

Abstract
Combinatory antibody library display technologies have been invented and successfully implemented for the selection and engineering of therapeutic antibodies. Precise targeting of important epitopes on the protein of interest is essential for such isolated antibodies to serve as effective modulators of molecular interactions. We developed a strategy to efficiently isolate antibodies against a specific epitope on a target protein from a yeast display antibody library using dengue virus envelope protein domain III as a model target. A domain III mutant protein with a key mutation inside a cross-reactive neutralizing epitope was designed, expressed, and used in the competitive panning of a yeast display naïve antibody library. All the yeast display antibodies that bound to the wild type domain III but not to the mutant were selectively sorted and characterized. Two unique clones were identified and showed cross-reactive binding to envelope protein domain IIIs from different serotypes. Epitope mapping of one of the antibodies confirmed that its epitope overlapped with the intended neutralizing epitope. This novel approach has implications for many areas of research where the isolation of epitope-specific antibodies is desired, such as selecting antibodies against conserved epitope(s) of viral envelope proteins from a library containing high titer, high affinity non-neutralizing antibodies, and targeting unique epitopes on cancer-related proteins.
Keywords: yeast display antibody library, competitive sorting, epitope specific targeting, dengue virus, human antibody library
Introduction
Protein-protein interactions are the molecular foundation for the functioning of life; antibodies have been discovered as modulators of molecular interactions and have emerged as useful drug candidates for many diseases such as cancer, immune diseases, and infectious diseases.1 The invention and implementation of combinatorial antibody library display technologies have dramatically facilitated the development of such therapeutic antibodies and proteins.2-7 Among those in vitro antibody display platforms, yeast display has become one of the most powerful platforms. It has been repeatedly and successfully used for the isolation, engineering, and epitope mapping of antibodies and proteins that have great potential in various biomedical applications, such as therapeutics, diagnostics, and research tools development.8,9
The conventional approach to isolate antibodies of interest from an in vitro display antibody library always starts with library panning, followed by single clone screening and further characterization of individual antibodies through epitope mapping and function evaluation, which is time-consuming and labor-intensive. Furthermore, the potency or usefulness of an antibody very often depends on its binding epitope on the target of interest. Therefore, efficiently isolating antibodies that precisely target important epitopes on a target protein of interest is crucial for their success as drugs or as other biomedical tools.
Among many features of yeast display, yeast cells can be quantitatively sorted through fluorescence-activated cell sorting (FACS) and consistently display multiple copies of antibodies on each cell are the most important ones. These important features offer a unique platform for a competitive sorting experiment design, in which a non-fluorescent, mutated protein of interest lacking the target epitope can compete with the fluorescent wild type protein of interest for target epitope-irrelevant antibodies and allow only target epitope-specific antibodies from the yeast display library to bind to the fluorescent wild type protein and be sorted. In this study, using dengue virus (DENV) envelope protein domain III as a model target, we validated a method to quickly isolate epitope-specific antibodies from a naïve antibody library through competitive yeast display library sorting.
DENVs belong to the Flavivirus genus of the Flaviviridae family. DENV-caused infectious disease is becoming a global public health threat.10 DENV strains are highly diversified as there are four serotypes (1, 2, 3, and 4) and numerous genotypes exist within each serotype. Previous studies demonstrated that cross-reactive neutralizing epitopes exist on the DENV envelope protein domain III.11-13 Isolating antibodies that specifically target those conserved neutralizing epitopes is crucial for the development of antibodies that can serve as effective anti-DENV drugs. Therefore, DENV envelope protein domain III was employed as an ideal model for the validation of our strategy in this study.
Results
Expression and purification of wild type and mutant DENV domain III Fc fusion proteins For easier and more efficient purification of the domain III proteins, human IgG1 Fc was fused with those domains. An Avi-tag was added to the C-terminus for site-specific biotinylation. The domain III Fc fusion proteins with c-Myc tag were also expressed and purified for ELISA binding assays to measure the binding activities of the isolated scFv-Fc antibodies. As shown in Figure 1, the four domain III Fc-Avi fusion proteins expressed in 293 free style cells were readily purified on Protein G columns. All four domain III Fc fusion proteins with the C-terminal c-Myc-tag were similarly expressed and purified (data not shown)
Figure 1.
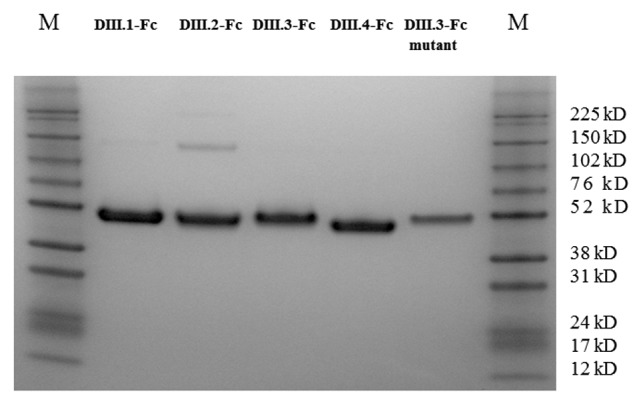
SDS-PAGE analysis of the purified DENV domain III.1–4 Fc fusion proteins (Avi-tagged) and the DIII.3-Fc-Avi mutant bearing the point mutation K310E. All five fusion proteins were readily purified on protein G column and analyzed under reducing conditions. M, Protein Marker.
Previous studies have shown that the lysine at position 310 on domain III is the key residue in the cross-reactive neutralizing epitope.14,15 The domain III mutant bearing the mutation K310E, constructed based on the domain III.3 consensus sequence, was also fused with human IgG1 Fc-Avi and readily expressed and purified (Fig. 1).
454 deep sequencing of antibody libraries
We used 454 deep sequencing to characterize the sequence diversities of the two naïve libraries constructed from 10 16 and 59 individuals,17 which contributed to a larger portion of diversity in the yeast display naïve library that was used for the competitive library sorting. The rearranged antibody sequences were typically more than 300 nt in length and unique at the amino acid level with productive functionality as determined by IMGT/V-QUEST.18 There were 28,925 VH and 26,671 VL sequences in the library of 10 individuals, and 41,605 VH and 53173 VL sequences in the library of 59 individuals. We determined the germline usages in these two libraries by computing frequencies of heavy (IGHV) and light (IGKV and IGLV) subgroups, and found that all germline families were present as shown in Figure S1A and B. We observed a total of 27,735 unique heavy chain complementarity determining regions 3 (HCDR3s) in the library of 10 individuals, and 34,200 unique HCDR3s the library of 59 individuals. The length distribution of HCDR3 regions for these libraries is plotted in Figure S1C.
Naïve library sorting on AutoMACS
AutoMACS is an efficient platform that can be used to quickly sort large number of yeast cells and downsize a naïve library to make it feasible for FACS-based cell sorting. Biotinylated domain III.3-Fc was used for three rounds of sorting of a yeast display naïve antibody library on AutoMACS. Excessive amount of an unrelated m102.4 IgG1 with the same Fc fragment as domain III.3-Fc fusion protein was mixed in the library to deplete the potential Fc specific binders as described in Materials and Methods. Three rounds of sorting on AutoMACS dramatically enriched the domain III.3-Fc binders (Fig. 2). The binding of the entire double positive population to the domain III.3-Fc fusion protein could be competed by a domain III.3 only protein, indicating the binding of the enriched pool was domain III specific and depletion of potential Fc binders using m102.4 IgG1 was efficient (data not shown).
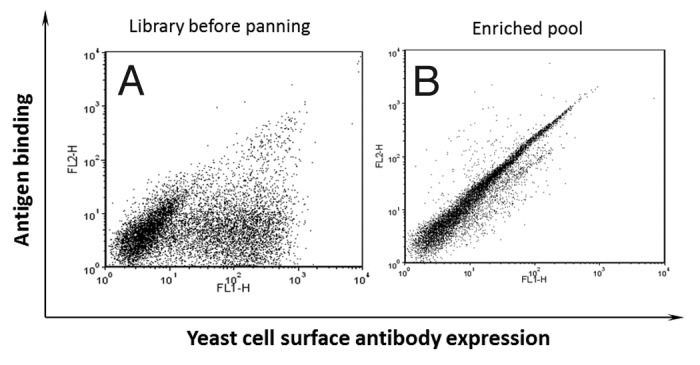
Figure 2. Enrichment of yeast display antibodies with specific binding to biotinylated domain III.3-Fc after three rounds of AutoMACS-based sorting of the naïve library against biotinylated domain III.3-Fc. Antibody expression on the yeast cell surface was detected with mouse anti-c-Myc antibody and Alexa Fluor 488 conjugated goat anti-mouse antibody as the secondary antibody; binding of the yeast surface displayed antibody to the biotinylated antigen was detected via R-Phycoerythrin (PE)-conjugated streptavidin. (A) The original naïve yeast library before sorting and (B) the yeast pool after three rounds of sorting was incubated with 1 μg/ml of biotinylated domain III.3-Fc and 2 μg/ml of mouse anti-c-Myc antibody before staining with Alexa Fluor 488 conjugated secondary antibody and PE-conjugated streptavidin.
Competitive library sorting using the designed DIII.3 mutant as a competitor
The downsized pool showed specific binding to the biotinylated domain III.3-Fc, with the target antigen at 1 μg/ml (Fig. 2). When 10 μg/ml of the unbiotinylated domain III.3-Fc mutant competitor and 1 μg/ml of the biotinylated wild type domain III.3-Fc were mixed with the yeast pool, the FACS profile obtained from the cell sorter showed the pool divided into several populations, as each was affected by the mutant competition to a different extent (Fig. 3). The cell population at the top represents those antibody expressing cells that could not bind to the unbiotinylated domain III.3-Fc mutant, but still bound well to the biotinylated wild type domain III.3-Fc. The cell population at the bottom represents those cells that bound well to epitopes shared by both the wild type and mutant domain III.3 (Fig. 3). Cells showing no competition by the mutant were sorted out and plated on a SDCAA plate for single clone screening.
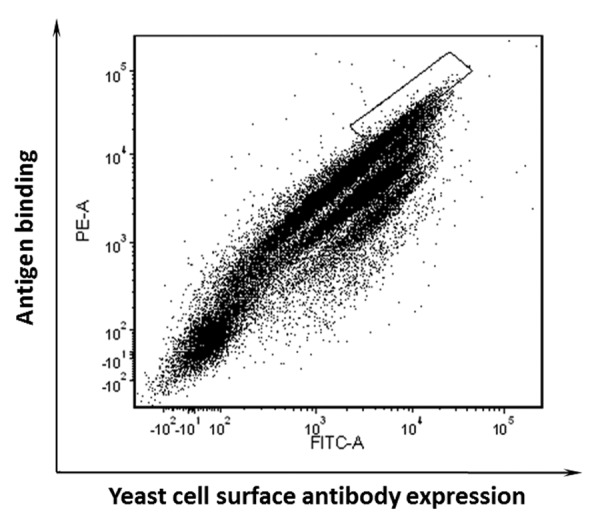
Figure 3. Competitive sorting of enriched yeast pool using the domain III.3 mutant K310E as a competitor. Sorting gate was set to sort out the cells that could not be competed by an excessive amount of the mutant.
Identification and characterization of monoclonal antibodies from the competitively sorted pool
Flow cytometry analysis was performed to screen 24 random clones from the sorted yeast pool that could not be competed by the designed mutant K310E. Domain III proteins from all four serotypes were used for the monoclonal yeast antibody screening. Plasmids were extracted from the cross-reactive yeast clones and sequenced. Two unique clones, designated as D6 and D7, were identified and their scFv inserts were cloned into the pSectag vector bearing the Avi-tagged human IgG1 Fc portion at the C-terminus for soluble expression.
Both D6 and D7 scFv-Fc fusion proteins were expressed and purified with high yields (2 mgs each) from 100 ml of transiently transfected 293 free style cell culture medium (Fig. 4A). The ELISA binding assay of the two clones that bound to all four serotypes’ domain IIIs (domain III.1–4-Fc) showed clone D6 bound to all the domain IIIs from all 4 serotypes, although relatively weaker to serotypes 1 and 4 (Fig. 4B), while clone D7 bound equally well to domain III.1–3-Fc but did not show any detectable binding to domain III.4-Fc (Fig. 4C).

Figure 4. Purification and ELISA binding assay of isolated domain III specific binders D6 and D7. (A) SDS-PAGE analysis of D6 and D7 scFv-Fc fusion proteins. Lane 1-D6 scFv-Fc; Lane 2-D7 scFv-Fc. ELISA binding assessment of (B) D6 scFv-Fc and (C) D7 scFv-Fc to c-Myc tagged domain III.1–4-Fc proteins. D6 scFv-Fc and D7 scFv-Fc were each coated on plate at 2 μg/ml; DENV domain III.1–4-c-Myc tag was added as antigens and binding was detected via mouse anti-c-Myc-HRP. (D) Competition ELISA showing the binding epitopes of D6 scFv-Fc and D7 scFv-Fc overlap. D6 scFv-Fc was coated on plate, serially diluted c-Myc tagged domain III.3-Fc only or mixed with a constant amount of competitor (K310E mutant or D7 scFv-Fc) was added to each well and binding of the domain III.3-Fc was detected by mouse anti-c-Myc-HRP antibody.
A competition ELISA was also performed to validate the competitive sorting approach. In agreement with our initial sorting experiment, the K310E mutant could not compete with the wild type domain III.3-Fc for binding to D6 scFv-Fc. In addition, since D6 and D7 were identified from the same sorted pool that initially could not be competed by the excessive amount of K310E mutant, D6 and D7 scFv binding epitopes should overlap, which was further confirmed by the fact that D7 scFv-Fc competed well with D6 scFv-Fc for binding to domain III.3-Fc. (Fig. 4D).
Epitope mapping of the isolated antibody D7 on DENV envelope protein domain III.2
To validate our strategy, we also attempted to locate the epitope of one of the two isolated antibodies, D7, on the DENV envelope protein domain III.2. The domain III consensus gene derived for serotype 2 was used as the template for error-prone PCR; this mutant gene repertoire was then cloned into the yeast display vector through gap repairing and a yeast display domain III.2 mutant library with the size of 2x108 was constructed. Two rounds of sorting were performed to enrich the cells that expressed domain III.2 on their surfaces but lacked binding to the isolated antibody D7. As shown in Figure 5A 2.9% of the whole cell population of the initial mutant library expressed domain III.2 but could not bind to D7 scFv-Fc. After one round of sorting, the percentage of the binding escape mutants dramatically increased to 53.5% (Fig. 5A). Sequence analysis showed all 48 single clones from the binding escape mutants pool had the complete and correct reading frame, which is in agreement with the fact that all the sorted clones showed antigen expression on the cell surface. Furthermore, all clones bear at least one mutation at the protein level, with up to 5 mutations for some clones (data not shown). All the mutants bearing mutations on cysteines and solvent inaccessible residues were excluded from the sequence analysis. All mutation positions are shown in red on the domain III.2 crystal structure (Fig. 5B). Overall, these residues were clustered around the initial mutation site 310K (shown in green). Although there are some mutated residues located far from the mutation site, these might be due to structurally silent mutations carried over from those mutants bearing multiple mutations or to mutations causing conformational changes. These data are in agreement with our initial design to isolate antibodies against the specific conserved neutralizing epitope on the DENV envelope protein domain III.
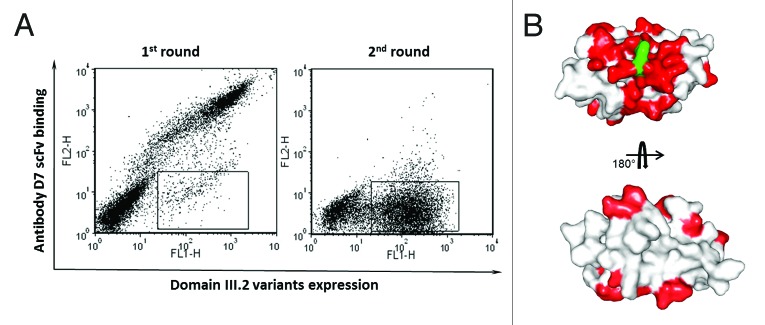
Figure 5. Epitope mapping of binder D7 on DENV envelope protein domain III.2. (A) Sorting of the yeast display domain III.2 mutant library. Expression of DENV domain III.2 variants on the yeast cell surfaces was detected with mouse anti-c-Myc antibody and Alexa Fluor 488 conjugated goat anti-mouse antibody as the secondary antibody; binding of the yeast surface displayed domain IIIs to the biotinylated D7 scFv-Fc was detected via PE-conjugated streptavidin. The sorting gate was set to specifically sort out the yeast cells expressing domain III.2 mutants but lacking the binding to biotinylated D7 scFv-Fc. Dramatic enrichment is seen after one round of sorting. (B) Mutated residues derived from the sequence analyses of domain III.2 mutants unable to bind to D7 were shown in red on the crystal structure of DENV envelope protein domain III.2 (PDB code: 2R69), with the original mutation site 310K of the mutant that was used in competitive sorting highlighted in green.
Discussion
The major goal of this study was to demonstrate that a highly efficient yeast display library sorting strategy can be used to identify site-specific antibodies to a target antigen from a large library with diverse binders. Using the highly diverse DENV envelope protein domain IIIs as model targets and the competitive sorting technique, two cross-reactive antibodies against the conserved neutralizing epitopes on these targets were quickly identified from a diverse yeast display antibody library. The binding epitopes were also experimentally confirmed. Thus, this novel sorting strategy to quickly and efficiently identify epitope specific antibodies from the complex antibody library was fully validated.
Although a competitive panning strategy has previously been reported,19 our approach allows for quick isolation of antibodies that more precisely target the specific epitope(s) of interest. This approach is especially useful in the isolation of antibodies targeting conserved sites among highly diversified proteins, such as HIV envelope proteins, from naïve or immune antibody libraries that contain high affinity and high titer of non-neutralizing antibodies to the target. Such non-neutralizing antibodies very often outcompete the site-specific antibodies during the library panning process in a regular setting, which make the monoclonal antibody screening and epitope mapping process an extreme burden. Furthermore, our approach can be slightly modified to isolate antibodies with a precisely defined binding footprint. Multiple mutants each bearing a single point mutation in the desired binding epitope or footprint can be combined for the competitive panning and quickly narrow down those that precisely target the designed footprint.
One of the key elements of this approach is the design of the mutant competitor. It is very important to select the spot(s) of special interest for mutant construction. It is more important, however, to ensure that the introduced mutation will make local structural modification(s) dramatic enough to destroy the epitope of the protein, but sufficiently limited leave the overall structure of the target protein intact. Mutations that dramatically change the overall folding of the target protein will decrease the competition sensitivity during the sorting process and eventually create false positive clones. Alternatively, appropriate natural mutants can be chosen for the competition because the overall structure of natural mutants is well-conserved to preserve the biological function of the proteins.
Similarly, this approach can be readily applied to the isolation of unique antibodies that can bind differentially to desired isoforms among members of a complex protein family or homogenous proteins from different species.
In addition, as shown in Figure 3, the few cell populations at the bottom represent antibodies that can bind to the epitopes shared by both the wild type and the mutant antigens. Therefore, another gate can be set to specifically isolate these cross-reactive binders. By replacing the wild type and mutant domain III with another pair of protein/molecule isoforms, this approach can be also applied to the isolation of cross-reactive antibodies targeting different isoforms from the same protein family.
It is likely that there are a few dominant clones preferentially enriched from the sorting or gene amplification (PCR) during the course of the library construction; we should also note that there are a significant number of binders distributed between those major populations, which might indicate the diversity of the enriched pool of binders. It is also known that the domain III is very immunogenic in vivo. Previously, we also tried to pan a phage display naïve antibody library against the dengue envelope protein domain III, from which a panel of antibodies was isolated. None of the antibodies bound to neutralizing epitopes; therefore, to some extent those data indicate that there is relatively high diversity of the domain III binders in the yeast library because the antibody gene repertoire from the phage library was also cloned into the yeast library used in this study. The overall diversity of those libraries was confirmed from the 454 deep sequencing data. We found all of the germline families were present, although with biased gene usages for certain families as typical for human antibody repertoires.20,21 The rearranged HCDR3 is a result of the V-(D)-J rearrangements, which largely determine the diversity of expressed VH repertoires. The HCDR3 lengths ranged from 4 to 32 following a Poisson distribution. Based on the statistics obtained from the deep sequencing analysis, the diversity of a yeast display naïve library composed of these two naïve libraries from 10 and 59 individuals would lead to heavy and light paring recombination in the order of 106, and somatic diversification and other secondary recombination events will further increase the diversity exponentially. Therefore, the total numbers of unique binders existing in the yeast library could lead to more combinatorial diversity.
Numerous studies have shown that antibodies targeting different epitopes on the same target can have very different biological functions. In the case of the validated cancer target HER2, the marketed antibody trastuzumab (Herceptin®) can block the signal transduction mediated by HER2 in cancer cells and inhibit cancer cell growth. HER2-specific antibodies with different binding epitopes, however, can enhance the growth of the cancer cells expressing high levels of HER2 (references 22, 23; Z Zhu and D Dimitrov, unpublished data). In addition, recent clinical data showed an antibody targeting a new epitope on CD20 offers better clinical outcomes than rituximab (Rituxan®).24,25 In the therapeutic antibody development field, biosimilar or biosuperior product development through the isolation of antibodies targeting fully validated targets is becoming a trend.26,27 Targeting such epitopes offers the most beneficial results, and avoiding ineffectual epitopes is critical for the success of those efforts. Our approach can be a perfect fit for the efficient isolation of such biosimilar or biosuperior antibodies.
In conclusion, we developed and validated a highly efficient yeast display antibody library sorting strategy to quickly identify antibodies that precisely target specific epitopes on any protein target of biomedical interest. This strategy and its modified version have great implications for the efficient isolation of site-specific antibodies from a complex library and for the isolation of cross-reactive antibodies targeting different protein isoforms/variants. The fully human antibodies isolated in this model experiment also offer great potential for anti-DENV therapeutics development.
Materials and Methods
Yeast display naïve scFv antibody library, domain III genes, antibodies, biotinylation kit, cells
A large yeast display naïve scFv antibody library (~5x109) was constructed using a collection of antibody gene repertoires extracted from peripheral blood mononuclear cells and various immune organs from more than 60 individuals, including the genes used for the construction of two phage display Fab libraries.16,17 Particularly, the diversities of antibody genes from two independent libraries constructed from donors of 10 16 and 59 individuals,17 which formed the basis of most of the diversity in the large yeast display naïve library, were characterized by 454 deep sequencing. Four DENV envelope protein domain III consensus genes (one per serotype) were synthesized at Genscript, Inc. with appropriate restriction enzyme recognition sites at the flanking sequence for cloning purposes. Mouse monoclonal anti-c-Myc antibody was purchased from Roche. PE-conjugated streptavidin and Alexa Fluor-488-conjugated goat anti-mouse antibody were purchased from Invitrogen. Protein G column was purchased from GE Healthcare. Avi-tag specific biotinylation kit was purchased from Avidity, LLC. Yeast plasmid extraction kit was purchased from Zymo Research. The 293 free style protein expression kit was purchased from Invitrogen Inc. AutoMACS was purchased from Miltenyi Biotec.
Mutant design, DENV envelope protein domain III Fc fusion protein expression, purification, and biotinylation
All four DENV envelope protein domain III consensus genes were cloned into the pSectag vector (Invitrogen) with an Avi or c-Myc tagged human IgG1 Fc portion fused at the C-terminal. A point mutation at K310E in the conserved neutralizing epitope was introduced into domain III.3 (serotype 3) through overlapping PCR and cloned into pSectag with Fc-Avi-tag at the C-terminus. All the constructs were transfected into 293 free style cells for expression following the protocol provided by the manufacturer and purified using protein G column. Biotinylation of the purified proteins with Avi-tag (excluding the mutant) was performed using the kit from Avidity, LLC. following the instructions from the manufacturer.
AutoMACS sorting to downsize the initial yeast display scFv antibody library
Biotinylated domain III.3-Fc was used as the target for three rounds of sorting to downsize the initial library. Five x 1010 cells from the initial naïve library and 10 μg of biotinylated domain III.3-Fc were incubated in 50 ml PBSA at room temperature (RT) for 2 h with rotation; 1 mg of unrelated antibody, m102.4 IgG1, with the same Fc as domain III.3-Fc was also added to the mixture to prevent any Fc-specific antibodies from binding to Fc portion of the biotinylated domain III.3-Fc. The cells were washed three times with PBSA and then incubated with 100 μl of streptavidin conjugated microbeads from Miltenyi Biotec for 30 min. The cells and beads mixture was washed once with PBSA, and loaded onto the AutoMACS system for the first round of sorting. The sorted cells were amplified in synthetic dextrose casamino acids (SDCAA) media (20 g dextrose, 6.7 g Difco yeast nitrogen base w/o amino acid, 5 g Bacto casamino acids, 5.4 g Na2HPO4 and 8.56 g NaH2PO4.H2O in 1 L water) at 30°C and 250 rpm for 24 h and subsequently induced in SGCAA media (20 g galactose, 20 g Raffinose, 1 g dextrose, 6.7 g Difco yeast nitrogen base w/o amino acid, 5 g Bacto casamino acids, 5.4 g Na2HPO4 and 8.56 g NaH2PO4.H2O in 1 L water) at 20°C and 250 rpm for another 16–18 h. The same amounts of antigen, control antibody m102.4 IgG1, and incubation volume were used for the next two rounds of sorting. The cell numbers used for these two subsequent rounds of sorting were set at 100 times of the size of the sorted pool from the previous round of sorting to maintain the diversity of each sorted pool.
Competitive library panning and monoclonal yeast screening by flow cytometry analysis
After 3 rounds of sorting against biotinylated domain III.3-Fc, the downsized library was further sorted on a FACSAria II cell sorter against the same antigen with the addition of the unbiotinylated domain III.3-Fc mutant, bearing the point mutation (K310E) in the neutralizing epitope, as a competitor. A total of 5x108 cells were incubated with 1 μg/ml of biotinylated domain III.3 Fc fusion protein, 10 μg/ml of unbiotinylated domain III.3 mutant Fc fusion protein, and 2 μg/ml of mouse anti-c-Myc antibody in 5 ml of PBSA with rotation for 2 h at RT, followed by three times washing with PBSA. 1:100 diluted PE-conjugated streptavidin and Alexa Fluor-488-conjugated goat anti-mouse antibody in PBSA were then mixed with the cells and incubated at RT for 30 min in the dark, followed by another two times washing with PBSA. The stained cells were then loaded on the cell sorter for sorting. The gate was set to collect the population with the brightest PE/Alexa 488 signal. The collected cells were amplified in SDCAA media and induced in SGCAA medium as described above. The induced cells were stained and sorted one more time following the same process, and the sorted cells were further analyzed by flow cytometry analysis to confirm the specificity of the sorting. The sorted pool containing only the cells that could not be competed by the mutant was plated on a SDCAA plate for single yeast clone analysis. Twenty-four random clones from the plate were each inoculated into 2 ml SDCAA medium and amplified at 30°C and 250 rpm for 24 h and then induced each in 2 ml SGCAA medium at 20°C and 250 rpm for 16 h. Induced monoclonal yeast cells were stained as described above with DIII.1–4 Fc fusion proteins, but without the mutant competitor and analyzed by flow cytometry analysis to select the positive binders with cross-reactive binding activities.
Expression and purification of scFv-Fc-Avi proteins
Plasmids were extracted from the identified positive yeast clones using the kit from Zymo Research and following the instructions from the manufacturer. The extracted plasmids were transformed into chemically competent E. coli strain 10G cells from Lucigen for further amplification; the plasmids extracted from the bacteria were used for DNA sequencing to obtain the antibody sequences of the positive binders. The scFv inserts of the two unique clones, D6 and D7, were digested with SfiI and ligated into the pSectag vector bearing the same set of SfiI sites and C-terminal Fc-Avi tag for soluble expression. These constructs were transfected into 293 free style cells for expression following the protocol from the manufacturer. After 72 h of growth, the scFv-Fc fusion proteins from the cell culture medium were purified using protein G columns.
ELISA binding assays
The purified D6 and D7 scFv-Fc proteins were each diluted into PBS at 2 μg/ml; 50 μl of the diluted antibodies were coated in a 96-well plate at 4°C overnight. The Fc-c-Myc tagged domain III proteins from all four serotypes were each serially diluted in 3% milk-PBS and added to the antibody-coated wells for 1 h after prior blocking with 3% milk-PBS at RT. After washing, 1:2000 diluted HRP-conjugated anti-c-Myc antibody in 3% milk-PBS was added for 1 h at RT. After washing, 3, 3, 5, 5′-tetramethylbenzidine (TMB) substrate was added and O.D. was read at 450 nm.
A competition ELISA was further performed, where the D6 scFv-Fc fusion protein was coated as described above, the serially diluted antigen of domain III.3-Fc-c-Myc with and without either D7 scFv-Fc or the mutant domain III.3-Fc-Avi was added, and the bound antigen was detected and recorded similarly as above.
Epitope mapping of D7 through domain III.2 mutant library sorting and mapping of D7 binding escape mutants to the structure of DENV domain III.2
Random point mutations were introduced into the DENV envelope protein domain III.2 (serotype 2) gene through error-prone PCR using the mutagenesis kit from Stratagene and following the protocol provided by the manufacturer. The gel purified mutant gene repertoire was re-amplified using primers YDRDF and YDRDR under regular PCR conditions to add the flanking sequences for in vivo recombination through Gap repairing process. SfiI digested and gel purified yeast display vector pYD7 was mixed with the re-amplified mutant gene repertoire and transformed into electroporation competent yeast cells. Yeast display domain III.2 mutant library amplification and induction were performed as described above. The induced mutant library (5x108 cells) was incubated with 1 μg/ml biotinylated D7 scFv-Fc and 2 μg/ml mouse anti-c-Myc antibody at RT for 2 h, followed by three times washing and incubation with the mix of two secondary antibodies as described above. The stained cells were loaded onto the cell sorter and the cells which lacked binding to the antibody but still expressed the mutant antigen on surface demonstrated by negative PE staining and positive Alexa Fluor 488 staining, referred to as D7 binding escape mutants, were sorted. The sorting process was repeated once under the same conditions. Yeast cells bearing the binding escape mutants after the second round of sorting were collected and the plasmid from this pool was extracted and transformed into E. coli 10G cells. Plasmids from 48 random colonies were extracted and sequenced.
The fine mapping of mutated residues derived from the sequence analyses of D7 binding escape mutants was labeled onto the crystal structure of DENV envelope protein domain III.2 as found in the antibody Fab 1A1D-2 complex.28 The molecular surface was rendered with PyMOL (DeLano, W.L. The PyMOL Molecular Graphics System (2002) DeLano Scientific, San Carlos, CA, USA.)
Supplementary Material
Acknowledgments
We thank Professor Dane Wittrup, Massachusetts Institute of Technology, for providing the yeast display vector and yeast strain. We also thank members of our groups for helpful discussions. This project was supported by the Intramural Research Program of the National Institute of Health (NIH), National Cancer Institute (NCI), Center for Cancer Research, and by federal funds from the NCI, NIH, under contract N01-CO-12400, as well as by the NIAID Biodefense Program to DSD.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/25211
References
- 1.Dimitrov DS, Marks JD. Therapeutic antibodies: current state and future trends--is a paradigm change coming soon? Methods Mol Biol. 2009;525:1–27, xiii. doi: 10.1007/978-1-59745-554-1_1. [xiii.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–74. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- 3.Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol. 2005;23:1105–16. doi: 10.1038/nbt1126. [DOI] [PubMed] [Google Scholar]
- 4.Groves MA, Osbourn JK. Applications of ribosome display to antibody drug discovery. Expert Opin Biol Ther. 2005;5:125–35. doi: 10.1517/14712598.5.1.125. [DOI] [PubMed] [Google Scholar]
- 5.Ladner RC, Sato AK, Gorzelany J, de Souza M. Phage display-derived peptides as therapeutic alternatives to antibodies. Drug Discov Today. 2004;9:525–9. doi: 10.1016/S1359-6446(04)03104-6. [DOI] [PubMed] [Google Scholar]
- 6.Brekke OH, Løset GA. New technologies in therapeutic antibody development. Curr Opin Pharmacol. 2003;3:544–50. doi: 10.1016/j.coph.2003.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Osbourn J, Jermutus L, Duncan A. Current methods for the generation of human antibodies for the treatment of autoimmune diseases. Drug Discov Today. 2003;8:845–51. doi: 10.1016/S1359-6446(03)02803-4. [DOI] [PubMed] [Google Scholar]
- 8.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553–7. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 9.Shibasaki S, Ueda M. Development of yeast molecular display systems focused on therapeutic proteins, enzymes, and foods: functional analysis of proteins and its application to bioconversion. Recent Pat Biotechnol. 2010;4:198–213. doi: 10.2174/187220810793611545. [DOI] [PubMed] [Google Scholar]
- 10.Whitehead SS, Blaney JE, Durbin AP, Murphy BR. Prospects for a dengue virus vaccine. Nat Rev Microbiol. 2007;5:518–28. doi: 10.1038/nrmicro1690. [DOI] [PubMed] [Google Scholar]
- 11.Shrestha B, Brien JD, Sukupolvi-Petty S, Austin SK, Edeling MA, Kim T, et al. The development of therapeutic antibodies that neutralize homologous and heterologous genotypes of dengue virus type 1. PLoS Pathog. 2010;6:e1000823. doi: 10.1371/journal.ppat.1000823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guzman MG, Hermida L, Bernardo L, Ramirez R, Guillén G. Domain III of the envelope protein as a dengue vaccine target. Expert Rev Vaccines. 2010;9:137–47. doi: 10.1586/erv.09.139. [DOI] [PubMed] [Google Scholar]
- 13.Gromowski GD, Barrett ND, Barrett AD. Characterization of dengue virus complex-specific neutralizing epitopes on envelope protein domain III of dengue 2 virus. J Virol. 2008;82:8828–37. doi: 10.1128/JVI.00606-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajamanonmani R, Nkenfou C, Clancy P, Yau YH, Shochat SG, Sukupolvi-Petty S, et al. On a mouse monoclonal antibody that neutralizes all four dengue virus serotypes. J Gen Virol. 2009;90:799–809. doi: 10.1099/vir.0.006874-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sukupolvi-Petty S, Austin SK, Engle M, Brien JD, Dowd KA, Williams KL, et al. Structure and function analysis of therapeutic monoclonal antibodies against dengue virus type 2. J Virol. 2010;84:9227–39. doi: 10.1128/JVI.01087-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu Z, Dimitrov DS. Construction of a large naïve human phage-displayed Fab library through one-step cloning. Methods Mol Biol. 2009;525:129–42, xv. doi: 10.1007/978-1-59745-554-1_6. [xv.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen W, Zhu Z, Liao H, Quinnan GV, Jr., Broder CC, Haynes BF, et al. Cross-Reactive Human IgM-Derived Monoclonal Antibodies that Bind to HIV-1 Envelope Glycoproteins. Viruses. 2010;2:547–65. doi: 10.3390/v2020547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giudicelli V, Brochet X, Lefranc MP. IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb Protoc; 2011:695-715. [DOI] [PubMed] [Google Scholar]
- 19.Zhang MY, Choudhry V, Sidorov IA, Tenev V, Vu BK, Choudhary A, et al. Selection of a novel gp41-specific HIV-1 neutralizing human antibody by competitive antigen panning. J Immunol Methods. 2006;317:21–30. doi: 10.1016/j.jim.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci U S A. 2009;106:20216–21. doi: 10.1073/pnas.0909775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arnaout R, Lee W, Cahill P, Honan T, Sparrow T, Weiand M, et al. High-resolution description of antibody heavy-chain repertoires in humans. PLoS One. 2011;6:e22365. doi: 10.1371/journal.pone.0022365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kita Y, Tseng J, Horan T, Wen J, Philo J, Chang D, et al. ErbB receptor activation, cell morphology changes, and apoptosis induced by anti-Her2 monoclonal antibodies. Biochem Biophys Res Commun. 1996;226:59–69. doi: 10.1006/bbrc.1996.1311. [DOI] [PubMed] [Google Scholar]
- 23.Yip YL, Smith G, Koch J, Dübel S, Ward RL. Identification of epitope regions recognized by tumor inhibitory and stimulatory anti-ErbB-2 monoclonal antibodies: implications for vaccine design. J Immunol. 2001;166:5271–8. doi: 10.4049/jimmunol.166.8.5271. [DOI] [PubMed] [Google Scholar]
- 24.Robak T, Robak E. New anti-CD20 monoclonal antibodies for the treatment of B-cell lymphoid malignancies. BioDrugs. 2011;25:13–25. doi: 10.2165/11539590-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 25.Bornstein GG, Quéva C, Tabrizi M, van Abbema A, Chavez C, Wang P, et al. Development of a new fully human anti-CD20 monoclonal antibody for the treatment of B-cell malignancies. Invest New Drugs. 2010;28:561–74. doi: 10.1007/s10637-009-9291-z. [DOI] [PubMed] [Google Scholar]
- 26.Reichert JM. Next Generation and Biosimilar Monoclonal Antibodies: Essential Considerations Towards Regulatory Acceptance in Europe; February 3-4, 2011; Freiburg, Germany. MAbs; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ledford H. ‘Biosimilar’ drugs poised to penetrate market. Nature. 2010;468:18–9. doi: 10.1038/468018a. [DOI] [PubMed] [Google Scholar]
- 28.Lok SM, Kostyuchenko V, Nybakken GE, Holdaway HA, Battisti AJ, Sukupolvi-Petty S, et al. Binding of a neutralizing antibody to dengue virus alters the arrangement of surface glycoproteins. Nat Struct Mol Biol. 2008;15:312–7. doi: 10.1038/nsmb.1382. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.