

Abstract
Despite recent advances in medical procedures, cardiovascular disease remains a clinical challenge and the leading cause of mortality in the western world. The condition causes progressive smooth muscle cell (SMC) dedifferentiation, proliferation, and migration that contribute to vascular restenosis. The incidence of disease of the internal mammary artery (IMA), however, is much lower than in nearly all other arteries. The etiology of this IMA disease resistance is not well understood. Here, using paired primary IMA and coronary artery SMCs, serum stimulation, siRNA knockdowns, and verifications in porcine vessels in vivo, we investigate the molecular mechanisms that could account for this increased disease resistance of internal mammary SMCs. We show that the residue-specific phosphorylation profile of the retinoblastoma tumor suppressor protein (Rb) appears to differ significantly between IMA and coronary artery SMCs in cultured human cells. We also report that the differential profile of Rb phosphorylation may follow as a consequence of differences in the content of cyclin-dependent kinase 2 (CDK2) and the CDK4 phosphorylation inhibitor p15. Finally, we present evidence that siRNA-mediated CDK2 knockdown alters the profile of Rb phosphorylation in coronary artery SMCs, as well as the proliferative response of these cells to mitogenic stimulation. The intrinsic functional and protein composition specificity of the SMCs population in the coronary artery may contribute to the increased prevalence of restenosis and atherosclerosis in the coronary arteries as compared with the internal mammary arteries.
Keywords: CDK2, cell proliferation, cell migration, coronary artery, internal mammary artery, smooth muscle cells, retinoblastoma protein phosphorylation
Introduction
Although the internal mammary artery (IMA) is anatomically located near the coronary artery (CA) and carries blood under the same pressure, this artery is uniquely resistant to restenosis and atherosclerosis. The internal mammary artery SMC layer has a significantly lower susceptibility to intimal hyperplasia as compared with the coronary artery and nearly all other arteries. The reasons for such resistance are still not completely elucidated and might be of great importance for our strategies to prevent restenosis and atherosclerosis.
SMCs as a main cell component in restenosis and atherosclerotic lesions are believed to be critical in the development of vascular disease. Mature vascular smooth muscle cells from different vascular territories exhibit distinct intrinsic properties and can perform both contractile and synthetic functions.1-4 Several studies related to the role of SMC in the disease process emphasize SMC apoptosis and lipid deposition. It has been proposed that the low cell migration capacity of the internal mammary SMCs in response to mitogenic factors might be one of the reasons for the disease “immunity”.5 The molecular mechanisms that underlie the arterial territory-specific SMCs propensity to either maintain the contractile phenotype or to proliferate and migrate in response to mitogenic factors are not well understood and are the subject of intense research.
The earliest event in the vascular medial cell layer, leading to SMC proliferation, is likely to be the altered expression of genes that control cell cycle transitions.4 Experiments with actively proliferating cell culture models and neointimal SMCs have revealed that the accumulation of several cyclins and CDKs is a critical event that serves as molecular trigger of cell cycle reentry.6,7 When cells are stimulated by extracellular mitogenic stimuli, active cyclin–CDK complexes accumulate and phosphorylate the retinoblastoma protein (Rb), an inhibitor of cell cycle progression, altering its affinity for binding to transcription factors. Physical interactions of hypophosphorylated Rb with the transcription factor YY1, for example, affect the differentiation of primary human coronary artery SMC in cell cultures.8,9 Rb also interacts with other transcription factors to repress their activity in the diverse processes of cell proliferation.10 The diverse functions of Rb are regulated by phosphorylation at more than 16 unique sites.10 Presently, the precise Rb phosphorylation profile, as well as the cell cycle progression proteins that convey the Rb coronary artery SMC and the Rb internal mammary artery SMC phosphorylation codes, are not well known. Additionally, vascular territory-specific proliferative and cell migration responses to mitogenic stimuli remain only partly characterized.11,12
The abillity of CDKs to phosphorylate Rb is regulated not only by interactions with cyclins, but also by association with 2 major classes of phosphorylation inhibitors, the INK4s and p21Cip1, p27Kip1, and p57Kip2.12-19
To date, there is little evidence that human SMCs from internal mammary and coronary arteries differ in content of proteins from the cell cycle control system that could partly explain their selective propensity to proliferate and migrate in response to mitogenic factors. The possibility of the presence of differential phospho-Rb species in internal mammary and coronary artery SMCs, which could provide hints for possible cell cycle specificities in regulation, has also not been investigated.
In experiments with paired human primary IMA and CA SMC cell cultures, we compared their proliferative and cell migration response to serum stimulation. We investigated the specific contents of cell cycle-regulatory proteins, phosphorylated Rb variants, and the transcription factor YY1 in the 2 cell types. We also used porcine IMA and CA tissue sections to verify some differences that we had observed in the cell culture experiments. Using CDK2-deficient cells, we established and compared the mechanistic role of CDK2 on the propensity of coronary artery SMCs to proliferate and migrate in response to serum stimulation. We propose that a CDK2-dependent control system, that features differential phosphorylation patterns of Rb at specific residues, alters the proliferative and migration response to mitotic stimuli and could explain, at least in part, the arterial territory-specific susceptibility to restenosis and atherosclerosis.
Results
Coronary artery SMCs exhibit greater proliferative and migration capacity than internal mammary artery SMCs in primary cell cultures
Based on presently available knowledge and our previously published data, we hypothesized that human SMCs derived from the internal mammary and coronary arteries would differ in their proliferative and cell migration response to mitogenic stimuli. Therefore, we compared the impact of serum stimulation on human SMCs from the aforementioned vessels. Cellular proliferation was measured with the 3H-thymidine incorporation assay in serum-starved (no FBS, growth arrested) and serum-stimulated (10% FBS in the medium) cells. In these paired primary cell culture studies, coronary artery SMCs exhibited a higher proliferation rate (11.98 ± 2.10-fold higher 3H-thymidine incorporation, n = 4) as compared with low-serum media (Fig. 1A). In contrast, serum stimulation of internal mammary SMCs showed only a 2.46 ± 0.91-fold higher 3H-thymidine incorporation rate (n = 4) as compared with cells in low-serum media. Comparison of the response to serum stimulation of coronary artery SMCs vs. internal mammary SMCs demonstrated an average 3.4-fold higher rate of proliferation of the former.
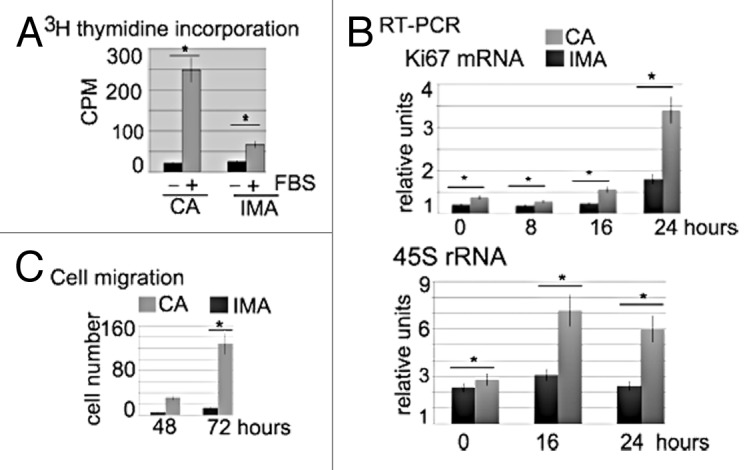
Figure 1. Proliferative and migration activity of coronary artery SMCs and internal mammary artery SMCs. (A) Comparison of [3H] thymidine incorporation in cultured SMCs from the coronary (CA) and internal mammary (IMA) arteries 24 h after FBS stimulation. Cells that are cultured without FBS (−) serve as a control for serum-stimulated cells (FBS +). Each bar represents the amount of [3H] thymidine incorporated into DNA (y-axes, as measured by scintillation counting) for each cell type as shown. Values represent the mean +/− SEM of 4 experiments (4 cell preparations from 4 different donors) conducted in triplicate. Error bars represent the standard error of the mean.(B) Serum stimulated coronary artery (CA) and internal mammary (IMA) artery SMCs differ in Ki67 mRNA and 45S rRNA content at different time points after serum stimulation, as indicated in the panels. Expression levels of these genes were measured by RT-PCR from total RNA preparations (5 × 104 cells) using gene-specific primers. Each bar represents the fold-change in specific RNA content, using the ARPP0 mRNA level as an endogenous reference gene that does not change in response to treatment. Values represent the mean +/− SEM of 4 experiments (on 4 separate cell preparations from 4 different donors) conducted in triplicates. (C) Cell migration as measured by the number of cells (on the vertical axis) that are found to migrate into the cell free zone 48 and 72 h after scratching. The results are consistent between 4 independent experiments (on 4 separate cell preparations from 4 different donors). The P values are shown above the bars. Error bars represent the standard error of the mean.
Using real-time PCR analysis (RT-PCR), we also tested if internal mammary SMCs and coronary artery SMCs would differ in the expression of other markers of proliferation, including Ki67 and 45S rRNA (Fig. 1B). When the data of ARPP0 was considered as a reference value, serum treatment significantly increased the expression levels of Ki-67 mRNA, especially after 24 h of serum treatment of coronary artery SMCs. The same effect was seen (albeit to a lesser extent) in stimulated internal mammary artery SMCs. The relative value of Ki-67 mRNA in coronary artery SMCs after 24 h was 1.54-fold higher (± 0.20; P < 0.05, n = 4) as compared with the value for internal mammary SMCs. In addition, coronary artery SMCs exhibited a significantly higher 45S rRNA content, which peaked at 16 h after serum stimulation.
SMC migration and proliferation are known to be important processes involved in disease-related intimal thickening, and we compared the migration propensity of SMCs from the coronary artery and the internal mammary artery. The scratch assay was conducted with both cell cultures (Fig. 1C); the number of migrating cells was counted 48 and 72 h after scratching the plate. Results showed a clear difference in the number of migrating cells between the 2 cell types. While > 25 (+/−7, n = 4) coronary artery SMCs were found to migrate back into the cleared area, less than 5 internal mammary SMC (+/− 2, n = 4) were found in the scratched zone after 48 h. At 72 h the difference was also highly pronounced.
The LDH release assay did not reveal significant difference in the viability of growth-arrested and serum-stimulated coronary artery and the internal mammary artery SMCs (data not shown).
Combined, these data suggest a higher migration and proliferative capacity for coronary artery SMC as compared with internal mammary SMC in paired human primary cell cultures.
Differences between internal mammary SMCs and internal mammary SMCs in regard to transcription factor YY1 and Rb phosphorylation variants in the SMC of internal mammary and coronary arteries
The transcription factor YY1 is one of the major factors that trigger DNA synthesis in human coronary artery SMCs.8,9 It functions in cell cycle transitions and is, at least in part, regulated by interactions with hypo-phosphorylated Rb.8 We hypothesized that the observed difference in the proliferative responses of internal mammary and coronary artery SMCs to serum stimulation could be due to cell type-specific differences in the total YY1 and/or Rb protein content as well as in the Rb phosphorylation profile. We first measured the total protein content of both factors by western blotting in total cell lysates from growth-arrested and serum-stimulated internal mammary and coronary arteries SMC cultures over 24 h (Fig. 2). The specific protein content was quantified by the intensity of the immunological reactions in total cell lysates with equal amount of protein (0.05 mg). Total YY1 and Rb protein content did not significantly differ between cell types and could not explain the difference in the proliferative response to serum stimulation. Therefore, the total Rb content was used as an internal control for equal protein loading. SMCs from the internal mammary and coronary arteries may, however, differ in their relative content of phosphorylated Rb species. Therefore, we compared the effect of serum stimulation on the phosphorylation profile of Rb in the 2 arteries by western blot assay (Fig. 2). We used commercially available antibodies that recognize 6 different Rb species phosphorylated at S807, S795, S780, S612, S608, and T373. Statistically significant (P < 0.04, n = 3) increases were observed in the level of Rb phosphorylation at the S807, S608, and T373 sites in coronary artery SMCs, but not in internal mammary artery SMCs, in response to serum stimulation. In contrast, serum stimulation was associated with increased content of S612 and S780 variants in cells from both arteries (P < 0.05). Notably, we did not observe any Rb phosphorylation species whose content was higher in internal mammary SMC. These results were consistent between 3 independent experiments with paired human cell cultures. We conclude that in internal mammary SMCs, Rb appears to exist mostly as hypo-phosphorylated species and responds far less robustly to serum stimulation than that in coronary artery SMCs.

Figure 2. Content of transcription factor YY1, total Rb protein, and Rb phosphorylation variants in internal mammary SMCs and coronary artery SMCs. (A) Immunoblot analysis of IMA and CA cell lysates with anti-YY1, anti-total Rb, anti-pRb-S795, anti-pRb-S608, anti-pRb S780, anti-pRb S807, anti p-Rb S612, and anti-pRb-T373 antibodies as indicated above the plots. The identity of the cells is shown at the right. Cells were serum-starved for 24 h, then subsequently treated with 10% FBS, from 0 up to 24 h as indicated in the bottom panel. On the right side of the panel, the pixel intensity measurement is presented in pixel units normalized to total Rb. Black bars are used for internal mammary SMC values and gray bars represent the values of coronary artery SMCs. The results are representative of 3 independent experiments (3 paired cell cultures from 3 different donors). Error bars represent the standard error of the mean. The asterisks above the bars indicate P < 0.05. (B) The table summarizes the Rb phospho-variants distribution in internal mammary SMCs and coronary artery SMCs, as well as the CDKs and cyclins that have been shown to phosphorylate Rb at the tested amino acids in other cell lines and tissues: (+) indicates immunologically present variants; +/− indicates low content; (−) indicates absence of the tested variant.
Thus, although total YY1 and Rb protein contents are similar between coronary and internal mammary arteries, the pattern of overall higher Rb phosphorylation appears to be specific to the arterial origin of the SMC, as summarized in Figure 2B. The significant association of specific Rb phospho-variants with SMC cell type led us to hypothesize that differences in the artery-specific content of cyclins, cyclin-dependent kinases, and/or their inhibitors could, in turn, explain the variations in the profile of Rb phosphorylation.
Protein content of cyclins, CDKs, and CDK inhibitors in serum-stimulated internal mammary SMC and coronary artery SMC cultures
Site-specific phosphorylation of Rb is performed by different kinases throughout the cell cycle. Therefore, we hypothesized that the observed difference in the profile of Rb phosphorylation in the 2 cell types is due to differences in the content of cell cycle-regulatory factors. Since Rb phosphorylation occurs during the transition from growth arrest (G0/G1 phase) to the S phase, we compared the total protein contents of cyclin D, cyclin E, CDK2, and CDK4 at different time points after serum stimulation of growth-arrested cells over 24 h (Fig. 3A). The relative levels of each protein were quantified by the intensity of the immunological reactions in a western blot assay with 0.05 mg protein in each lane. In these experiments, we consistently observed higher levels of cyclin D (> 30%; P = 0.05, n = 4) in SMCs from the coronary artery as compared with those from internal mammary artery at 12 and 16 h after serum stimulation. Over the tested 24 h, the cyclin E content, however, was consistently higher at 0, 20, and 24 h (> 20%; P = 0.05, n = 3) in the internal mammary SMCs as compared with coronary artery SMCs. We next compared the total protein contents of CDK4 and CDK2, both of which act concurrently with cyclin D1 and cyclin E throughout the cell cycle, in the 2 cell types. The western blot experiments revealed a generally low content of CDK4, and nearly undetectable CDK2, in internal mammary artery SMCs during the test period of 24 h. In addition, serum stimulation did not have a measurable effect on the level of either of these kinases. In coronary artery SMCs, however, CDK2 was elevated >30% (P < 0.03, n = 3) at 12 h after serum addition. CDK4 content was found to increase >30%, at 12–20 h of serum stimulation in these cells as well.

Figure 3. Expression of cell cycle proteins in internal mammary SMCs and coronary artery SMCs. Cellular protein content was analyzed by western blotting with specific antibodies in equal amount (0.05 mg/lane) of cell lysates at different time points after serum stimulation. Cells are incubated with serum for the indicated time periods from 0 to 24 h (as shown at the bottom.) (A) The identity of the tested antibodies to human cyclin D, cyclin E, CDK2, CDK4 protein is shown above the western blots. The identity of the cells is shown at the right. (B) The identity of the tested antibodies to human p15, p16, p27, p21, and β-actin is shown above the western blots. The identity of the cells is shown at the right. On the right site of the western blot panels in (A and B) the pixel intensity measurement is presented in pixel units, normalized to β-actin as a loading control. Results are representative of 3 independent experiments (3 paired coronary artery SMCs and internal mammary artery SMCs cultures from 3 different donors). Error bars represent the standard error of the mean. *P = 0.05, **P < 0.03. (C) Artery (IMA and CA) specific distribution of cell cycle regulators, their cell cycle position (G1 and S phase), and participation in the degree of Rb phosphorylation (phosphorylated amino acids, red dots; empty dots, absence of Rb phosphorylation). The cell cycle regulators with high and artery specific content are shadowed in colors; **P < 0.05.
Significant differences in the content of the cyclin-dependent kinase inhibitor p15 and to some extend p16 and p21 were observed in internal mammary SMCs and coronary artery SMCs as well. Internal mammary SMCs consistently displayed nearly undetectable p16, higher content of p15 (>55%, P < 0.03, n = 3), and slightly more p21 (>10%, P = 0.05, n = 3) as compared with coronary artery SMCs (Fig. 3B). Although the cellular content of immunologically detectable p16 in coronary artery SMCs was relatively low, it showed cell cycle-dependent distribution, increasing >30% at 12 h after stimulation, which corresponds to the S phase of the cell cycle. Basal levels of p27 in both cell types were similar, and significantly higher (>2 times) in the first 4 h after serum stimulation in each. β-actin levels served as a control for equal loading in the western blot experiments as well as to normalize the individual band intensity during the test period.
The diagrams shown in Figure 3C summarize the observed artery specific distribution of cell cycle regulators, their cell cycle position, and participation in the profile of Rb phosphorylation. Overall, serum stimulation resulted in a significantly higher content of CDK2 in the coronary artery SMCs (and to lesser extent, cyclin D1 and CDK4) as compared with internal mammary SMCs. In contrast, the CDK2 inhibitor p15 predominates in the internal mammary SMCs independently of the serum stimulation. No significant differences in cell type specific levels were observed for cyclin E and p27.
The SMC in the porcine internal mammary artery and the coronary artery differ in protein content of CDK2, phospho Rb species, and expression of cell cycle regulators
To test whether the SMCs layers in the left anterior descending artery (LAD) and the internal mammary artery differ in content of CDK2 and phospho-Rb species in vivo (as we observed in the cell culture experiments), we looked for an appropriate in vivo model system. Since work in rodents is not representative of restenosis and atherosclerosis in humans, we used porcine vessels from 4 healthy 3-month-old male Ossabaw pigs.21
We used 2 assays to compare CDK2 and phospho-Rb species content between the 2 cell types. First, we compared the relative levels of CDK2 together with the Rb S807, T373, and S612 by western blotting using total tissue lysates from paired left internal mammary artery and LAD (Fig. 4A). The CDK2 content and the presence of RbT373 and RbS807 consistently showed a difference in the 4 pigs: their immunologically detectable content is higher in LAD than in the internal mammary artery lysates. YY1 and RbS612 are consistently present in both arteries. Fluorescent staining of LAD and internal mammary artery tissue sections with anti-CDK2 antibody verified the difference in the content of CDK2; the protein was immunologically present in the LAD SMC layer and nearly undetectable in the internal mammary artery (Fig. 4B). YY1 was present in the vascular bed of both arteries, and the intensity of staining revealed no significant difference in its levels in either artery type. Since the commercially available antibodies to p15, p16, p27, p21, and cyclin D produced neither a detectable nor a specific reaction in the pig tissue. We next conducted RT-PCR to compare their expression mRNA levels together with CDK2 and YY1 in both arteries. The relative level of CDK2 mRNA transcripts in LAD were >4 times higher than in the paired IMA (P < 0.05; n = 4 pigs). The relative level of p15 was determined to be >2.5 times higher in IMA than in LAD (P < 0.05, n = 4 pigs). The levels of YY1, p16, p21, and p27 mRNAs in LAD and IMA were nearly identical (P < 0.05; n = 4) relative to ARPPO (Fig. 4C).

Figure 4. Vascular bed distribution of CDK2 and YY1 in porcine internal mammary artery and LAD. (A) Western blot analysis with porcine total IMA and LAD lysates and RbT373, RbS708, RbS780, YY1, and CDK2 specific antibodies. All lanes received 0.05 mg of protein. The results from parallel experiments with LAD (CA) and IMA lysates from foul pigs (1, 2, 3, 4) are shown. (B) Immunofluorescence with porcine IMA and LAD (CA) paraffin sections (indicated at the top of the panels) was used to visualize the CDK2 and YY1 content and distribution within the vascular wall. The white single arrows indicate the position of the endothelial layer and the double arrows indicate the intimal layer with the smooth muscle cells. Specific rabbit anti-CDK2 and chicken anti-YY1 reactions were visualized with fluorescently labeled secondary antibodies (red). Nuclear DNA is stained with DAPI (blue). The smooth muscle cells layers are visualized with smooth muscle α actin-specific antibodies (green). The results are consistent between sections from 4 different pigs. (C) The CDK2 and YY1 mRNA levels as well as cyclin D, p16, p15, p21, p27 mRNAs are measured by RT-PCR from total IMA and CA RNA preparations with gene-specific primers. The bars represent the mRNA content of the target gene relative to ARPPO (ARPP0 level adjusted to 1): black, IMA; gray, CA. Values represent the mean+/− SEM of four experiments with paired RNAs from 4 different pigs. Error bars represent the standard error of the mean. **P < 0.01 (n = 4, 4 different pigs).
Of note, the smooth muscle cell layer in the LAD expresses more CDK2 protein than the SMC in the IMA. The p16 mRNA content in the LAD is only slightly higher than in the IMA, whereas p15, and to a lesser extent p21, mRNA predominate in the IMA.
CDK2 knockdown alters coronary artery SMC proliferation and migration
The expression of CDK2 in internal mammary SMCs vs. coronary artery SMCs represents a notable contrast. This protein is present in very low, nearly undetectable levels in the internal mammary SMCs, but has a stronger signal in the serum stimulated cells from the coronary artery. Moreover, we observed that some CDK2-related Rb phospho-variants predominate in the coronary artery SMC cultures. Based on our data and previous observations that reduced expression of CDK2 in actively proliferating cells causes growth arrest, we hypothesized that CDK2 knockdown by siRNA in the SMCs of the coronary artery would cause these cells to behave more similarly to internal mammary SMCs, i.e., exhibiting lower Rb phosphorylation and reduced proliferative propensity.22
We first tested for cytotoxicity in response to coronary artery SMC transfection with different amounts of CDK2 siRNAs and control siRNAs. A lactose dehydrogenase (LDH)-based cytotoxicity assay was performed on cells that were electroporated with 1, 2, 5, or 10 nM of siRNA. The assay revealed significant cytotoxicity in cells that received 10 nM of CDK2 siRNA (Fig. 5A), and this dose level was not further applied for subsequent experiments. We next tested if treatment with the nontoxic doses of 1, 2, or 5 nM CDK2 siRNA would alter the cellular CDK2 protein content (panel B). Transfection of SMCs with CDK2 siRNA, followed by 48 h cultivation in medium with 10% serum, consistently resulted in a specific decrease of CDK2 protein as revealed by western blot assay with total cell lysates (Fig. 5B). A dosage of 5 nM siRNA was found to be the most optimal, resulting in significantly reduced CDK2 protein content (>85%; P < 0.05; n = 3) 60 to 84 h post-transfection. We next employed the 5-nM dose to investigate the effect of CDK2 knockdown on the accumulation of the S807, T373, and S780 Rb variants. Western blotting revealed significantly reduced content of the S807 and T373 Rb species (>95%; P < 0.02; n = 3) in coronary artery SMC lysates in CDK2-deficient cells (Fig. 5C). The level of Ser780, however, did not change significantly. Notably, the presence of the S807 and T373 Rb species in the internal mammary SMCs remains very low before and after siRNA treatment (not shown).

Figure 5. CDK2 knockdown in SMCs. (A) LDH-based cytotoxicity assay of coronary artery SMCs treated with various doses of CDK2 siRNA. Cells received 1, 2, 5, and 10 nM of siRNA as indicated below the bars: gray bars represent cells that received control siRNA; black bars represent cells that received CDK2siRNA. The LDH value is shown on the vertical axis.(B) Depletion of CDK2 in total cell lysates (0.05 mg protein/lane) as shown by western blotting with CDK2-specific antibodies. SMCs were electroporated with increasing concentrations of CDK2 or control (C) siRNA, and cells were harvested and analyzed 78 h after electroporation. The amount of siRNA (0, 1, 2, and 5 nM) is shown below the lines. Migration of the immunoreactive CDK2 protein is shown on the right. (C) Western blots for content of phospho-T373, -S807, and -S780 variants (shown on the left) in lysates (0.05 mg protein/lane) from coronary artery SMC that received 5 nM control siRNA (C) or CDK2siRNA as shown below the lines. (D) A [3H]-thymidine incorporation assay was conducted with 3 × 104 siRNA treated internal mammary artery SMCs and coronary artery SMC from three pigs in triplicate. Doses of siRNA (0, 1, 2, and 5 nM) received in each treatment are shown below the bars. The bars represent the measured CPMs (on the vertical) and the values represent the mean+/− SEM of 3 independent experiments (3 paired coronary artery SMCs and internal mammary artery SMCs cultures from 3 different donors) (P < 0.05, n = 3). The identity of the bars is shown below the bars plot.
We next obtained evidence that CDK2 knockdown has an effect mainly on coronary artery SMC DNA replication. Using the [3H] thymidine incorporation assay, we compared the DNA replication rate of SMCs from the CDK2 siRNA and control siRNA-treated cells (Fig. 5D). This assay demonstrated that treatment with CDK2 siRNA dose-dependently reduces the rate of DNA synthesis in serum-stimulated coronary artery SMCs; in cells treated with 5 nM of CDK2 siRNA, thymidine incorporation dropped by >45% (P < 0.009, n = 3), significantly reducing the gap in the proliferative response to serum treatment between the coronary and siRNA-treated internal mammary SMCs. The rate of [3H] thymidine incorporation in the internal mammary SMC culture remains low and independent of the CDK2 knockdown (Fig. 5D).
These results indicate that the phosphorylation of Rb at T373 and S807 in coronary artery SMCs is controlled, at least in part, by CDK2. Knockdown of CDK2 revealed that it is a target for cell proliferation and cell migration control in the coronary artery SMCs. Coronary artery SMCs with reduced content of CDK2, T373, and S807-phosphorylated Rb, like the internal mammary SMCs, are less responsive to mitogenic stimuli. Of note, the internal mammary SMCs rate of proliferation remains low and does not change significantly in response to siRNA treatment.
Discussion
The principal novel finding of the present study is that SMCs from the coronary and the internal mammary arteries differ significantly in their content of CDK2 protein and specific phospho-Rb species. We also established that these variations are contributing factors for the observed divergent proliferative and migratory SMC properties. We have shown that coronary artery SMCs exhibit significantly lower proliferative capacity when the CDK2 content is reduced to the level in the internal mammary SMCs.
CDK2 appears to be critical for the coronary artery SMC response to serum stimulation. Similarly, the lower contents of CDK2 in the internal mammary SMCs may explain the weak proliferative response of these cells to serum stimulation. Our siRNA knockdown results directly demonstrate a requirement for CDK2 in the proliferation and migration of coronary SMCs. In the internal mammary SMCs, the cellular CDK2 content is low, and this coincides with the consistently reduced proliferative and migratory response to serum stimulation. The relatively low content of CDK2 in the porcine IMA smooth muscle layer (as compared with the matched coronary artery) further corroborated the significance of these findings. It appears that the expression of CDK2 is reduced not only in the human internal mammary SMC cultures, but also in the porcine IMA, suggesting a direct correlation between CDK2 levels and the low proliferative and migratory behavior of the SMC layer in the IMA from both species. Considering the established involvement of CDK2 in serum-mediated cell proliferation and migratory activity, it is quite plausible that low CDK2 may contribute significantly to the low proliferative IMA incidences in vivo.12,13 The remarkable resistance of IMA grafts to stenosis in patients could be partly attributed to the low content of CDK2. The reason for the lower CDK2 content in the internal mammary SMC and the porcine artery compared with LAD is presently not known and remains to be established.
CDK2, together with the cyclins D and E, is known to exert their role in cell cycle regulation by phosphorylating Rb at various sites. While existing evidence strongly indicates that phosphorylation inactivates Rb and is mediated by one or more CDKs, the Rb phosphorylation profile in SMCs from the internal mammary and coronary arteries has remained unexplored. Although in both cell types the total pRb protein content is nearly identical, we have seen a different pattern of Rb phosphorylation between them; for the most part, phosphorylation at sites T373, Ser608, S780, and S807 is detectable only in the coronary artery SMCs. The relatively high activity of CDK2 and CDK4 and their associated cyclins in the coronary artery SMCs may be causally related to this cell-specific Rb phosphorylation pattern, and is in stark contrast to the immunologically undetectable CDK2 in internal mammary SMCs. The higher p15 content in the internal mammary SMCs may also contribute to the decreased phosphorylation at sites T373 and S807 in these cells.
Furthermore, CDK2 knockdown coincides with a reduced content of the specific phospho-Rb-S807 variant in the coronary artery SMCs. Presently we do not know how exactly the Rb S807 variant contributes to the proliferative and the migratory SMCs response to mitogenic stimulation. Distinct patterns of Rb phosphorylation could have a profound effect on Rb activity as a cell cycle regulator by changing its binding affinity to different transcription factors. For example, Rb phosphorylation at S608/S612 has been shown to inhibit binding to E2F and subsequent activation of the latter in tumor cells.25,26,27 We have previously shown that only the hypophosphorylated form of Rb interacts with YY1.8 This interaction is an important determinant of the subcellular localization of YY1 and its gene expression regulatory functions in DNA replication and ribosomal 45SRNA synthesis. At present, we do not know the exact mechanism through which cell-specific patterns of phosphorylated Rb variants may contribute to the proliferative and the migratory capacity of coronary artery SMCs and internal mammary artery SMCs. Distinct phosphorylation events may control specific interactions with YY1 or other transcription factors that, together with CDK2 and the CDK inhibitors p15 and p16 (INK4s), shape the proliferative and migratory phenotype in a vascular territory-specific manner.16 Consequently, the Rb phosphorylation map might act as a code by which distinct phosphorylation events could control arterial territory-specific SMC behavior in response to mitogenic stimuli.27 The higher p15 protein content in internal mammary SMCs could, at least in part, explain their low proliferative response to serum stimulation when compared with the coronary artery SMCs. On the other hand, the low p15 content in coronary artery SMCs, together with the relatively high CDK4 and cyclin D levels, could explain the observed higher proliferation and migration in these cells. Although levels of the other INK4 inhibitor p16 are higher in coronary artery SMCs, the proliferative response of these cells still remains relatively high. Such a discrepancy is suggestive of a coronary artery SMC-specific p16 function, or perhaps interactions of p16 with other artery-specific factors that remain to be determined. The INK4 proteins are structurally redundant and equally potent cell cycle inhibitors that could harbor non-overlapping properties and distinct regulatory modes.23 Of note, the INK4 gene locus is only slightly distal to the coronary artery disease (CAD) region at the human Chr9p21.24
While the INK4 family members p15 and p16 mainly inhibit the activity of CDK4 and CDK6, the Cip/Kip members p21 and p27 inhibit a broader spectrum of cyclin–CDK complexes. Although the p21 level in the coronary artery SMC is only slightly higher in the internal mammary SMC, by acting simultaneously in cell cycle regulation and in gene transcription, p21 could also contribute to the observed vascular territory-specific difference in the proliferative and migratory response.28
The use of paired populations of equal passage human coronary artery and internal mammary artery SMCs was a strength of our study that let us control for interdonor variation in the results. An unavoidable limitation of the study, however, was the assumption that the behavior of SMCs in cell culture is comparable with that in the vascular wall. Given that the CDK2 content in the pig internal mammary artery SMCs layer is significantly lower than in the matched LAD is an important verification that the observed differences may also exist in patients.
Although the proliferation and migration of arterial SMC is thought to play a central role in vascular remodeling, we still do not fully understand the initial molecular processes that trigger neointimal formation. Our findings provide evidence that the signaling pathways that are involved in the proliferative and migratory response to mitogenic stimulation appear to differ according to the content of CDK2 and phospho-Rb species in the vascular territory from which the SMCs originate. The low CDK2 level, together with the profile of Rb phosphorylation and possibly the higher content of p15 in the internal mammary artery SMCs, may play a beneficial role in the prevention of vascular remodeling. These observations also suggest that CDK2 and specific phospho-Rb species may regulate the formation of the neointima in vascular injury and disease. If this is true, then alterations in their content may have a therapeutic potential for treatment of restenosis and atherosclerosis. Further functional studies are needed to explore the possible relationship between CDK2 expression and content of specific phospho-Rb variants in the arterial territory-specific susceptibility to restenosis and atherosclerosis.
Materials and Methods
Cell culture
Coronary artery SMC and internal mammary artery SMC from healthy, male Caucasians were obtained from Lonza, Cell Applications, Inc, and Coriell Institute for medical research. Cells were cultured in M199 medium (Sigma) containing endothelial cell growth supplement (Sigma, 0.02 mg/mL), Heparin (Sigma, 0.05mg/ml), L-glutamine (Invitrogen, 2 mM) and 10% fetal bovine serum (FBS) (Invitrogen). Cells were cultured at 37 °C in a humidified atmosphere and 5% CO2. After 24 h of serum starvation for cell cycle synchronization, cells were treated for 24 h with cell culture medium containing either 10% FBS for growth stimulation or 0.1% FBS for growth arrest. All experiments were conducted with paired cell preparations from 4 individual donors where coronary artery SMCs and internal artery SMC were obtained from the same donor and where their passage numbers did not differ.
Cell proliferation, migration, and toxicity assays
DNA synthesis was estimated by measuring [3H] thymidine incorporation. Cells were grown in 12-well tissue culture dishes. After 24 h of serum starvation for cell cycle synchronization, cells were treated for 24 h with cell culture medium containing either 10% FBS or 0.1% FBS as a control. [3H]thymidine (5μCi/ml) (Perkin Elmer) was added during the last 6 h of the 24 h treatment period. Cells were treated with 5% trichloroacetic acid for 30 min at 4 °C and subsequently harvested, and radioactivity was measured in a liquid scintillation counter (Beckman). The cell migration assay was conducted as previously reported.20 The Pierce Cytotoxicity Lactate Dehydrogenase (LDH) Assay Kit was used to measure the cytotoxicity of the siRNA treatment. The assay was conducted as recommended by the supplier (Thermo Scientific).
RT-PCR
Total RNA was extracted from human CASMCs and IMASMCs or whole porcine coronary (LAD) and left mammary arteries using the RNeasy mini Kit (Qiagen) according to the manufacturer's instructions. Three sets of control siRNA and CDK2siRNA SMC treatment and paired arterial isolations were assayed in triplicates with gene-specific primers (Invitrogene). Relative gene expression values were calculated after normalization to ARPP0 mRNA that did not change significantly in either the arteries, or in response to siRNA treatment. The value of the ARPP0 mRNA content was adjusted as 1. Results obtained in individual experiments were expressed as mean ± SEM.
Western blotting and immunohistochemistry
Total cell lysates were prepared with RIPA buffer. Western blotting was prepared with the total cell lysates (0.05 mg of protein/lane). In order to avoid differences related to the experimental conditions, the total proteins recovered from paired cell cultures were separated by SDS-PAGE together on 2 parallel gels (10-well gels, BioRad) in the same chamber, transferred onto nitrocellulose, and incubated together with the relevant antibodies. ImageJ was used to quantify average values for the relative protein abundance (in pixels) between the analyzed cell lysates. The total Rb protein content or β-actin were used as the loading controls. For immunohistochemistry staining, 5-μm-thick paraffin-embedded tissue sections of pig coronary artery and the internal mammary artery were used. Sections were immunostained with rabbit anti-CDK2 (Cell Signaling Technology) and chicken anti-YY1 antibodies (MediMabs, cat. #MM-0205-P). The following antibodies were used in the western blotting experiments: total anti-Rb antibody (Cell Signaling Technology), anti-phospho-Rb antibody S795, S608, S780, S807 (Cell Signaling Technology), S612 (Invitrogen), T373 (Santa Cruz), anti-cyclin D1 antibody (Cell Signaling Technology), anti-cyclin E antibody (Santa Cruz), anti-CDK4 antibody (Cell Signaling Technology), anti-CDK2 antibody (Cell Signaling Technology), anti-p27 antibody (Santa Cruz), anti-p21 antibody (Santa Cruz), anti-p16 INK4A antibody (Cell Signaling Technology), anti-p15 INK4B antibody (Cell Signaling Technology), chicken anti-YY1 antibody (MediMabs, Canada), anti-smooth-muscle-α-actin (SMAA) antibody (Sigma) and anti-β-actin antibody (Sigma) overnight at 4 °C.
Gene silencing with small interfering RNA (siRNA)
siRNA duplexes targeting CDK2 and control siRNA were obtained from Ambion. siRNAs were transfected into cells with the TRANS-II transfection reagent for siRNA (MediMabs) and the Amaxa Nucleofector transfection system. Cells were incubated for 84 h before sample collection. Successful knock-down of CDK2 at protein level was confirmed by immunoblotting analysis using anti-CDK2 antibody.
Glossary
Abbreviations:
- CA
coronary artery
- CAD
coronary artery disease
- CASMC
coronary artery smooth muscle cell
- CDK
cyclin-dependent kinase
- FBS
fetal bovine serum
- IMA
internal mammary artery
- IMASMC
internal mammary artery smooth muscle cell
- LAD
left anterior descending artery
- LDH
lactate dehydrogenase
- Rb
retinoblastoma protein
- siRNA
small interfering RNA
- SMC
smooth muscle cell
- YY1
ying yang 1
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Endnotes
This work was supported, in whole or in part, by National Institute of Health Grants R01HL062458 (to AU), R01HL46716, R01HL69024 (to FWS.), Training grant 5T32-HL076134 (to AL), American Heart Association Grant-in-Aid Program 11GRNT5250000 (to CB), and from the Thoracic Surgery Foundation for Research and Education Fellowship (to AL).
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27056
References
- 1.Majesky MW. Vascular smooth muscle diversity: insights from developmental biology. Curr Atheroscler Rep. 2003;5:208–13. doi: 10.1007/s11883-003-0026-x. [DOI] [PubMed] [Google Scholar]
- 2.Mekontso-Dessap A, Kirsch M, Guignambert C, Zadigue P, Adnot S, Loisance D, Eddahibi S. Vascular-wall remodeling of 3 human bypass vessels: organ culture and smooth muscle cell properties. J Thorac Cardiovasc Surg. 2006;131:651–8. doi: 10.1016/j.jtcvs.2005.08.048. [DOI] [PubMed] [Google Scholar]
- 3.Frischknecht K, Greutert H, Weisshaupt C, Kaspar M, Yang Z, Luscher TF, Carrel TP, Tanner FC. Different vascular smooth muscle cell apoptosis in the human internal mammary artery and the saphenous vein. Implications for bypass graft disease. J Vasc Res. 2006;43:338–46. doi: 10.1159/000093606. [DOI] [PubMed] [Google Scholar]
- 4.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–9. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 5.Mahadevan VS, Campbell M, McKeown PP, Bayraktutan U. Internal mammary artery smooth muscle cells resist migration and possess high antioxidant capacity. Cardiovasc Res. 2006;72:60–8. doi: 10.1016/j.cardiores.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 6.Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73:1059–65. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- 7.Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–4. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 8.Petkova V, Romanowski MJ, Sulijoadikusumo I, Rohne D, Kang P, Shenk T, Usheva A. Interaction between YY1 and the retinoblastoma protein. Regulation of cell cycle progression in differentiated cells. J Biol Chem. 2001;276:7932–6. doi: 10.1074/jbc.M007411200. [DOI] [PubMed] [Google Scholar]
- 9.Hiromura M, Choi CH, Sabourin NA, Jones H, Bachvarov D, Usheva A. YY1 is regulated by O-linked N-acetylglucosaminylation (O-glcNAcylation) J Biol Chem. 2003;278:14046–52. doi: 10.1074/jbc.M300789200. [DOI] [PubMed] [Google Scholar]
- 10.Tamrakar S, Rubin E, Ludlow JW. Role of pRB dephosphorylation in cell cycle regulation. Front Biosci. 2000;5:D121–37. doi: 10.2741/Tamrakar. [DOI] [PubMed] [Google Scholar]
- 11.Gerthoffer WT. Mechanisms of vascular smooth muscle cell migration. Circ Res. 2007;100:607–21. doi: 10.1161/01.RES.0000258492.96097.47. [DOI] [PubMed] [Google Scholar]
- 12.Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–63. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 13.Hunter T, Pines J. Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell. 1994;79:573–82. doi: 10.1016/0092-8674(94)90543-6. [DOI] [PubMed] [Google Scholar]
- 14.Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–61. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 15.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–7. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 16.Hirai H, Roussel MF, Kato JY, Ashmun RA, Sherr CJ. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol Cell Biol. 1995;15:2672–81. doi: 10.1128/mcb.15.5.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–16. doi: 10.1016/0092-8674(93)90499-G. [DOI] [PubMed] [Google Scholar]
- 18.Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 19.Matsuoka S, Edwards MC, Bai C, Parker S, Zhang P, Baldini A, Harper JW, Elledge SJ. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 1995;9:650–62. doi: 10.1101/gad.9.6.650. [DOI] [PubMed] [Google Scholar]
- 20.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–33. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 21.Vilahur G, Padro T, Badimon L. Atherosclerosis and thrombosis: insights from large animal models. J Biomed Biotechnol. 2011;2011:907575. doi: 10.1155/2011/907575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charron T, Nili N, Strauss BH. The cell cycle: a critical therapeutic target to prevent vascular proliferative disease. Can J Cardiol. 2006;22(Suppl B):41B–55B. doi: 10.1016/S0828-282X(06)70986-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cánepa ET, Scassa ME, Ceruti JM, Marazita MC, Carcagno AL, Sirkin PF, Ogara MF. INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life. 2007;59:419–26. doi: 10.1080/15216540701488358. [DOI] [PubMed] [Google Scholar]
- 24.Holdt LM, Teupser D. Recent studies of the human chromosome 9p21 locus, which is associated with atherosclerosis in human populations. Arterioscler Thromb Vasc Biol. 2012;32:196–206. doi: 10.1161/ATVBAHA.111.232678. [DOI] [PubMed] [Google Scholar]
- 25.Burke JR, Deshong AJ, Pelton JG, Rubin SM. Phosphorylation-induced conformational changes in the retinoblastoma protein inhibit E2F transactivation domain binding. J Biol Chem. 2010;285:16286–93. doi: 10.1074/jbc.M110.108167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis R, Pillai S, Lawrence N, Sebti S, Chellappan SP. TNF-α-mediated proliferation of vascular smooth muscle cells involves Raf-1-mediated inactivation of Rb and transcription of E2F1-regulated genes. Cell Cycle. 2012;11:109–18. doi: 10.4161/cc.11.1.18473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubin SM. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem Sci. 2013;38:12–9. doi: 10.1016/j.tibs.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 2003;13:65–70. doi: 10.1016/S0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]