular, in NER p21 was found to disrupt the association of p300 with PCNA,15 thereby removing the inhibitory effect on the acetylation of histones,16 but also of NER factors, such as XPG.17 Other lines of evidence indicated that after DNA damage, p21 was degraded by the CUL4A-DDB1Ctd2 complex through a PCNA-dependent mechanism.18,19 DDB2 was also found to be important for the ubiquitination mediating p21 proteasomal degradation;20,21 however, the molecular interplay between DDB2, p21 and PCNA has never been elucidated.
In the present study, we have investigated whether PCNA may directly interact with DDB2 and mediate its binding to p21; in addition, we have studied whether this association may influence proteolytic degradation of DDB2. We provide evidence that DDB2 co-localized with both p21 and PCNA to the DNA damage sites; however, this phenomenon was not observed in cells expressing a p21 mutant protein unable to interact with PCNA (p21PCNA–), thus indicating that p21 could not interact directly with DDB2. These results were also confirmed by immunoprecipitation and pull-down experiments with cell extracts, and with recombinant proteins, respectively. DDB2 was found to bind directly to PCNA through a conserved PCNA-interacting protein (PIP) box present in the N-terminal region of DDB2 sequence. Finally, we demonstrate that this direct interaction is important to DDB2 degradation, since a PIP-box mutant DDB2 protein was impaired in PCNA binding, was not degraded, and accumulated in cells exposed to UV irradiation. Similarly, depletion of PCNA by RNA interference (RNAi) significantly prevented proteolytic degradation of DDB2.
Results
DDB2 co-localizes with p21 and PCNA at DNA damage sites
In this work, a pcDNA3.1-DDB2 wild-type construct6 was expressed in HeLa cells, and nuclear localization and expression levels were confirmed by immunofluorescence microscopy and western blotting, respectively (data not shown). To study the recruitment of the exogenous DDB2 to DNA damage sites, HeLa cells were transfected, then exposed to local UV-C (100 J/m2) irradiation through filters with 3-μm pores. Five, 10, and 30 min later, the cells were processed for in situ hypotonic lysis, fixed, and immunostained with the antibody to CPDs. Confocal sections of green (CDPs) and red (DDB2) fluorescence signals show that DDB2 was localized to the irradiated area at least up to 30 min after UV-C irradiation (Fig. 1A). The merged image demonstrates that the exogenous DDB2 protein was able to bind damaged DNA, as visualized by the co-localization of DDB2 with CDPs-positive foci. The profiles of the pixel intensity of the green and red channels along the white line drawn in the merged image show the co-localization of DDB2 with CDPs at the damaged sites (Fig. 1B). Similar recruitment of DDB2 to DNA repair sites was observed at all time points examined (data not shown).

Figure 1. Co-localization of DDB2-NER proteins at DNA damaged sites. HeLa cells were co-transfected with DDB2 and p21wt-GFP constructs. For color images, refer to the online version of the article. (A) Twenty-four h after transfection, cells were exposed to local UV-C irradiation (100 J/m2) through filters with 3-μm pores, extracted in situ and then fixed for immunofluorescence staining with anti-DDB2 (red fluorescence) and anti-CDPs or anti-PCNA antibody (green fluorescence; p21 protein is detectable thanks to fusion with GFP). DNA was counterstained with Hoechst 33258 (blue fluorescence). Representative confocal merged images show the co-localization with the DDB2 protein. (B) The distribution profiles of the pixel intensity of the green and red channels along the white line drawn in the merged image show the co-localization of DDB2 with CDPs at the DNA damaged sites. (C) Confocal microscopy sections of irradiated HeLa cells showing DDB2 (red signal), PCNA (blue signal), p21 (green signal). The merged image proves the co-localization of the 3 proteins. (D) The distribution profiles of the pixel intensity for each fluorescence signal (blue, green, and red) analyzed by confocal microscopy are shown. (E) HeLa cells co-transfected with p21wt-GFP and DDB2 expression vectors and 24 h later exposed to UV-C radiation (100 J/m2). The cells were extracted in situ and fixed for determining the percentage of cells showing co-localization of DDB2-PCNA, DDB2-p21, and DDB2-PCNA-p21. Cells were analyzed at 5 min (empty bars), 10 min (gray bars), and 30 min (black bars) after UV-C irradiation.
To investigate the co-localization of DDB2 with p21, HeLa cells were transfected with DDB2 and p21wt-GFP constructs, due to the low levels of the latter protein in this cell line. Thus, following co-transfection with pcDNA3.1-DDB2 and p21wt-GFP plasmids, HeLa cells were locally irradiated, stained for DDB2 and PCNA, and analyzed by confocal microscopy. Figure 1C shows confocal sections of red, green, and blue fluorescence images of DDB2, p21, and PCNA, respectively, localized to UV-C exposed areas. Figure 1D shows the spatial profiles of red, green and blue fluorescence signals along the white line drawn in the merged image (Fig. 1C), demonstrating the co-localization of DDB2, p21, and PCNA proteins at UV-C-exposed areas. According to our previous results, the chromatin-bound p21wt-GFP was detectable in about 65% of transfected cells.22 Intriguingly, the results here obtained demonstrate that, after UV irradiation, the percentage of cells showing co-localization of DDB2 with p21wt-GFP was limited to about 30% and remained constant at all recovery time points considered (Fig. 1E). In contrast, a small but statistically significant increase (from about 70% up to about 90%) in the percentage of cells showing co-localization of DDB2 and PCNA associated to DNA damage sites was observed with time. The percentage of cells showing co-localization of the 3 proteins after irradiation ranged from about 40 to 50%, at all time points (Fig. 1E). These results indicate that the 3 proteins may be present together at the same DNA damage sites, but that DDB2 could co-localize with PCNA more frequently than with p21wt-GFP.
DDB2 interacts with p21 and PCNA proteins
In order to verify whether co-localization was accompanied by an interaction between DDB2, PCNA, and p21 proteins, HeLa cells expressing exogenous DDB2 and p21wt-GFP proteins were UV-irradiated, and the cell extracts were fractionated into the soluble and the chromatin-bound fractions. After immunoprecipitation with anti-DDB2 antibody, the chromatin-bound proteins were analyzed by western blot to detect the presence of proteins typically interacting with DDB2, such as DDB1, CUL4A, in addition to PCNA and p21. The CUL4A ubiquitin ligase was previously shown to target the chromatin-bound form of DDB2 under physiological conditions, as well as post-UV irradiation.23,24 Figure 2 shows that DDB2 was able to interact with CUL4A and, therefore, to participate, with DDB1 in the formation of UV-DDB complex. Interestingly, the results demonstrated that DDB2 recruited at chromatin interacts specifically with PCNA and p21, both before and after UV irradiation. These interactions remain well detectable at all considered time points.
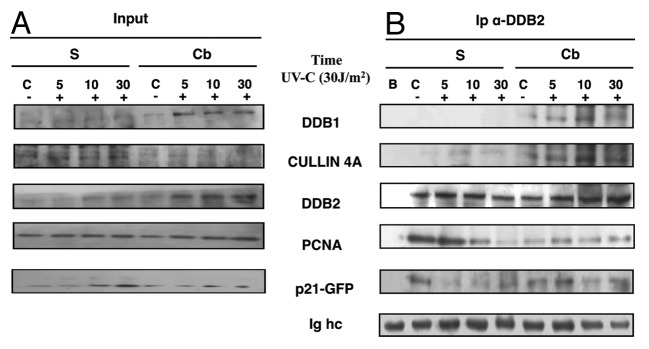
Figure 2. DDB2 interacts with NER proteins. HeLa cells co-transfected with pcDNA3.1-DDB2 and p21-GFP plasmids were grown for 24 h and collected before or 5, 10, and 30 min after UVC irradiation (30 J/m2), as described in “Materials and Methods”. Cells were fractionated in soluble (S) and chromatin-bound (Cb) samples for immunoprecipitation and analysis by western blot. (A) Input load: 1/30 of cell extract. (B) Immunoprecipitation (Ip) with anti-DDB2 antibody on fractionated cell extracts, indicated above. Not transfected cells (sample B) were used as a negative control.
Interaction of DDB2 with p21 is mediated by PCNA
To understand whether p21 is able to bind directly DDB2, HeLa cells were transfected with DDB2 construct and p21PCNA–-HA plasmid, driving the expression of a p21 mutant form unable to bind PCNA.25 In UV-C locally irradiated cells, p21PCNA– mutant was not recruited to the DNA damage sites, and did not co-localize with PCNA, as previously demonstrated,22 whereas DDB2 and PCNA clearly co-localized at sites of DNA damage, as indicated by violet spots in the merged image (Fig. 3A). To clarify this aspect, HA-tagged p21wt or p21PCNA– proteins were expressed in HeLa cells. Thirty min after UV-C irradiation, the cells were collected and fractionated in soluble and chromatin-bound samples. After immunoprecipitation with DDB2 antibody, it was found that DDB2 interacted with PCNA and p21wt both in soluble and in chromatin-bound fractions (Fig. 3B). Instead, in the sample transfected with the mutant p21PCNA– construct, no significant interaction between DDB2 and p21 was detectable in the chromatin-bound fraction, while the association between DDB2 and PCNA was observed in the soluble fraction.
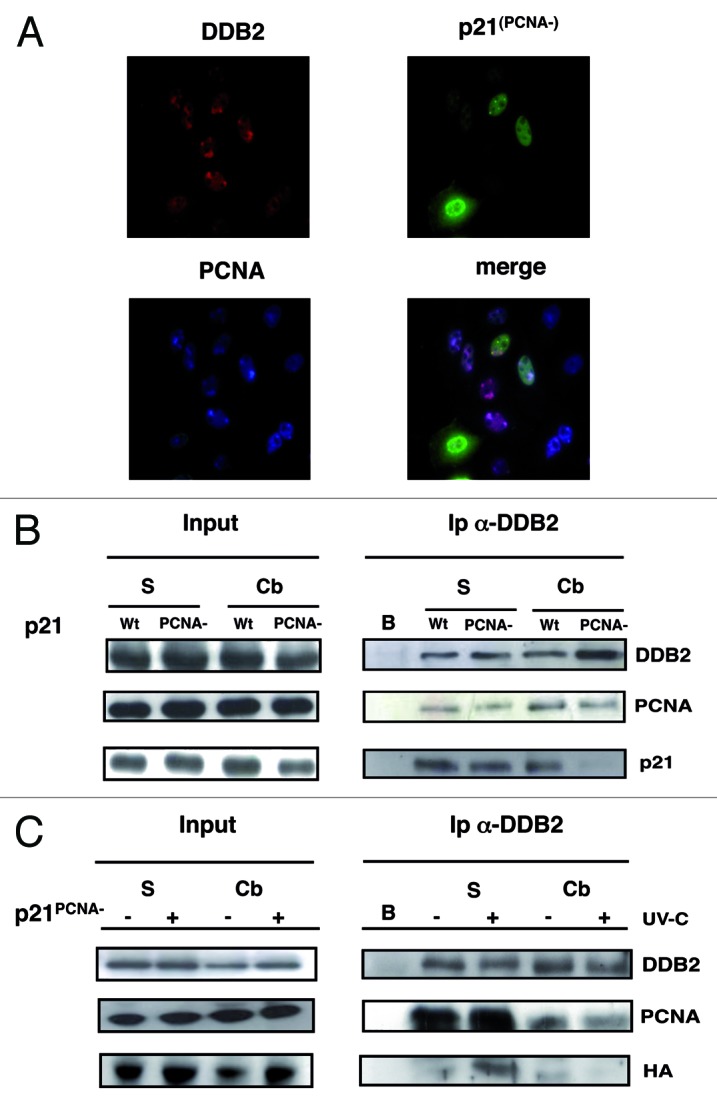
Figure 3. Interaction of DDB2 with p21 is mediated by PCNA. For color images, refer to the online version of the article. (A) HeLa cells grown on coverslips were lysed in situ and fixed 30 min after UV irradiation, as described in “Materials and Methods” for immunofluorescence analysis of chromatin-bound proteins: DDB2 (red fluorescence), PCNA (blue fluorescence), and HA-p21PCNA– (green fluorescence). (B) HeLa cells expressing exogenous DDB2 protein and HA-p21wt or HA-p21PCNA– were collected 30 min after UV-C irradiation (30 J/m2). Immunoprecipitation (Ip) with anti-DDB2 antibody from soluble (S), chromatin-bound (Cb) fractions. Not transfected cells (sample B). Input load: 1/30 of cell extracts. (C) Immunoprecipitation (Ip) with anti-DDB2 antibody of soluble (S), chromatin-bound (Cb) samples obtained before or after 30 min UV-irradiation (30 J/m2) from cells expressing DDB2 exogenous protein and HA-p21PCNA–. Not transfected cells (sample B). Input load: 1/30 of cell extracts.
For a better understanding of this interaction, HA-tagged p21 mutant protein was co-expressed with DDB2 in HeLa cells and analyzed before and after UV irradiation. Figure 3C shows that, after immunoprecipitation with anti-DDB2 antibody, p21PCNA– protein (as revealed by the anti-HA antibody) was detectable in the soluble, but not in the chromatin-bound, fraction in UV-irradiated cells. These results may be explained by considering that some p21PCNA– protein may still interact with PCNA through its CDK-binding domain.26 Remarkably, the interaction between DDB2 and PCNA was again found to occur independently of cell exposure to UV damage.
DDB2 physically associates with PCNA
To investigate in more detail whether the association between DDB2 and PCNA or p21 was of direct or indirect nature, recombinant His-tagged p21wt was compared with a mutant form (GST-p21ASM19) that does not interact with PCNA27 in a binding assay with recombinant PCNA and DDB2 proteins. Figure 4A shows the results obtained from an immunoprecipitation assay using anti-DDB2 antibody, as reported in the “Materials and Methods”. The first lane shows that p21 recombinant protein was unable to bind DDB2, while PCNA was able to do so, as shown by the presence of the band in lane 2. In lane 3, the results of the immunoprecipitate show that DDB2 interacted with p21 in the presence of PCNA. However, the interaction with the p21ASM19 mutant that cannot bind PCNA was not detectable even in the presence of PCNA (lane 4).

Figure 4. Direct interaction between PCNA and DDB2. (A) The analysis of interaction between proteins has been performed by immunoprecipitation, as reported in “Material and Methods”. DDB2 recombinant protein was present in each sample: p21wt-His protein was added in sample 1, PCNA recombinant protein was added in sample 2, both p21 and PCNA recombinant proteins were added in sample 3, and both PCNA and p21 mutant proteins were added in sample 4. (B) Identification and phylogenetic comparison of PIP-box in the DDB2 amino acidic sequence. The sequence is conserved from zebrafish to man. (C) Sequence alignment of the PIP-box motif of DDB2 with conserved PIP-box motifs of p21, another PCNA-interacting protein. The consensus PIP-box sequence is indicated. Underlined amino acids have been mutated in DDB2 gene. (D) In vitro binding assay of DDB2wt-PCNA (lane 1 and 3) and DDB2mut-PCNA (lane 2 and 4). On the left panel, immunoprecipitation (Ip) was performed with anti-DDB2 antibody; on the right, with anti-PCNA antibody. Immunoprecipitation was performed as described in “Materials and Methods”.
DDB2-PCNA interaction is important for DDB2 protein degradation
To understand the role of the interaction between PCNA and DDB2 proteins, we analyzed the DDB2 sequence and identified a possible PIP-box that appears to be well conserved in eukaryotes (Fig. 4B). To demonstrate the functionality of this PIP-box, we performed a site-directed mutagenesis in this region to produce a protein unable to interact with PCNA (DDB2mut) (Fig. 4C). After production and purification of recombinant DDB2wt and DDB2mut, these proteins were used to perform a new in vitro binding assay. The results showed that the interaction between DDB2 and PCNA was detectable only with the wild-type, but not with the mutant protein, demonstrating for the first time that DDB2 is a PCNA-binding protein through a typical PIP-box (Fig. 4D).
To investigate the functional significance of DDB2-PCNA interaction, we performed a time-course experiment with HeLa cells transfected with pcDNA3.1-DDB2wt or pcDNA3.1-DDB2mut, irradiated and collected at 0.5, 4, 7, and 14 h after DNA damage. The western blot analysis (Fig. 5A) showed that after UV-irradiation DDB2wt was degraded, as expected. This result was detectable at all time points after UV irradiation, and at 7 and 14 h the degradation was almost complete. On the contrary, in samples transfected with DDB2mut construct, the mutant protein was accumulated in the cells, and its degradation was evident only after 7 h from irradiation, suggesting that PCNA interaction was necessary for DDB2 degradation. To confirm this hypothesis, HeLa cells transfected with pcDNA3.1-DDB2wt were treated with the proteasome inhibitor MG132. As shown in Figure 5A (right panels), the DDB2wt protein was degraded more slowly in the presence than in the absence of MG132. In addition, the time course of DDB2wt degradation with MG132 was similar to that of the mutant form of DDB2. Densitometric analysis of the data, reported in Figure 5B, confirmed a statistically significant difference in the extent of degradation between the 2 forms of DDB2 protein.

Figure 5. DDB2 degradation is dependent on PCNA binding. (A) HeLa cells expressing DDB2wt or DDB2mut proteins were irradiated (30 J/m2) and collected 0.5, 4, 7, and 14 h later. Parallel samples of cells transfected with DDB2wt plasmid were also treated with MG132 proteasomal inhibitor and collected at the same times. The samples were fractionated, and the soluble form was analyzed by western blot for DDB2 content. In the same samples, actin levels, reported as loading control, are also shown. (B) DDB2 protein levels in similar experiments, as those shown in (A), were quantified by densitometric analysis and normalized to actin values, as standard for protein loading. The mean values (± s.d.), obtained from 3 independent experiments are reported. Statistical analysis was performed by t test. *P < 0.05; **P < 0.01. (C) After transfection with PCNA siRNA, HeLa cells were UV irradiated, incubated for 4 h, and analyzed for DDB2 degradation.
A further demonstration of the role of PCNA in DDB2 degradation was obtained by using siRNA technology to suppress PCNA protein expression, as shown in Figure 5C. In cells transfected with non-targeting siRNA oligos, DDB2 protein was degraded after UV irradiation, as expected, since the intensity of the band was lower than that of the respective unirradiated control sample. In contrast, in PCNA siRNA irradiated cells, where the total level of PCNA was clearly decreased, DDB2 protein accumulated after DNA damage, the intensity of the band not being significantly different from that of the unirradiated control cells as quantified by densitometric analysis.
This finding is in line with the other results obtained and confirms that PCNA plays a role in the degradation of DDB2 protein.
Discussion
After UV irradiation, cells activate different proteins involved in NER mechanism to repair DNA damage; among these, we have previously shown that p21 is actively involved in the DNA repair process, thanks to its interaction with PCNA.14,22,28 A functional relationship between DDB2 and p21 has been reported in knockout mice,5,29 while no information was available about any interaction of DDB2 with PCNA. In this study we have investigated the association between DDB2, PCNA, and p21 proteins, providing evidence that DDB2 directly interacts with PCNA, and that this association is determinant for DDB2 degradation after UV damage.
We have found that DDB2, PCNA, and p21 proteins co-localize at the DNA damaged sites after local UV-C irradiation (Fig. 1A). The specific time course of co-localization between DDB2 and p21, or PCNA showed that the first 2 proteins co-localized only in a limited percentage of cells (about 30%), while the extent of co-localization of DDB2 and PCNA was much higher (Fig. 1E), suggesting that their interaction persisted longer. The immunoprecipitation results showed that DDB2 interacts with p21 and PCNA, both before and after UV irradiation (Fig. 2B), indicating that their association is required not only after DNA damage, but also in normal conditions. These data are in agreement with previous results showing that p21 co-immunoprecipitates with DDB2, even in the absence of DNA damage.20 Similarly, the association of p21 with DDB1-CUL4A has been shown to occur not only after DNA damage,18,19 but also in undamaged cells.30 In fact, DDB2 plays a role in targeting p21 for ubiquitination, as shown by the accumulation of p21 in DDB2-deficient cells.20 We show here that DDB2-p21 interaction is mediated by PCNA, because a p21 form mutated in the PCNA binding domain, and unable to bind PCNA, also failed to associate with DDB2 (Fig. 3). These results are in accordance with the ubiquitination process mediated by CUL4A-DDB1Ctd2 that occurs via a PCNA-dependent mechanism.31,32 Thus, p21 may be also targeted for degradation by a CRL4 complex formed by CUL4A-DDB1DDB2 in a PCNA-dependent manner.
The co-localization of DDB2 with PCNA, as well as the direct interaction between these proteins, shown not only after DNA damage but also in non-irradiated cells, suggested a specific biological role for this association. Thus, we inspected the DDB2 sequence to identify a possible region involved in this interaction. Remarkably, our findings (Fig. 4) indicate that the direct binding between DDB2 and PCNA is mediated by the conserved sequence termed PCNA-interacting protein box (PIP-box)33 that is present in most of the PCNA partners known so far.34,35 In DDB2 protein, the PIP-box (not previously described) is located in the N-terminal region (residues 87–94) near the first 66 residues important for DDB1 interaction.9 The whole region (aa 1–102) may be also accessible to PCNA to form the DDB1-CUL4A-PCNA complex32 or, alternatively, may undergo conformational changes to accommodate PCNA interaction. This PIP-box is functional, since its mutation abolished the interaction between DDB2 and PCNA, as clearly demonstrated in immunoprecipitation experiments with recombinant proteins.
Since PCNA has been shown to be monoubiquitinated by a CRL4Cdt2 complex,36 in addition to the RAD6/RAD18 complex,37 the direct association of DDB2 with PCNA could be explained in terms of PCNA being the substrate of the ubiquitin ligase complex CUL4A-DDB1 including DDB2 instead of Cdt2.32 However, the DDB2–PCNA interaction may have an alternative explanation, attributing to PCNA the role of substrate recognition factor for the E3 ligase CRL4 complex,31 in a way similar to that occurring for p21 and Cdt1 degradation.32 In support of this hypothesis, our results have shown that DDB2 mutated in the PIP-box (unable to bind PCNA, Fig. 5B) was accumulated in the cells, because its degradation after DNA damage was impaired. This finding was clearly confirmed by the data obtained from PCNA siRNA experiments, thus indicating that interaction with PCNA is important for DDB2 proteolytic turnover following DNA damage. Studies on dynamics of NER proteins recruitment showed a distinct binding rate of factors implicated in different stages of the NER reaction. In particular, PCNA showed a slower recruitment rate after DNA damage, compared with DDB2, XPC, or XPA proteins.38 This is in agreement with our data, in which the interaction between DDB2 and PCNA was more evident at 30 min after DNA damage, suggesting that this association occurs at a late time, after the early DNA lesion recognition step performed by DDB2. Very recently, it has been reported that Poly(ADP-ribosyl)ation of DDB2 is required to reduce the DDB2 affinity toward the DNA damaged sites.39,40 This observation does not exclude our data interpretation about the involvement of PCNA in driving DDB2 to degradation.
Based on the above lines of evidence, we propose a model, illustrated in Figure 6, giving evidence to the PCNA-mediated association of p21 with DDB2, and to the binding to PCNA through the DDB2 PIP-box identified in this study (Fig. 6A). This interaction, which is required to mediate DDB2 degradation, occurs after the initial recognition step of DNA damage (Fig. 6B).

Figure 6. Schematic representation of the interactions between DDB2, p21 and PCNA. (A) DDB2 interacts directly with PCNA through a conserved PIP-box, while the binding to p21 is mediated by PCNA. For simplification, other NER factors have not been drawn here. (B) DBB2 is first recruited to DNA damage sites and, after the recognition step, interacts with PCNA which then promotes its proteolytic degradation.
In conclusion these results indicate that: (1) DDB2 protein is not able to interact directly with p21; (2) DDB2 has a PIP-box in its N-terminal region mediating interaction with PCNA; and (3) this association is required to drive DDB2 degradation.
Materials and Methods
Plasmids and constructs
Expression constructs coding for p21wt or a mutant protein (p21PCNA–) deficient for PCNA interaction25 were cloned in pEGFP-N1 (Clontech), or pcDNA3 vectors, for the expression of p21-GFP or p21-HA fusion proteins, respectively, as previously described.26 Expression construct coding for DDB2 was kindly provided by Dr Q Wang.6 To obtain DDB2 mutated in the PIP-box sequence (DDB2mut), the DDB2 For 3M: CCATCTGTCG CCCAGGGGGC CCAGCAGTCC GCCTTGCAC and DDB2 Rev 3M: GTGCAAGGCG GACTGCTGGG CCCCCTGGGC GACAGATGG primers were used. The mutated gene was cloned in the same expression vector of the wt form and the sequence was verified. The p21wt, DDB2wt and DDB2mut, and PCNAwt were cloned in pET45 vector (Novagen) to produce his-tagged recombinant proteins in BL21 (DE3) bacteria. pGEX-p21ASM19 was kindly provided by B Ducommun.
Cell culture, transfection, and irradiation
HeLa S3 cell line was grown in Dulbecco modified Eagle medium (DMEM, Sigma) supplemented with 10% fetal bovine serum (FBS, Gibco BRL), 4 mM L-glutamine (Gibco BRL), 100 U/ml penicillin, 100 μg/ml streptomycin in a 5% CO2 atmosphere. Cells seeded on coverslips or petri dishes were transiently transfected 24 h later with Effectene transfection reagent (Qiagen) (about 70% confluence), and irradiation was usually performed 24 h (about 70% confluence) after transfection. Cell exposure to UV-C was performed with a lamp (Philips TUV-9) emitting mainly at 254 nm, at doses of 30 or 100 J/m2, as measured with a DCRX radiometer (Spectronics). Localized irradiation was performed by laying Isopore polycarbonate filters (Millipore) with 3-μm pores on top of the cells.41
Immunofluorescence and confocal microscopy
HeLa cells seeded on coverslips were transfected as described above. After 24 h, the cells were locally irradiated and re-incubated in whole medium for the indicated period of time (5, 10, or 30 min). The cells on coverslips were then washed twice in cold PBS, fixed, and lysed in buffer containing freshly made 2% paraformaldehyde, 0.5% Triton X-100 in PBS, 0.2 mM phenylmethylsulfonyl fluoride (PMSF), and 0.2 mM Na3VO4 for 30 min at 4 °C. Thereafter, the samples were washed twice with cold PBS and then treated with 2 M HCl at 37 °C for 10 min to denature the DNA, followed by a PBS rinse to remove HCl.42 After re-hydration, the samples were blocked in PBST buffer (PBS, 0.2% Tween 20) containing 1% bovine serum albumin (BSA), and then incubated for 1 h with specific antibodies: goat polyclonal anti-DDB2 goat (1:100 Santa Cruz), mouse monoclonal TDM anti-CPDs (1:3000, MBL), and PC10 anti-PCNA (1:100, Dako), and rabbit polyclonal anti-p21 C-terminal polyclonal (1:100, Santa Cruz), all diluted in PBST buffer/BSA. After washing, each reaction was followed by incubation for 30 min with anti-mouse or anti-rabbit antibody conjugated with Alexa 488, anti-goat Alexa Fluor 594, anti-mouse Alexa 350 (Molecular Probes). After immunoreactions, the cells were incubated with Hoechst 33258 dye (0.5 μg/ml) for 2 min at RT and washed in PBS. The slides were mounted in Mowiol (Calbiochem) containing 0.25% 1,4- diazabicyclo-octane (Aldrich) as antifading agent. Images of fixed cells were taken with a Nikon Eclipse E400 fluorescence microscope equipped with a Canon Power Shot A590 IS digital camera. Fluorescence signals were acquired with a Leica TCS SP2 or TCS SP5 II Leica confocal microscope, at 0.3-μm intervals through the Z-stack. Image analysis was performed on confocal slices using the LAS AF software.
Western blot and pull down
For blot analysis, the cells were directly lysed in SDS sample buffer (65 mM Tris-HCl pH 7.5, 1% SDS, 30 mM dithiothreitol [DTT], 10% glycerol, 0.02% Bromophenol Blue), or fractionated in soluble and chromatin-bound fraction, as previously described43 with minor modifications. The cells were lysed in hypotonic buffer containing 10 mM Tris-HCl (pH 7.4), 2.5 mM MgCl2, 1 mM PMSF, 0.5% Nonidet NP-40, 0.2 mM Na3VO4, and a mixture of protease and phosphatase inhibitor cocktails (Sigma). After 10 min on ice, the cells were pelleted by low-speed centrifugation (200 g, 1 min), and the detergent-soluble fraction was recovered. Lysed cells were washed once in hypotonic buffer, followed by a second wash in 10 mM Tris-HCl buffer (pH 7.4), containing 150 mM NaCl, and protease/phosphatase inhibitor cocktails. Cell pellets were then incubated with DNase I (20 U/106 cells) in 10 mM Tris-HCl (pH 7.4), 5 mM MgCl2, and 10 mM NaCl for 15 min at 4 °C. After a brief sonication on ice, the samples were centrifuged again (13 000 g, 1 min), and the supernatant containing the chromatin-bound fraction was collected. For immunoprecipitation, about 107 cells were re-suspended in 1 ml lysis buffer and fractionated as above. Equal amounts of each extract were incubated with anti-DDB2 rabbit polyclonal antibody (Santa Cruz), pre-bound to protein G Dynabeads (Invitrogen). Half the amount of each antibody was used for chromatin-bound fractions. The reactions were performed for 3 h at 4 °C under constant agitation. The samples were then centrifuged at 14 000 g (30 min, 4 °C), and immunocomplexes were washed with ice-cold 50 mM Tris-HCl (pH 7.4) containing 150 mM NaCl, 0.5% Nonidet NP-40. Immunoprecipitated peptides were eluted in SDS sample buffer and resolved by SDS-PAGE (SDS-PAGE) on NuPage pre-casted gels (Life Technologies). Proteins were electrotransferred to nitrocellulose, then membranes were blocked for 30 min in 5% non-fat milk in PBST buffer and probed with the following primary antibodies: anti-DDB1 (1:1000, Genetex) anti-DDB2 (1:500, Santa Cruz), anti-PCNA (1:1000, Dako), anti-p21 (1:500, Santa Cruz), anti-CUL4A (1:500, Rockland). The membranes were then washed in PBST, incubated for 30 min with appropriate HRP-conjugated secondary antibodies (Amersham), and revealed using enhanced chemiluminescence. All the pull-down experiments were performed at least 3 times.
Recombinant proteins and in vitro assay
The his-tag recombinant proteins (p21wt, DDB2wt, DDB2mut, and PCNA) were purified using commercial Ni-conjugated resin (Sigma) according to the manufacturer’s instructions. GST-p21ASM19 mutant that does not bind PCNA was purified as previously described.43 GST-p21ASM19 has the same mutation of p21PCNA– construct used to transfect HeLa cells. Equimolar concentrations of p21wt, p21 mutant, PCNA (1 μM each) were incubated with 250 ng of DDB2 protein for 1 h at 4°C in 50 mM Tris-HCl buffer (pH 8.0), containing 2.5 mM MgCl2, 75 mM KCl, 10% glycerol, 1 mM DTT, and 0.2 mM PMSF. Fifty microliters of Protein G coupled to magnetic beads (Dynabeads, Life Technologies) were pre-incubated with DDB2 antibody for 1 h at 4 °C and added to each sample. Then, they were kept under agitation for 2 h, washed and eluted by SDS-denaturing buffer. The interactions between DDB2 and the 3 proteins were analyzed by western blot.
DDB2wt and DDB2mut proteins were used to perform in vitro immunoprecipitation assay with PCNA recombinant protein. Anti-DDB2 (or anti-PCNA) antibody was bound to the beads, then each sample was added, and the same protocol described above was used.
Analysis of DDB2 degradation
HeLa cells were transfected with DDB2wt or DDB2mut, as previously described. Twenty-four h later, the cells were irradiated and collected after 0.5, 4, 7, or 14 h. The same experiment was performed in HeLa cells transfected with DDB2wt and treated with 50 µM MG132 proteasome inhibitor. All the samples were lysed and the soluble fractions were quantified and separated in SDS-PAGE and analyzed by western blot. Specific antibodies revealed DDB2 and actin (1:1000, Sigma) proteins, and the densitometric analysis was performed using the public software ImageJ (http://rbs.info.nih.gov/nih-image). Results are expressed as mean ± standard deviation. Statistical significance was calculated using the Student t test.
siRNA treatment
HeLa cells were treated with PCNA siRNA or non-targeting siRNA (Dharmacon) for 48 h 44, then transfected with DDB2 wild-type, and after 24 h irradiated with UV-C. The cells were then washed twice in PBS and lysed as previously described. Samples were separated in SDS-PAGE and analyzed by western blotting following the protocol as above described.
Acknowledgments
This work was supported by AIRC IG grants (no. 5126 and no. 11747 to EP). We gratefully thank Q Wang (Ohio University) for gift of plasmid DDB2-pcDNA3, B Ducommun (Université Sabatier, Toulouse, France) for gift of plasmids pGEX-p21ASM19, and p21-HA constructs and S Sabbioneda for gift of non-targeting siRNA pool and PCNA siRNA. We thank also P Vaghi (Centro Grandi Strumenti, Università di Pavia) for help in confocal microscopy analysis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26987
References
- 1.Fujiwara Y, Masutani C, Mizukoshi T, Kondo J, Hanaoka F, Iwai S. Characterization of DNA recognition by the human UV-damaged DNA-binding protein. J Biol Chem. 1999;274:20027–33. doi: 10.1074/jbc.274.28.20027. [DOI] [PubMed] [Google Scholar]
- 2.Wakasugi M, Kawashima A, Morioka H, Linn S, Sancar A, Mori T, Nikaido O, Matsunaga T. DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. J Biol Chem. 2002;277:1637–40. doi: 10.1074/jbc.C100610200. [DOI] [PubMed] [Google Scholar]
- 3.Wittschieben BØ, Iwai S, Wood RD. DDB1-DDB2 (xeroderma pigmentosum group E) protein complex recognizes a cyclobutane pyrimidine dimer, mismatches, apurinic/apyrimidinic sites, and compound lesions in DNA. J Biol Chem. 2005;280:39982–9. doi: 10.1074/jbc.M507854200. [DOI] [PubMed] [Google Scholar]
- 4.Stoyanova T, Roy N, Bhattacharjee S, Kopanja D, Valli T, Bagchi S, Raychaudhuri P. p21 cooperates with DDB2 protein in suppression of ultraviolet ray-induced skin malignancies. J Biol Chem. 2012;287:3019–28. doi: 10.1074/jbc.M111.295816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–74. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 6.Barakat BM, Wang QE, Han C, Milum K, Yin DT, Zhao Q, Wani G, Arafa SA, El-Mahdy MA, Wani AA. Overexpression of DDB2 enhances the sensitivity of human ovarian cancer cells to cisplatin by augmenting cellular apoptosis. Int J Cancer. 2010;127:977–88. doi: 10.1002/ijc.25112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alekseev S, Kool H, Rebel H, Fousteri M, Moser J, Backendorf C, de Gruijl FR, Vrieling H, Mullenders LH. Enhanced DDB2 expression protects mice from carcinogenic effects of chronic UV-B irradiation. Cancer Res. 2005;65:10298–306. doi: 10.1158/0008-5472.CAN-05-2295. [DOI] [PubMed] [Google Scholar]
- 8.Alekseev S, Luijsterburg MS, Pines A, Geverts B, Mari PO, Giglia-Mari G, Lans H, Houtsmuller AB, Mullenders LH, Hoeijmakers JH, et al. Cellular concentrations of DDB2 regulate dynamic binding of DDB1 at UV-induced DNA damage. Mol Cell Biol. 2008;28:7402–13. doi: 10.1128/MCB.01108-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yeh JI, Levine AS, Du S, Chinte U, Ghodke H, Wang H, Shi H, Hsieh CL, Conway JF, Van Houten B, et al. Damaged DNA induced UV-damaged DNA-binding protein (UV-DDB) dimerization and its roles in chromatinized DNA repair. Proc Natl Acad Sci U S A. 2012;109:E2737–46. doi: 10.1073/pnas.1110067109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H, Zhai L, Xu J, Joo HY, Jackson S, Erdjument-Bromage H, Tempst P, Xiong Y, Zhang Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol Cell. 2006;22:383–94. doi: 10.1016/j.molcel.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 11.Kapetanaki MG, Guerrero-Santoro J, Bisi DC, Hsieh CL, Rapić-Otrin V, Levine AS. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci U S A. 2006;103:2588–93. doi: 10.1073/pnas.0511160103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luijsterburg MS, Lindh M, Acs K, Vrouwe MG, Pines A, van Attikum H, Mullenders LH, Dantuma NP. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J Cell Biol. 2012;197:267–81. doi: 10.1083/jcb.201106074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuss JO, Tainer JA. XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA Repair (Amst) 2011;10:697–713. doi: 10.1016/j.dnarep.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cazzalini O, Perucca P, Savio M, Necchi D, Bianchi L, Stivala LA, Ducommun B, Scovassi AI, Prosperi E. Interaction of p21(CDKN1A) with PCNA regulates the histone acetyltransferase activity of p300 in nucleotide excision repair. Nucleic Acids Res. 2008;36:1713–22. doi: 10.1093/nar/gkn014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cazzalini O, Scovassi AI, Savio M, Stivala LA, Prosperi E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat Res. 2010;704:12–20. doi: 10.1016/j.mrrev.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 16.Hong R, Chakravarti D. The human proliferating Cell nuclear antigen regulates transcriptional coactivator p300 activity and promotes transcriptional repression. J Biol Chem. 2003;278:44505–13. doi: 10.1074/jbc.M303138200. [DOI] [PubMed] [Google Scholar]
- 17.Tillhon M, Cazzalini O, Nardo T, Necchi D, Sommatis S, Stivala LA, Scovassi AI, Prosperi E. p300/CBP acetyl transferases interact with and acetylate the nucleotide excision repair factor XPG. DNA Repair (Amst) 2012;11:844–52. doi: 10.1016/j.dnarep.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 18.Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 2008;22:2496–506. doi: 10.1101/gad.1676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T, Tsurimoto T. CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J Biol Chem. 2008;283:29045–52. doi: 10.1074/jbc.M806045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stoyanova T, Yoon T, Kopanja D, Mokyr MB, Raychaudhuri P. The xeroderma pigmentosum group E gene product DDB2 activates nucleotide excision repair by regulating the level of p21Waf1/Cip1. Mol Cell Biol. 2008;28:177–87. doi: 10.1128/MCB.00880-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stoyanova T, Roy N, Kopanja D, Bagchi S, Raychaudhuri P. DDB2 decides cell fate following DNA damage. Proc Natl Acad Sci U S A. 2009;106:10690–5. doi: 10.1073/pnas.0812254106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perucca P, Cazzalini O, Mortusewicz O, Necchi D, Savio M, Nardo T, Stivala LA, Leonhardt H, Cardoso MC, Prosperi E. Spatiotemporal dynamics of p21CDKN1A protein recruitment to DNA-damage sites and interaction with proliferating cell nuclear antigen. J Cell Sci. 2006;119:1517–27. doi: 10.1242/jcs.02868. [DOI] [PubMed] [Google Scholar]
- 23.Nag A, Bondar T, Shiv S, Raychaudhuri P. The xeroderma pigmentosum group E gene product DDB2 is a specific target of cullin 4A in mammalian cells. Mol Cell Biol. 2001;21:6738–47. doi: 10.1128/MCB.21.20.6738-6747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen X, Zhang Y, Douglas L, Zhou P. UV-damaged DNA-binding proteins are targets of CUL-4A-mediated ubiquitination and degradation. J Biol Chem. 2001;276:48175–82. doi: 10.1074/jbc.M106808200. [DOI] [PubMed] [Google Scholar]
- 25.Cayrol C, Ducommun B. Interaction with cyclin-dependent kinases and PCNA modulates proteasome-dependent degradation of p21. Oncogene. 1998;17:2437–44. doi: 10.1038/sj.onc.1202189. [DOI] [PubMed] [Google Scholar]
- 26.Cazzalini O, Perucca P, Riva F, Stivala LA, Bianchi L, Vannini V, Ducommun B, Prosperi E. p21CDKN1A does not interfere with loading of PCNA at DNA replication sites, but inhibits subsequent binding of DNA polymerase delta at the G1/S phase transition. Cell Cycle. 2003;2:596–603. doi: 10.4161/cc.2.6.502. [DOI] [PubMed] [Google Scholar]
- 27.Podust VN, Podust LM, Goubin F, Ducommun B, Hübscher U. Mechanism of inhibition of proliferating cell nuclear antigen-dependent DNA synthesis by the cyclin-dependent kinase inhibitor p21. Biochemistry. 1995;34:8869–75. doi: 10.1021/bi00027a039. [DOI] [PubMed] [Google Scholar]
- 28.Cazzalini O, Donà F, Savio M, Tillhon M, Maccario C, Perucca P, Stivala LA, Scovassi AI, Prosperi E. p21CDKN1A participates in base excision repair by regulating the activity of poly(ADP-ribose) polymerase-1. DNA Repair (Amst) 2010;9:627–35. doi: 10.1016/j.dnarep.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 29.Liu L, Lee S, Zhang J, Peters SB, Hannah J, Zhang Y, Yin Y, Koff A, Ma L, Zhou P. CUL4A abrogation augments DNA damage response and protection against skin carcinogenesis. Mol Cell. 2009;34:451–60. doi: 10.1016/j.molcel.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 2008;22:2507–19. doi: 10.1101/gad.1703708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Havens CG, Shobnam N, Guarino E, Centore RC, Zou L, Kearsey SE, Walter JC. Direct role for proliferating cell nuclear antigen in substrate recognition by the E3 ubiquitin ligase CRL4Cdt2. J Biol Chem. 2012;287:11410–21. doi: 10.1074/jbc.M111.337683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Havens CG, Walter JC. Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 2011;25:1568–82. doi: 10.1101/gad.2068611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Warbrick E. The puzzle of PCNA’s many partners. Bioessays. 2000;22:997–1006. doi: 10.1002/1521-1878(200011)22:11<997::AID-BIES6>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 34.Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–79. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 35.Prosperi E. The fellowship of the rings: distinct pools of proliferating cell nuclear antigen trimer at work. FASEB J. 2006;20:833–7. doi: 10.1096/fj.05-5469hyp. [DOI] [PubMed] [Google Scholar]
- 36.Terai K, Abbas T, Jazaeri AA, Dutta A. CRL4(Cdt2) E3 ubiquitin ligase monoubiquitinates PCNA to promote translesion DNA synthesis. Mol Cell. 2010;37:143–9. doi: 10.1016/j.molcel.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–41. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 38.Vermeulen W. Dynamics of mammalian NER proteins. DNA Repair (Amst) 2011;10:760–71. doi: 10.1016/j.dnarep.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 39.Robu M, Shah RG, Petitclerc N, Brind’Amour J, Kandan-Kulangara F, Shah GM. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc Natl Acad Sci U S A. 2013;110:1658–63. doi: 10.1073/pnas.1209507110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, Hensbergen P, Deelder A, de Groot A, Matsumoto S, et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J Cell Biol. 2012;199:235–49. doi: 10.1083/jcb.201112132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katsumi S, Kobayashi N, Imoto K, Nakagawa A, Yamashina Y, Muramatsu T, Shirai T, Miyagawa S, Sugiura S, Hanaoka F, et al. In situ visualization of ultraviolet-light-induced DNA damage repair in locally irradiated human fibroblasts. J Invest Dermatol. 2001;117:1156–61. doi: 10.1046/j.0022-202x.2001.01540.x. [DOI] [PubMed] [Google Scholar]
- 42.Wei Q, Lee JE, Gershenwald JE, Ross MI, Mansfield PF, Strom SS, Wang LE, Guo Z, Qiao Y, Amos CI, et al. Repair of UV light-induced DNA damage and risk of cutaneous malignant melanoma. J Natl Cancer Inst. 2003;95:308–15. doi: 10.1093/jnci/95.4.308. [DOI] [PubMed] [Google Scholar]
- 43.Riva F, Savio M, Cazzalini O, Stivala LA, Scovassi IA, Cox LS, Ducommun B, Prosperi E. Distinct pools of proliferating cell nuclear antigen associated to DNA replication sites interact with the p125 subunit of DNA polymerase delta or DNA ligase I. Exp Cell Res. 2004;293:357–67. doi: 10.1016/j.yexcr.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 44.Niimi A, Brown S, Sabbioneda S, Kannouche PL, Scott A, Yasui A, Green CM, Lehmann AR. Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells. Proc Natl Acad Sci U S A. 2008;105:16125–30. doi: 10.1073/pnas.0802727105. [DOI] [PMC free article] [PubMed] [Google Scholar]