

Abstract
We developed a method for deep mutational scanning of antibody complementarity-determining regions (CDRs) that can determine in parallel the effect of every possible single amino acid CDR substitution on antigen binding. The method uses libraries of full length IgGs containing more than 1000 CDR point mutations displayed on mammalian cells, sorted by flow cytometry into subpopulations based on antigen affinity and analyzed by massively parallel pyrosequencing. Higher, lower and neutral affinity mutations are identified by their enrichment or depletion in the FACS subpopulations. We applied this method to a humanized version of the anti-epidermal growth factor receptor antibody cetuximab, generated a near comprehensive data set for 1060 point mutations that recapitulates previously determined structural and mutational data for these CDRs and identified 67 point mutations that increase affinity. The large-scale, comprehensive sequence-function data sets generated by this method should have broad utility for engineering properties such as antibody affinity and specificity and may advance theoretical understanding of antibody-antigen recognition.
Keywords: antibody engineering, cetuximab, EGFR, mammalian display, next generation sequencing
Introduction
Protein engineering has proven successful at dramatically altering the function and utility of many proteins across multiple species and protein classes. In particular, the ability to alter binding of antibodies to their antigens has been shown to have substantial effects on their biological function and has led to the creation of numerous antibody therapeutics.1,2
Established display-based methods for engineering protein affinity involve generating large libraries of variants of a starting sequence, followed by multiple rounds of affinity-based selection. Such methods are capable of impressive affinity increases, but yield information for only the small number of higher affinity variants that dominate the final rounds of selection. Expression, folding and other biases can result in potential loss of useful variants, and information on neutral or lower affinity mutations is missing entirely. Alternatively, it would be desirable to efficiently and comprehensively determine the effect on affinity of all possible point mutations in a protein binding domain. Beneficial point mutations could be combined to achieve higher affinities, while information on neutral and lower affinity variants could inform engineering efforts aimed at other properties.
Recent advances in next-generation DNA sequencing (NGS),3 which can generate gigabases of sequence from millions of DNA templates in parallel at low cost, have revolutionized genomic research and are increasing being used as tools for molecular engineering. In particular, the use of NGS to analyze the results of sorted protein display libraries in deep mutational scanning approaches promises to be the method of choice for generation of very large sequence-function fitness landscapes.4
We devised a deep mutational scanning method in which NGS is used to determine the effect on affinity of every possible point mutation in an antibody binding domain. In this method, a DNA library comprising all possible single amino acid substitutions in the complementarity-determining regions (CDRs) is constructed and cloned into a vector that expresses the variants as fusion proteins tethered to the surface of mammalian cells via a trans-membrane anchor. The library is then transfected into cells and incubated with excess fluorescently-tagged antigen at a concentration approximately equal to the dissociation constant (KD) of the wild type interaction, so that the amount of antigen bound to each cell is proportional to the affinity of the displayed variant. The cells are sorted by flow cytometry into two subpopulations, with the first containing all cells expressing antibody above background and the second containing the subset of the first subpopulation with the highest amount of antigen bound. Plasmid DNA from the cells in the two subpopulations is recovered and sequenced using massively parallel pyrosequencing. Finally, the frequency of each mutant in each subpopulation is tabulated, and analysis of how the frequency of each mutant varies between the different subpopulations is used to generate an affinity ranking of the entire library.
Results
Humanization of the anti-EGFR antibody 225
The model system for this approach was the anti-epidermal growth factor receptor (EGFR) mouse antibody 225,5 the parent of cetuximab, which is approved in the US for the treatment of metastatic colorectal cancer and squamous cell cancer of the head and neck.6 A humanized form, hu225, was generated by structure-aided design (Fig. 1), expressed as an IgG1/kappa antibody and tested for affinity to EGFR. In a flow cytometry-based assay for binding to EGFR-expressing A431 cells, hu225 affinity was equivalent to cetuximab; however, affinity measurements using recombinant EGFR extracellular domain showed a ~4- to 5-fold loss of affinity for hu225 compared with a chimeric 225 that we prepared similarly or to cetuximab (Table 1).

Figure 1. Humanization of murine antibody 225 to create hu225. heavy (A) and light (B) variable domains of 225, hu225 and human acceptor frameworks 60P2 and NOV. CDRs (Kabat definition) are underlined in 225. Framework positions in hu225 where murine residues were retained are shown in bold and underlined. At VH position 12 double underlined and bold indicates where the infrequent Ile found in 60P2 was changed to Val as found in the human VH3 family consensus.
Table 1. Affinity measurements of chimeric and humanized anti-EGFR antibodies, as determined by surface plasmon resonance (Biacore), luminescent oxygen-channeling immunoassay (AlphaLISA) and flow cytometry.
| BIAcore | AlphaLISA | A431 FACS binding | ||||||
|---|---|---|---|---|---|---|---|---|
| ka | kd | KD | WT/x | EC50 | WT/x | EC50 | WT/x | |
| (M−1s−1) | (s−1) | (M) | (nM) | (ug/ml) | ||||
| cetuximab | 6.5E+05 | 1.1E-03 | 1.7E-09 | 1.0 | 0.09 | 1.00 | 2.01 | 1.00 |
| ch225 | 6.0E+05 | 1.1E-03 | 1.8E-09 | 0.9 | 0.08 | 1.13 | ND | ND |
| hu225 | 5.4E+05 | 4.6E-03 | 8.5E-09 | 0.2 | 0.33 | 0.27 | 1.79 | 1.12 |
Ratio of activity for chimeric (ch225) or humanized (hu225) variants compared with cetuximab is given as WT/x. ND, not determined.
Library construction, cell surface expression and fluorescence-activated cell sorting
Two libraries (one each for the heavy (VH) and light (VL) chain variable regions) comprising all possible single amino acid substitutions in the CDRs of hu225 were prepared by cloning 59 synthetic gene fragments with NNK degeneracy. The two libraries jointly contained 1121 single amino acid substitutions at 59 positions (32 VH and 27 VL positions). The libraries were cloned into pYA206, an Epstein-Barr virus-derived plasmid for mammalian cell surface display of IgG molecules,7 and transfected into 293c18 cells. Control experiments demonstrated that the library outgrowth conditions were sufficient to ensure plasmid segregation such that each cell expressed a single clonal variant (Fig. S1). FACS titration of displayed hu225 with Alexa Fluor 647-labeled EGFR extracellular domain-Cλ fusion protein (AF647-EGFR-Cλ) established a dissociation constant (KD) of 1.9 nM for the wild type interaction (Fig. S2). The VH and VL libraries were incubated with 1.0 nM AF647-EGFR-Cλ and phycoerythrin (PE)-labeled anti-human kappa to normalize for IgG expression variation and analyzed by flow cytometry (Fig. 2A−C). For cells expressing wild type hu225, a typical pattern was seen with a population of non-expressing cells in the lower left quadrant, while hu225 expressing cells formed a diagonal on the right half of the diagram (Fig. 2A). In contrast the diagonal was substantially broadened in the VH and VL libraries, indicating the presence of variants with both higher and lower affinities in the libraries (Fig. 2B and C). Flow cytometric gates were drawn as shown. The first gate (expression gate, or “Exp”) collected all cells expressing surface-displayed IgG regardless of antigen affinity, while the second gate (binding gate, or “Bind”) collected the brightest few percent of cells above the diagonal. Between 1.0 million and 2.4 million cells were collected from the expression and binding gates for sequence analysis.
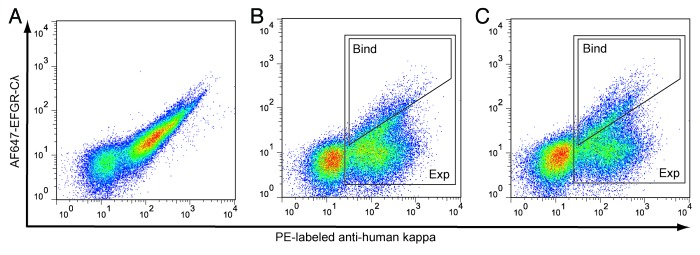
Figure 2. FACS sorting of 293c18 cells expressing surface-displayed hu225 and libraries. (A) wild type hu225 and (B-C) hu225 VH and VL libraries, respectively, were stained with AF647-EGFR-Cλ fusion protein and anti-human kappa PE (to normalize for IgG surface expression). Approximately 108 cells from each library were sorted on a MoFlo MLS instrument using high affinity binding (Bind) and expression (Exp) gates are indicated.
Pyrosequencing and data analysis
Cells collected from the FACS gates were lysed and plasmids recovered. PCR amplicons covering the variable regions were prepared from the plasmids, pooled and sequenced on a single run of a Roche/454 GS FLX instrument. From a single run, 831,032 total sequences that were well-distributed between the heavy and light chains, forward and backward reads, and both the expression and binding gates were obtained. Average read lengths were over 300 base pairs (Table 2).
Table 2. Hu225 pyrosequencing summary.
| FACS gate | type | number | Ave. length | 3 CDRs |
|---|---|---|---|---|
| bp | % | |||
| Binding | VH/F | 105,866 | 310 | 79 |
| “ | VH/R | 83,792 | 315 | 81 |
| “ | VL/F | 97,846 | 311 | 84 |
| “ | VL/R | 108,662 | 309 | 85 |
| Expression | VH/F | 112,431 | 315 | 80 |
| “ | VH/R | 95,051 | 321 | 87 |
| “ | VL/F | 116,288 | 311 | 86 |
| “ | VL/R | 111,096 | 308 | 86 |
| Total | 831,032 |
Distribution of reads between the two FACS gates, heavy (VH) and light (VL) chains, forward (F) and reverse (R) reads. Average read length and the fraction of reads for which all 3 CDRs were identified are indicated.
Data analysis was restricted to the CDRs, the only regions where sequence variation was designed into the libraries. Hu225 CDR sequences were identified from each read based on exact matches to 9 base pairs of wild type flanking sequences. All 3 CDRs were successfully identified for the majority (> 80% on average) of the reads (Table 2). Each CDR sequence was further analyzed for the presence and type of mutation (Table 3). From the library design, each CDR sequence should be either wild type (from a clone containing a mutation in a different CDR) or contain a single NNK-encoded point mutation. Of the mutant CDRs, 73% contained a single variant codon, most of which (~92%) were NNK-encoded as designed. Very few (3.8%) of the CDR sequences contained more than one mutated codon. A larger fraction (23.2%) of the CDR sequences were found to contain insertions or deletions; however, the pyrosequencing platform used is prone to frame shift misreads8 and error rate analysis comparing Sanger sequencing of library clones to the apparent pyrosequencing error rate shows 50-fold more frameshift mutations in the pyrosequencing data than exist in the starting library (Fig. S3). CDRs sequences containing frame shift mutations, non-NNK codons, or more than one mutant codon were eliminated from further analysis.
Table 3. Hu225 pyrosequencing mutant analysis.
| FACS Gate | CDR | total reads | WT | Mutant | indel (%) | 1 mutant (%) | > 1 mutant (%) |
|---|---|---|---|---|---|---|---|
| binding | H1 | 169,515 | 151,867 | 17,648 | 2,466 (14.0) | 14,627 (82.9) | 555 (3.1) |
| “ | H2 | 157,383 | 51,265 | 106,118 | 14,361 (13.5) | 87,747 (82.7) | 4,010 (3.8) |
| “ | H3 | 156,949 | 145,565 | 11,384 | 4,489 (39.4) | 6,667 (58.6) | 228 (2.0) |
| “ | L1 | 185,058 | 72,533 | 112,525 | 15,704 (14.0) | 90,092 (80.1) | 6,729 (6.0) |
| “ | L2 | 186,919 | 170,671 | 16,248 | 4,707 (29.0) | 11,338 (69.8) | 203 (1.2) |
| “ | L3 | 183,699 | 84,961 | 98,738 | 50,269 (50.9) | 46,682 (47.3) | 1,787 (1.8) |
| expression | H1 | 191,396 | 171,585 | 19,811 | 2,643 (13.3) | 16,665 (84.1) | 503 (2.5) |
| “ | H2 | 180,492 | 91,927 | 88,565 | 14,153 (16.0) | 69,995 (79.0) | 4,417 (5.0) |
| “ | H3 | 172,423 | 112,982 | 59,441 | 6,539 (11.0) | 49,653 (83.5) | 3,249 (5.5) |
| “ | L1 | 205,999 | 121,315 | 84,684 | 12,715 (15.0) | 68,185 (80.5) | 3,784 (4.5) |
| “ | L2 | 204,450 | 150,691 | 53,759 | 5,517 (10.3) | 46,805 (87.1) | 1,437 (2.7) |
| “ | L3 | 204,621 | 103,969 | 100,652 | 44,681 (44.4) | 53,600 (53.3) | 2,371 (2.4) |
| Totals | 2,198,904 | 1,429,331 | 769,573 | 178,244 (23.2) | 562,056 (73.0) | 29,273 (3.8) |
For each CDR identified the total number of reads is indicated along with the distribution of the reads between wild type and mutated and the further breakdown of mutant CDR sequences into frameshifts (indel), single mutant codons or more than one mutant codon.
The number of occurrences of each variant codon in the expression and binding gates was tabulated. The 1806 possible variant codons in the expression gate were read an average of 165 times each, with a reasonable distribution between the various codons and positions, although an apparent DNA synthesis error resulted in a significant underrepresentation of CDR H3 codons containing cytosine in the first or second position (Fig. S4, Table S1). In contrast, codon occurrences in the binding gate showed much greater variability, with certain variants dramatically enriched while others were entirely depleted from the population (Fig. S4, Table S2).
We also compared codon frequencies in the expression gate to a similarly sequenced aliquot of the non-transfected library to look for expression biases. In general, codons are found in the expressed library at similar frequencies to the untransfected library. No major bias against any particular class of amino acid substitutions was observed, even for cysteine or proline substitutions, although nonsense codons were depleted from the expression gate as expected (Fig. S5).
For each variant codon the enrichment ratio (ER), the ratio of its frequency in the binding gate to its frequency in the expression gate, was determined (Table S3). An arbitrary cutoff of 10 was used to eliminate variants with fewer than 10 occurrences in the expression gate, resulting in the loss of 144/1806 (8.0%) variant codons, primarily the previously mentioned CDR H3 codons with cytosine in the first or second positions. Due to redundancy in the genetic code, this loss was less severe at the protein level, with only 61/1121 (5.4%) amino acid variants not represented by at least one codon. The final data set comprised ERs for 1662 variant codons representing 1060 amino acid point mutations, which varied from a high of 14.2 to a low of zero (for variants never observed in the binding gate).
Affinity ranking and internal consistency analysis
The hu225 libraries were sorted such that higher affinity variants should be enriched in the binding gate while lower affinity variants should be depleted; thus, the ER values should define an affinity ranking of all variants, with the higher affinity variants having the highest ERs, lower affinity variants having the lowest ERs and neutral variants in the middle. We next analyzed this ER-based affinity ranking for internal consistency, by comparing the ERs of synonymous codons.
There are 71 synonymous wild type codons in the data set whose ERs can be used to define neutral binding in the affinity ranking. As expected, these silent wild type codons fall in the middle of the data set with an average ER of 1.04 +/− 0.39 (Fig. S6, Table S4).
We next compared 398 synonymous codon pairs of the general formula NNG/NNT (encoding Ala, Gly, Leu, Pro, Arg, Ser, Thr and Val) that encode identical proteins and thus would be expected to have identical ERs. The analysis is shown as a scatter plot in Figure 3; as expected good agreement was seen for these synonymous codon pairs (R2 = 0.891).
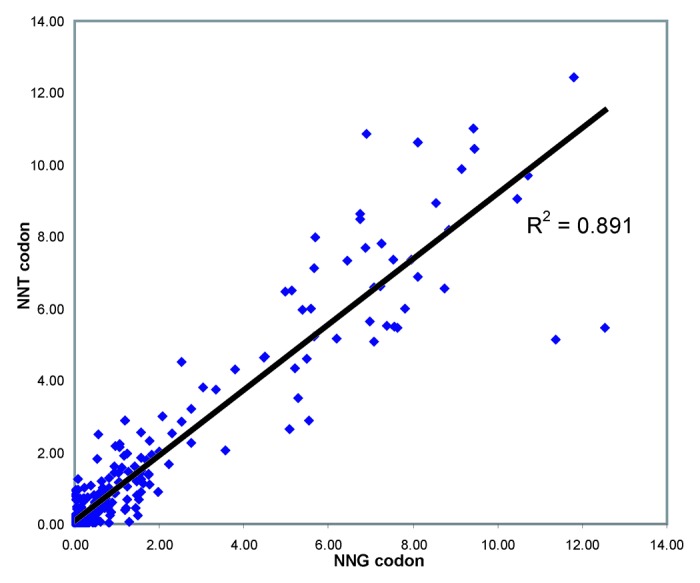
Figure 3. ER correlation for synonymous codon pairs. Scatter plot showing enrichment ratios for 398 synonymous NNT/NNG codon pairs encoding Ala, Gly, Leu, Pro, Arg, Ser, Thr and Val substitutions,. ER for the NNG codon is shown on the X axis, ER for the corresponding NNT codon is shown on the Y axis.
Agreement with results from previous affinity maturation experiments
The ER affinity ranking was compared with data from previously characterized higher affinity variants of antibody 225 CDRs. An in silico electrostatics-based prediction method identified 3 variants of cetuximab with 1.4- to 3.5-fold improved affinity for EGFR.9 These 3 variants are represented by 4 codons, all of which occur in the top 10.5% of the ranking, with ERs between 4.2 and 8.8 (Fig. S6, Table S5). A phage display-based affinity maturation of mAb 225 CDRs discovered a number of combinations of mutations with increased affinity for EGFR;10 the 33 codons for the 24 point mutations occurring in these combinations are markedly enriched in the higher ER range of the data set, with the majority (29/33 = 88%) occurring in the top 25% of the ranking (Fig. S6, Table S5). Comparison of our results to these previous experiments is complicated because the prior mutant CDRs were characterized in different framework contexts (murine or humanized); in addition, some of the phage display affinity maturation variants contained combinations of more than one mutation, not all of which necessarily behaved independently or contributed to increased affinity. Nonetheless a clear general trend was seen where most point mutations (33/37) from previously characterized higher affinity CDR variants were found in the upper portion of the ranking with ERs greater than the average of the synonymous wild type codons.
Structural analysis and positional tolerance to mutation
ERs (averaged for multiple codons where applicable) for 1121 amino acid variants were plotted as a heat map (Fig. 4A), providing a picture of general tolerance to mutation at each CDR position, with some locations (CDRH1, CDRH2, CDRL1 and CDRL3) seen as generally more tolerant of substitution compared with others (CDRH3, CDRL2). The average ERs for all point mutations at each position were color-coded and plotted onto the crystal structure of cetuximab Fab in complex with the extracellular domains of EGFR (Fig. 4B).11 Because humanization of murine antibodies retains the essential function of the murine CDRs in the context of human frameworks and crystal structures of humanized and parent antibodies show generally good retention of the murine CDR conformations,12 the cetuximab Fab structure should serve as a reasonable model of hu225. As would be expected, a number of previously identified centrally located residues of cetuximab with proposed functionally significant interactions with EGFR (VL:Y50, VH:D100, VH:Y99, VH:Y100a, VH:D58 and VL:W94) are notably resistant to substitution, while more tolerant positions are generally located to the periphery of the binding interface, in accordance with general features of protein-protein interfaces.13 The method thus gives essential structure-function insight into the interaction of a protein binding domain with its ligand, similar to the results of an alanine scan14 but at the level of resolution of all 20 amino acids.

Figure 4. Structural analysis and positional tolerance to mutation. (A) Heatmap showing color coded ERs for each hu225 point mutation (where applicable data are average of multiple codons). To right of the heatmap in parentheses are indicated 67 higher affinity point mutations confirmed by FACS-based assay. Blank spaces indicate variants for which fewer than 10 total occurrences were found in the expression gate and which were eliminated from the analysis. (B) Average ER for all codon variants at each position color-coded and plotted onto the cetuximab Fab / EGFR crystal structure.11 Color code: average ER 0–0.125 (red), 0.125–0.75 (orange), 0.75–1.5 (yellow) and greater than 1.5 (green).
Identification of higher affinity hu225 variants
To demonstrate the utility of the method for comprehensive identification of all higher affinity point variants in a binding domain, a number of predicted higher affinity variants from the ER ranking were selected for affinity characterization. We focused on variant codons with ERs greater than 4.0; these 179 codons represent 122 amino acid variants that comprise the top ~10% of all hu225 CDR variants and occur at 31 different positions within the CDRs. A number of the positions are hotspots where as many as 13 different substitutions with ER greater than 4 are found. We selected high ER variants from 28 different positions to cover a broad range of positions, rather than strictly by highest overall ER in the set. Details of the selection criteria and final choices for confirmation are shown in Figure S7. The final candidate pool for confirmation contained 67 variants, slightly more than half (55%) of all variants with ER greater than 4 and included 11 point mutations identical to those previously discovered from the in silico predictions and phage display affinity maturation, as well as 56 novel point mutations. To avoid expressing and purifying this many proteins, the individual clones for these 67 variants were analyzed by FACS in the mammalian display vector to confirm increased binding to 1.0 nM AF647-EGFR-Cλ compared with wild type hu225 (Fig. 5). All 67 variants exhibited higher binding to antigen, with most (50/67 = ~75%) 2-fold or greater in the FACS assay (Table S6). Thus, by focusing on the top ~10% of variants with ER greater than 4, we identified a small pool that is highly enriched for higher affinity variants, but there is no obvious correlation between ER and FACS affinity within this pool, even when looking at variants with multiple codons in agreement or those with the highest number of sequence reads (Fig. S8).
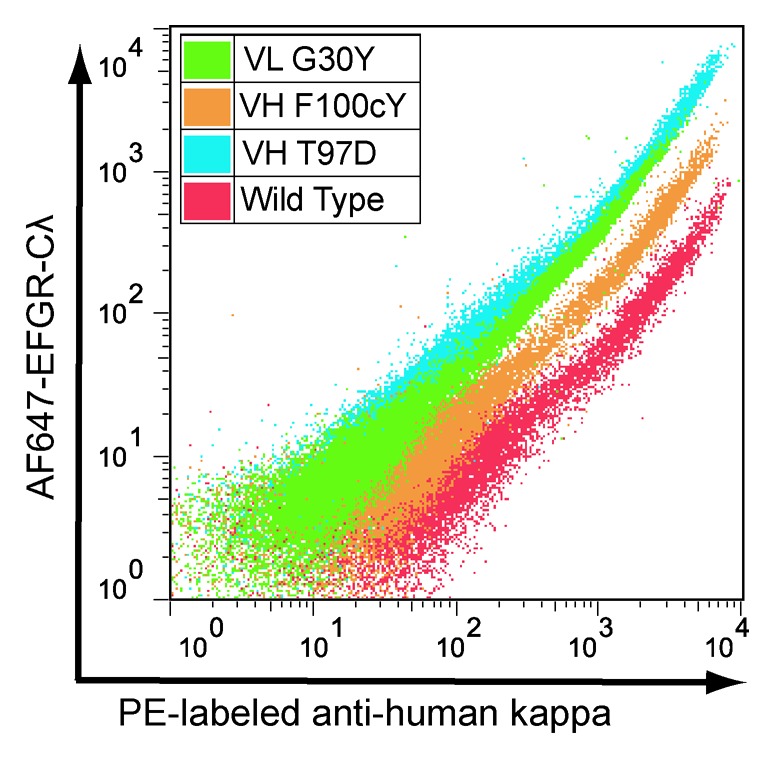
Figure 5. FACS analysis confirming higher affinity for selected hu225 variants. X axis = staining with anti-human kappa PE and Y axis = staining with AF647-EGFR-Cλ. A variant with vertical shift in relation to wild type indicates increased binding affinity.
Most beneficial mutations occurred in CDRH2, CDRL1 and CDRL3, with few or none in CDRH1, CDRH3 and CDRL2. Certain positions where four or more higher affinity variants were confirmed appear to be “hot spots” (VH:I51, VH:T57; VH:Y59, VL:A25, VL:G30 and VL:T97, Fig. 4 and Table S6). The cetuximab:EGFR structure was analyzed to identify contact residues with EGFR (defined as being within 5 Å distance, Table S3). The set of 67 higher affinity variants are found at 28 different positions, both contact and non-contact, with non-contact positions predominating (19/28).
Finally, a subset of 16 hu225 variants was expressed as soluble IgG1/kappa proteins and binding kinetics were characterized by surface plasmon resonance (Biacore) (Table 4). All 16 variants were confirmed to bind antigen with higher affinity, primarily through improvements to the off rate, with greater than 4-fold affinity improvement seen for a number of the variants.
Table 4. Affinity measurements for selected higher affinity hu225 variants.
| variant | ER for codon(s) | codon rank(s) | ka (M−1s−1) | kd (s−1) | KD (nM) | WT/x |
|---|---|---|---|---|---|---|
| hu225 | 5.4E+05 | 4.6E-03 | 8.5 | 1.0 | ||
| VH:Y32R | 7.22, 6.61, 5.90 | 79, 100, 117 | 5.7E+05 | 3.9E-03 | 6.8 | 1.3 |
| VH:G33D | 8.11 | 54 | 8.3E+05 | 1.6E-03 | 1.9 | 4.5 |
| VH:V50L | 10.87, 6.89. 6.46 | 15, 90, 104 | 5.2E+05 | 1.6E-03 | 3.1 | 2.7 |
| VH:V50Q | 12.73 | 3 | 5.5E+05 | 1.6E-03 | 2.9 | 2.9 |
| VH:I51G | 7.67, 6.88 | 64, 92 | 5.9E+05 | 2.1E-03 | 3.6 | 2.4 |
| VH:T57G | 7.09, 6.60 | 85, 101 | 5.6E+05 | 2.2E-03 | 3.9 | 2.2 |
| VH:T57P | 8.74, 6.56 | 42, 102 | 5.6E+05 | 2.1E-03 | 3.8 | 2.2 |
| VH:T97D | 6.4 | 108 | 7.0E+05 | 1.1E-03 | 1.6 | 5.3 |
| VH:Y98W | 4.82 | 151 | 6.6E+05 | 3.8E-03 | 5.8 | 1.5 |
| VH:F100cY | 14.19 | 1 | 6.3E+05 | 2.5E-03 | 4.0 | 2.1 |
| VL:A25V | 10.70, 9.71 | 17, 29 | 6.0E+05 | 1.8E-03 | 3.0 | 2.8 |
| VL:Q27Y | 10.47 | 21 | 5.6E+05 | 1.2E-03 | 2.1 | 4.0 |
| VL:G30Y | 11.51 | 11 | 6.1E+05 | 1.1E-03 | 1.8 | 4.7 |
| VL:T97C | 14 | 2 | 5.6E+05 | 2.0E-03 | 3.6 | 2.4 |
| VL:T97D | 5.98 | 112 | 6.1E+05 | 1.7E-03 | 2.8 | 3.0 |
| VL:T97S | 12.44, 11.80, 10.80 | 7, 8, 16 | 5.7E+05 | 1.5E-03 | 2.6 | 3.3 |
Discussion
Efforts to engineer desirable properties into protein binding domains are confounded by the enormous size of the potential sequence space, our inability to predict the effect of mutations on function and the limited throughput of existing methods to express and characterize variants. The development of simple, high throughput methods to assess the effect on affinity of all possible single amino acid substitutions in a binding domain will be of great practical importance to guide engineering of proteins for therapeutic and other applications.
Recent advances in next-generation DNA sequencing3 enable the acquisition of gigabases of DNA sequence information in a single experiment. In addition to sequencing genomes, the use and number of applications of NGS for molecular engineering is constantly expanding. NGS has been used to assess antibody diversity in organisms as well as in recombinant antibody repertoires,15-20 and has allowed deeper and earlier analysis of the output from sorting phage-displayed antibody, cDNA and peptide libraries.21-23 NGS has been coupled with protein display in approaches that have been collectively termed deep mutational scanning,4 which have been used to assess sequence-function relationships for ligand binding, as well as stability and expression on an unprecedented scale.24-27
We developed such a high throughput mutational analysis method and used it to generate a near comprehensive fitness landscape for the interaction of an antibody’s CDR regions with its antigen. In contrast to approaches that have used short (< 100 bp) reads from the Illumina platform to analyze short contiguous regions of proteins,25,27 we used the longer read capability of the Roche/454 platform to sequence the discontinuous CDRs in an antibody variable domain. In addition, our method uses mammalian display, which allows for mutational scanning of glycoproteins in the context of normal mammalian glycosylation.
We demonstrated this method on a humanized version of the anti-EGFR antibody 225, obtained binding data for 1060 point variants in a single experiment and generating a near comprehensive functional map of the antibody/antigen interface that shows good agreement with previous structural and functional characterization of this antibody. Although the data indicate that the majority of CDR substitutions are neutral or deleterious to binding (Fig. S6), we nonetheless identified 67 higher-affinity point mutations from our analysis.
Examination of the X-ray structure of the cetuximab:EGFR complex suggests possible mechanisms of action for some of the higher affinity variants shown in Table 4. Position Thr 97 of the heavy chain is proximal to Lys 465 on EGFR and thus the substitution to Asp could potentially create a beneficial electrostatic interaction. Light chain position Gly 30 is a “hotspot” where 13 codons (encoding 10 amino acids) with ERs greater than 4 occur. Gly 30 is not a contact residue in the structure, suggesting that a multiplicity of side chains can find ways to bridge the gap to EGFR for beneficial interactions. Especially interesting is position Thr 97 in the light chain, also a hot spot where fully 19 codons (encoding 13 amino acids) with ER > 4 occur. Thr 97 is also a non-contact residue located 9.2 Å from EGFR, so the effect of mutations at this position is unclear, although the large number of beneficial substitutions for Thr suggests that a common mechanism of removing a deleterious effect of the Thr side chain may be involved. Also of particular interest is the increased affinity of the Cys variant at this position, despite the relative scarcity of unpaired Cys residues in antibody CDRs. In fact, Cys residues seem to be not particularly disfavored in the hu225:EGFR interface, with an average ER of 1.03 across all 59 Cys substitution, not dissimilar to an overall average ER of 1.17 for all codon variants (Table S3).
A comparison of ERs for the 16 variants shown in Table 4 and their affinity by Biacore shows only a modest correlation (R2 = 0.287, Fig. S9) Thus while the method correctly identifies a population of higher affinity variants it lacks sufficient resolution to make precise affinity determinations within that population. For example, the hu225 variant with the highest ER (VH:F100cY) shows less affinity improvement than either VL:G30Y or VH:T97D, ranked 11 and 108, respectively (Table 4, Fig. 5). Possible sources of error include insufficient sequencing reads per variant, insufficient cells sorted, pyrosequencing errors and PCR errors from amplicon preparation. Significant improvement in the precision of the method should be readily achievable through ongoing advances in next-generation DNA sequencing data quality and quantity, as well as improvements in library design and construction.
Beneficial point mutations identified from NGS mutation analysis methods can serve as the input for focused combinatorial libraries to drive toward much higher affinities. Recently Fujino et al.28 used NGS to analyze selections from point mutant libraries of a single chain Fab anti-tumor necrosis factor receptor antibody using ribosome display. The NGS analysis was much shallower than in the present work, necessitating 3 rounds of selection to reduce diversity sufficiently to identity enriched mutants (with concomitant loss of resolution on neutral and deleterious variants), and the enriched single mutants were not characterized for affinity benefit. A combinatorial library focused on these enriched point mutations nonetheless yielded combinations with 400- to 2100-fold improved affinity. While the relative efficacy of NGS-enabled “find all good mutants and combine” affinity maturation strategies compared with traditional approaches will require much additional experimental work, ideally using the same model systems for head-to-head comparisons, the Fujino work clearly shows such strategies can yield results comparable to the best existing methods.
The general principles of this approach should be broadly applicable to different display systems, other classes of protein binding domains and other NGS platforms. We used this method to map and engineer the interactions of CTLA-4 with its ligands.29 The library sorting step need not be FACS-based binding, but could in principle be any property of interest as long as a suitable fractionation method could be devised for enriching variants. We found the output of the method useful for engineering therapeutic antibodies and other proteins by providing a complete menu of higher affinity mutations for affinity and cross-reactivity engineering. In addition, the list of neutral substitutions defines a functionally equivalent sequence space that can be sampled for variants with decreased immunogenicity30 and improved drug-like properties such as solubility, stability and pharmacokinetics. Equally important, the large comprehensive sequence-function data sets should help inform theoretical understanding of protein binding and contribute to improvements in rational protein design.
Materials and Methods
Proteins, DNA, cell lines, software
A fusion protein consisting of the extracellular domain of human epidermal growth factor receptor (EGFR) amino acids 1–616 fused to human Cλ constant domain (EGFR-Cλ) containing a Cys 214 to Ser mutation for monomeric expression was transiently expressed in HEK293 cells, purified by affinity chromatography on an immobilized anti-Cλ antibody column and labeled with Alexa Fluor 647 (Invitrogen) following the manufacturer’s recommendations. 293c18, 293T/17 and A431 cells were from ATCC. Oligonucleotide primers were from Eurofins MWG Operon. Gene synthesis was performed by DNA 2.0. Structural figures were generated using PyMol (Schrodinger LLC).
Humanization of Mab 225
A humanized form of the anti-EGFR antibody 225 was generated by the method of Queen.31 The computer programs ABMOD and ENCAD were used to construct a molecular model of the mouse variable regions.32 Framework regions derived from the human VH3 family member 60P233 and the VK3 family member NOV34 were used as acceptor sequences for grafting the mouse CDRs, and a small number of mouse framework back mutations were made where computer modeling predicted close contact between frameworks and CDRs. Retained mouse residues were at positions H28, H29, H30, H48, H49, H67, H70, H71, H78, H94, L1, L3 and L49.
Expression and purification of antibodies
Antibodies were expressed from an expression plasmid as IgG1/kappa under control of the CMV promoter. HEK 293/T17 cells were transfected with DNA in Lipofectamine 2000 (Invitrogen), grown in DMEM with 2% low IgG FBS (HyClone), and supernatants were harvested after 5 d. Antibodies were purified using immobilized protein A, dialyzed against PBS and concentrated. Final concentration was determined by OD280 and the purified antibodies stored in PBS at 4°C.
A431 competition binding assay
Relative binding affinity to EGFR-expressing A431 cells was determined by a competition FACS assay. A431 cells were grown to 2 × 105 cells per well of V-bottom 96-well plates and washed with PBS containing 0.5% PBS (FACS buffer). Biotinylated cetuximab was diluted to a final concentration of 0.5 μg/ml, mixed with competitor antibody at various concentrations, and the mixtures were transferred to 96-well plates containing the A431 cells. The plates were incubated on ice for 1 h, washed twice with FACS buffer, then incubated with 25 μl of strepavidin-R-phycoerythrin (streptavidin-RPE) conjugate (Biosource), diluted to 2.5 μg/ml in FACS buffer on ice for another 30 min in the dark. Cells were washed two times with FACS buffer, resuspended with 200 μl of 1% paraformaldehyde and read using a flow cytometer.
Surface plasmon resonance (Biacore) measurements
Affinity measurements were performed on Biacore model 2000 or T100 (Biacore, GE Healthcare) at 25°C using HBS-EP+ with 0.1 mg/mL BSA as running buffer. A CM5 sensor chip with goat anti-human Fc amine-coupled in all 4 flow cells at ~15,000 RU was used to capture test antibodies with EGFR in the mobile phase. Between 50 and 75 RUs of hu225 variants were captured on the CM5 chip. A 3-fold dilution series of EGFR from 90 to 0.37 nM was injected and dissociation was monitored for 15 min. Kinetic analysis was done by simultaneously fitting the association and dissociation phases of the sensorgram using the 1:1 Langmuir binding model in BIAevaluate software as supplied by the manufacturer. Double referencing was applied in each analysis to eliminate background responses from the reference surface and buffer only control.
Competition AlphaLISA
AlphaLISA acceptor beads (Perkin Elmer) were directly conjugated with goat-anti-human Cλ. Serial dilutions of the antibody of interest were mixed with a fixed amount of biotinylated cetuximab, then a fixed amount of EGFR-Cλ and anti-human Cλ acceptor beads were added. After a 2 h incubation, strepavidin-donor beads were added and incubated for 30 min, analyzed following the manufacturer’s instructions and reported as EC50 wild type/EC50 mutant.
Mammalian display vectors
Mammalian cell surface display of hu225 and libraries was performed using pYA206, a derivative of pYA104,7 an Epstein-Barr virus (EBV)-derived plasmid for display of full length IgG/lambda antibodies on the surface of 293c18 cells via a glycosylphosphatidylinositol (GPI) linkage signal. pYA104 was modified by (1) incorporating new flanking restriction sites for variable region cloning (NotI and XhoI for VL; NgoMIV and SacI for VH), (2) replacing human C lambda with human C kappa light chain constant regions and (3) replacing the GPI anchor with a transmembrane domain derived from human platelet-derived growth factor receptor to create pYA206.
Construction of synthetic genes and libraries
Synthetic DNA fragments encoding the hu225 VH and VL coding regions and flanking restrictions sites were subcloned into vector pYA206 to create surface expression plasmid pYA206-hu225. Fifty-nine synthetic DNA gene segments, each with NNK degeneracy at a different CDR position, were obtained from a commercial supplier (DNA 2.0) and cloned into pYA206-hu225 to create 59 positional sublibraries. To ensure oversampling of each variant, ligations and E. coli transformations were performed to ensure at least 10-fold coverage for each of the 32 NNK codons at each position. VH and VL positional sublibraries were pooled in equimolar ratios to make the final VH and VL libraries.
Transfection of libraries and cell sorting
Libraries were transfected into 293c18 cells using Lipofectamine 2000 (Invitrogen) with 0.5 ug plasmid and 100 ug carrier plasmid pACYC184/ER2420 (New England Biolabs) in DMEM media (HyClone) supplemented with 10% FBS (HyClone) and 0.25 mg/ml G418 (Mediatech Inc.). Cells were cultured for 2 d then transferred to selection media containing 0.8 ug/ml puromycin (Clontech) and grown an additional 35 or 36 d, splitting 1:3 every 2 to 4 d to ensure plasmids had segregated and every cell expressed a single clonal variant. For library sorting, approximately 1 × 108 cells were incubated with 1 nM EGFR-Cλ−AF647 in FACS buffer for 1 h at room temperature. Cells were washed, incubated with 1:500 dilution of goat-anti-human-kappa-PE (Southern Biotech) for 45 min at 4°C, washed three times with cold FACS buffer and resuspended in PBS with 20 mM HEPES and 20 mM EDTA. Stained cells were sorted on a MoFlo MLS (DakoCytomation). For the VH library, a total of 9.0 × 107 cells were sorted and 2.0 × 106 and 2.4 × 106 cells collected from the expression and binding gates, respectively. For the VL library a total of 1.1 × 108 cells were sorted and 1.0 × 106 and 1.9 × 106 cells collected from the expression and binding gates, respectively.
DNA recovery and pyrosequencing
Following sorting, cells were lysed, plasmids recovered by miniprep (Qiagen) and treated with restriction enzyme DpnI to digest carrier plasmids. A modified sequencing protocol using the Roche/454 GS FLX instrument and genomic reagent kit was used in which PCR was used to generate amplicons for sequencing with appended “A” and “B” adaptor sequences (vs. by ligation in the original protocol). Each amplicon was prepared in two versions, with the A and B adaptors in either orientation to allow sequencing from either end.
FACS confirmation of higher affinity hu225 point mutations
For high throughput confirmation of higher affinity hu225 variants, a flow cytometric assay was performed in which individual cell surface displayed variants (obtained by sequencing individual clones from the appropriate positional sublibrary or by gene synthesis) were similarly stained with 1 nM EGFR-Cλ-AF647 and goat-anti-human-kappa-PE as was done for the library sorting. Data are reported as the ratio of mean fluorescence intensity in the AF647 channel for the mutant compared with wild type hu225.
Supplementary Material
Acknowledgments
We would like to thank Susan Rhodes and Rick Powers (both of AbbVie) and Marie Cardenas and Wenge Zhang (both formerly of AbbVie) for molecular biology and protein chemistry support; Peter Lambert (AbbVie) for statistical analysis and advice; Steve Hartman (AbbVie) for assistance with the AlphaLISA assay; and Don Halbert (AbbVie) and Stan Falkow (Stanford) for critical reading and comments on the manuscript.
Disclosure of Potential Conflicts of Interest
The authors are present or former employees of AbbVie. This study was sponsored by AbbVie. Colleagues at AbbVie contributed to the study design, research, interpretation of data, writing and review of the manuscript and approval of the publication.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/24979
References
- 1.Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol. 2006;6:343–57. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- 2.Presta LG. Molecular engineering and design of therapeutic antibodies. Curr Opin Immunol. 2008;20:460–70. doi: 10.1016/j.coi.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 3.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 4.Araya CL, Fowler DM. Deep mutational scanning: assessing protein function on a massive scale. Trends Biotechnol. 2011;29:435–42. doi: 10.1016/j.tibtech.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sato JD, Kawamoto T, Le AD, Mendelsohn J, Polikoff J, Sato GH. Biological effects in vitro of monoclonal antibodies to human epidermal growth factor receptors. Mol Biol Med. 1983;1:511–29. [PubMed] [Google Scholar]
- 6.Vincenzi B, Zoccoli A, Pantano F, Venditti O, Galluzzo S. Cetuximab: from bench to bedside. Curr Cancer Drug Targets. 2010;10:80–95. doi: 10.2174/156800910790980241. [DOI] [PubMed] [Google Scholar]
- 7.Akamatsu Y, Pakabunto K, Xu Z, Zhang Y, Tsurushita N. Whole IgG surface display on mammalian cells: Application to isolation of neutralizing chicken monoclonal anti-IL-12 antibodies. J Immunol Methods. 2007;327:40–52. doi: 10.1016/j.jim.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 8.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lippow SM, Wittrup KD, Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol. 2007;25:1171–6. doi: 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beidler CB, Vasserot AP, Watkins JD. Anti-EGFR Antibodies. US Patent No. 7,723,484.
- 11.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–11. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 12.Holmes MA, Buss TN, Foote J. Conformational correction mechanisms aiding antigen recognition by a humanized antibody. J Exp Med. 1998;187:479–85. doi: 10.1084/jem.187.4.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 14.Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–5. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 15.Boyd SD, Marshall EL, Merker JD, Maniar JM, Zhang LN, Sahaf B, et al. Measurement and clinical monitoring of human lymphocyte clonality by massively parallel VDJ pyrosequencing. Sci Transl Med. 2009;1:12ra23. doi: 10.1126/scitranslmed.3000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer N. Sequencing antibody repertoires: the next generation. MAbs. 2011;3:17–20. doi: 10.4161/mabs.3.1.14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ge X, Mazor Y, Hunicke-Smith SP, Ellington AD, Georgiou G. Rapid construction and characterization of synthetic antibody libraries without DNA amplification. Biotechnol Bioeng. 2010;106:347–57. doi: 10.1002/bit.22712. [DOI] [PubMed] [Google Scholar]
- 18.Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci U S A. 2009;106:20216–21. doi: 10.1073/pnas.0909775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reddy ST, Ge X, Miklos AE, Hughes RA, Kang SH, Hoi KH, et al. Monoclonal antibodies isolated without screening by analyzing the variable-gene repertoire of plasma cells. Nat Biotechnol. 2010;28:965–9. doi: 10.1038/nbt.1673. [DOI] [PubMed] [Google Scholar]
- 20.Weinstein JA, Jiang N, White RA, 3rd, Fisher DS, Quake SR. High-throughput sequencing of the zebrafish antibody repertoire. Science. 2009;324:807–10. doi: 10.1126/science.1170020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Niro R, Sulic AM, Mignone F, D’Angelo S, Bordoni R, Iacono M, et al. Rapid interactome profiling by massive sequencing. Nucleic Acids Res. 2010;38:e110. doi: 10.1093/nar/gkq052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravn U, Gueneau F, Baerlocher L, Osteras M, Desmurs M, Malinge P, et al. By-passing in vitro screening--next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic Acids Res. 2010;38:e193. doi: 10.1093/nar/gkq789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.’t Hoen PA, Jirka SM, Ten Broeke BR, Schultes EA, Aguilera B, Pang KH, et al. Phage display screening without repetitious selection rounds. Anal Biochem. 2012;421:622–31. doi: 10.1016/j.ab.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 24.Ernst A, Gfeller D, Kan Z, Seshagiri S, Kim PM, Bader GD, et al. Coevolution of PDZ domain-ligand interactions analyzed by high-throughput phage display and deep sequencing. Mol Biosyst. 2010;6:1782–90. doi: 10.1039/c0mb00061b. [DOI] [PubMed] [Google Scholar]
- 25.Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, Stephany JJ, Baker D, et al. High-resolution mapping of protein sequence-function relationships. Nat Methods. 2010;7:741–6. doi: 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlinkmann KM, Honegger A, Türeci E, Robison KE, Lipovšek D, Plückthun A. Critical features for biosynthesis, stability, and functionality of a G protein-coupled receptor uncovered by all-versus-all mutations. Proc Natl Acad Sci U S A. 2012;109:9810–5. doi: 10.1073/pnas.1202107109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whitehead TA, Chevalier A, Song Y, Dreyfus C, Fleishman SJ, De Mattos C, et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol. 2012;30:543–8. doi: 10.1038/nbt.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujino Y, Fujita R, Wada K, Fujishige K, Kanamori T, Hunt L, et al. Robust in vitro affinity maturation strategy based on interface-focused high-throughput mutational scanning. Biochem Biophys Res Commun. 2012;428:395–400. doi: 10.1016/j.bbrc.2012.10.066. [DOI] [PubMed] [Google Scholar]
- 29.Xu Z, Juan V, Ivanov A, Ma Z, Polakoff D, Powers DB, et al. Affinity and cross-reactivity engineering of CTLA4-Ig to modulate T cell costimulation. J Immunol. 2012;189:4470–7. doi: 10.4049/jimmunol.1201813. [DOI] [PubMed] [Google Scholar]
- 30.Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2:256–65. doi: 10.4161/mabs.2.3.11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Queen C, Schneider WP, Selick HE, Payne PW, Landolfi NF, Duncan JF, et al. A humanized antibody that binds to the interleukin 2 receptor. Proc Natl Acad Sci U S A. 1989;86:10029–33. doi: 10.1073/pnas.86.24.10029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levitt M. Molecular dynamics of native protein. I. Computer simulation of trajectories. J Mol Biol. 1983;168:595–617. doi: 10.1016/S0022-2836(83)80304-0. [DOI] [PubMed] [Google Scholar]
- 33.Schroeder HW, Jr., Hillson JL, Perlmutter RM. Early restriction of the human antibody repertoire. Science. 1987;238:791–3. doi: 10.1126/science.3118465. [DOI] [PubMed] [Google Scholar]
- 34.Kipps TJ, Tomhave E, Pratt LF, Duffy S, Chen PP, Carson DA. Developmentally restricted immunoglobulin heavy chain variable region gene expressed at high frequency in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 1989;86:5913–7. doi: 10.1073/pnas.86.15.5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.