

Abstract
Bromodomains are gaining increasing interest as drug targets. Commercially sourced and de novo synthesized substituted [1,2,4]triazolo[4,3-a]phthalazines are potent inhibitors of both the BET bromodomains such as BRD4 as well as bromodomains outside the BET family such as BRD9, CECR2, and CREBBP. This new series of compounds is the first example of submicromolar inhibitors of bromodomains outside the BET subfamily. Representative compounds are active in cells exhibiting potent cellular inhibition activity in a FRAP model of CREBBP and chromatin association. The compounds described are valuable starting points for discovery of selective bromodomain inhibitors and inhibitors with mixed bromodomain pharmacology.
Introduction
The rapidly expanding field of epigenetics can be broadly divided into two levels of processes: DNA methylation and histone modification. Various post-translational modifications of histone proteins contribute to the epigenetic code including methylation, acetylation, phosphorylation, ubiquitinylation, and citrullination.1 Acetylation of lysine residues plays an important role in the regulation of chromatin structure and ultimately transcription due to the charge neutralization that occurs, leading to changes in protein conformation and protein–protein interactions. It is similar to phosphorylation in its prevalence and has been particularly studied on unstructured histone tails. Aberrant lysine acetylation frequently leads to alterations in gene expression, causing activation of pro-survival and proliferation-promoting pathways and inactivation of tumor suppressor functions. Insight into the regulation of ε-N-acetyl-lysine (Kac) marks is therefore desirable in the understanding of and development of novel drugs for cancer treatment. Consequently, enzymes that write (acetyltransferases, HATs) and erase (histone deacetylases, HDACs) these marks have emerged as interesting targets.2,3
The bromodomain family of protein interaction modules specifically recognizes the acetyl-lysine mark, and these acetyl-lysine reader domains have likewise gained interest of late as novel targets for pharmacological intervention.4,5 Bromodomain-containing proteins are key components in diverse biological processes and are involved in mediating the assembly of various nuclear protein complexes, including the recruitment of chromatin modifying enzymes and transcriptional regulators to acetylated chromatin. Chromosomal rearrangements, aberrant expression of bromodomain-containing proteins, and protein dysfunctions have been tightly linked to tumorigenesis,6 and new avenues for the development of antineoplastic drugs have recently been highlighted by the potent antitumor activity exhibited by inhibitors which selectively target bromodomains.7 Known bromodomain inhibitors mainly target the BET family of bromodomains, including BRD3 and BRD4, but many other bromodomain-containing proteins such as CREBBP, TIF1α, ATAD2, and SMARCA4 have been implicated in a variety of diseases.4
Potent BET inhibitors generally fall into three structural classes: isoxazoles,8 amides/ureas,9 and 1,2,4-triazoles.5 Most BET inhibitors described to date have a methyl group adjacent to a hydrogen bond acceptor which mimics the acetyl group of acetyl lysine. The structurally related thieno- and benzo-diazepine triazoles (+)-JQ1 and I-BET762 (Figure 1A) were shown to be potent inhibitors of the BET bromodomains and have potential for use in inflammatory disease,10 atherosclerosis,11 NUT-midline carcinoma,7 acute leukemia,12 lymphoma,13 and HIV infection.14 Two structurally related fused triazoles (compounds 3 and 4) have been exemplified in patents from GSK15 and Constellation16 as potent BET inhibitors.
Figure 1.

(A) Triazole-containing BET inhibitors. (B) The bromodomain family is made of eight subfamilies (large italic).17 Family members screened in this work are shown in larger typeface.
Given the success of the chemical probes (+)-JQ1 and I-BET762 in deciphering the role of the BET subfamily of bromodomains in disease, it is clear that there is an urgent need for inhibitors for the remaining subfamilies of bromodomain-containing proteins in order to investigate their biological function and therapeutic potential.
Results and Discussion
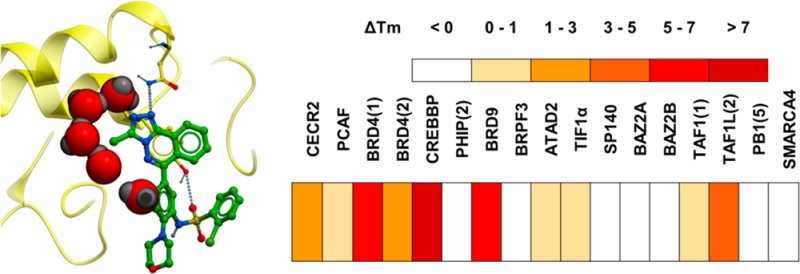
The selectivity of (+)-JQ1 and I-BET762 for the BETs has been attributed to the 4-chlorophenyl moiety which forms hydrophobic interactions with residues on the edge of the acetyl-lysine binding pocket including W81. It was hoped that by keeping the 3-methyl-[1,2,4]-triazolo motif but varying the fused ring and pendant substituents, novel compounds could be found that would maintain bromodomain potency with altered selectivity for non-BET proteins. To find new starting points for bromodomains outside the BET family, a number of triazole-containing commercial compounds were purchased and profiled against 17 BRDs in the bromodomain family tree by differential scanning fluorimetry (DSF) (Figure 1B).7,18 The [1,2,4]triazolo[4,3-a]phthalazines shown in Figure 2 were thought to be attractive potential BRD inhibitors due to the presence of the 5-methyl group adjacent to the triazole H-bond acceptor.19 It has been shown by Chung et al. that potent BRD inhibitors can be discovered by focusing on privileged substructures.20 By testing the potential inhibitors against bromodomains from the entire protein family by the operationally simple DSF assay in a platform discovery approach, a rapid assessment of BRD potency and selectivity was established.
Figure 2.

Commercial [1,2,4]triazolo[4,3-a]phthalazines are potent inhibitors of multiple bromodomains by DSF screening.
When tested in a panel of bromodomains, these initial compounds showed hits for BRD4(1), BRD9, CECR2, CREBBP, and TAF1L(2), with the greatest potencies against BRD9 (compounds 7 and 15) and CREBBP (compounds 9, 14, and 17). Very little activity was observed against BAZ2A, BAZ2B, PB1(5), and TIF1α. All of the commercially available triazolophthalazines shared an amide or sulfonamide substituent at the meta-position (R1) and a methyl group at the para-position (R2) of the pendant phenyl group. Sulfonamides were observed to have greater potency than amides (e.g., compounds 7 and 15 vs compound 5). N- and S-linked sulfonamides were found to have similar potencies (compound 7 vs 15). A variety of substituents were tolerated on the sulfonamide, both aryl and alkyl. To examine the effect of the omnipresent para-methyl group in the commercial compounds, compound 13 was synthesized (Scheme 1). This methyl group was shown to positively influence activity as the des-methyl analogue 13 was less active in the DSF assay against all bromodomains tested.
Scheme 1. The Synthesis of Compounds 13 and 24–33.

Reagents and conditions: (a) NH2NHAc, nBuOH, reflux (41%); (b) NH2NH2, THF; (c) R5CO2H, p-dioxane, reflux (32–42%); (d) Boc-glycine, THF, reflux (39%); (e) 35, Pd(PPh3)4, K2CO3, p-dioxane/H2O (29–80%); (f) HCl, EtOAc (100%); (g) 36, Pd(PPh3)4, K2CO3, p-dioxane/H2O (25%); (h) (i) 37, Pd(PPh3)4, K2CO3, p-dioxane/H2O, (ii) SnCl2, EtOH, reflux, (iii) PhSO2Cl, pyridine, THF (16% over 3 steps); (i) 39, Pd(PPh3)4, K2CO3, p-dioxane/H2O (35%); (j) (i) 38 or 40, Pd(PPh3)4, K2CO3, p-dioxane/H2O, (ii) KOH, MeOH (39–79% over 2 steps).
As these compounds showed binding to multiple bromodomains, compound 17 was chosen as a representative for docking into the bromodomain of CREBBP. Although there were other compounds as potent as compound 17, it was chosen for docking studies as it had a good combination of high potency and rigidity which provided a small number of calculated poses. All but the five water molecules previously shown to be important in CREBBP8e were removed from the published complex of CREBBP with a fragment ligand (PDB ID 3SVH), and compound 17 was docked into the protein using the ligedit functionality of ICM-Pro.21 As seen in Figure 3, compound 17 has an excellent fit to the bromodomain. The triazole forms two hydrogen bonds via adjacent nitrogen atoms to a structural water molecule and the conserved asparagine residue (N1168) found in most bromodomains. The triazole’s methyl group fits well into the cavity formed by the remaining water molecules. The meta-sulfonamide formed two hydrogen bonds to arginine (R1173) in CREBBP.
Figure 3.
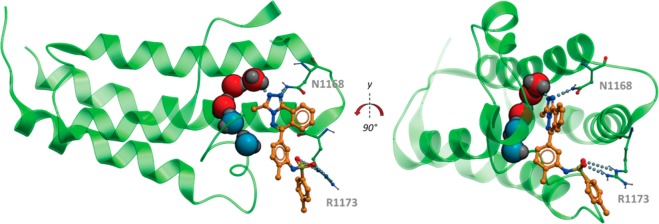
Docking of compound 17 (orange ball and stick) in bromodomain of CREBBP (PDB ID, 3SVH; protein, green ribbon and sticks; water molecules, red and blue spheres; top loop removed for clarity in first image). The triazole moiety forms two H-bonds (dashed line) to a conserved water (red sphere) and asparagine 1168 (N1168). The sulfonamide accepts two H-bonds from arginine 1173 (R1173).
To confirm the potencies initially established by DSF, AlphaScreen competition assays were used to test whether representative compounds 6–8 and 15 could displace labeled peptides from seven representative bromodomains (Table 1).22 These compounds were chosen as representatives as they all had similar structures but differed in their bromodomain inhibition profiles. With one exception (compound 8 with CREBBP), the pIC50 values measured for the representative compounds were consistent with DSF Tm shifts (ΔTm). The coefficient of determination (R2) between the two assay formats was high enough that the general and operationally simple DSF assay was felt to be a useful surrogate for the more complex AlphaScreen in further efforts to increase the potency of the compounds.
Table 1. pIC50 of Representative Compounds As Measured by AlphaScreen and ΔTm As Measured by DSF.
|
6 |
7 |
8 |
15 |
|||||
|---|---|---|---|---|---|---|---|---|
| target | ΔTm °Ca,b | pIC50 | ΔTm °Ca,b | pIC50 | ΔTm °Ca,b | pIC50 | ΔTm °Ca,b | pIC50 |
| BRD4(1) | 1.4 ± 0.40 (3) | 4.7 (4.6–4.8)c | 2.2 ± 0.15 (3) | 5.7 (5.6–5.8)c | 1.7 ± 0.08 (3) | 5.9 (5.9–5.9)c | 4.3 ± 0.16 (3) | 5.3 ± 0.54 (2)a |
| BRD9 | 2.1 ± 0.56 (3) | ND | 3.6 ± 0.31 (3) | 5.9 (5.7–6.1)c | 1.8 ± 0.22 (3) | 5.8 (5.6–6)c | 5.6 ± 0.30 (3) | 6.3 ± 0.12 (2)a |
| CECR2 | 1.5 ± 0.54 (3) | 5.3 (5.1–5.5)c | 2.4 ± 0.7 (3) | 5.5 (4.9–6.0)c | 3.2 ± 0.87 (3) | 6.6 (6.5–6.7)c | 3.3 ± 0.14 (3) | 6.3 ± 0.17 (2)a |
| CREBBP | 1.6 ± 0.10 (3) | 5.2 ± 0.48 (2)a | 1.9 ± 0.12 (3) | 5.8 (5.5–6.1)c | 0.7 ± 0.1 (3) | 5.9 (5.7–6.2)c | 2.8 ± 0.13 (3) | 4.8 ± 0.061 (3)a |
| BAZ2B | 0.03 ± 0.03 (3) | <4d | 0.23 ± 0.09 (3) | <4d | 0.20 ± 0.11 (3) | <4d | –0.013 ± 0.18 (3) | <4d |
| PB1(5) | 0.16 ± 0.03 (3) | <4d | –0.18 ± 0.13 (3) | <4d | –0.33 ± 0.05 (3) | <4d | –0.070 ± 0.087 (3) | <4d |
| TIF1α | –0.14 ± 0.04 (3) | <4d | –0.27 ± 0.05 (3) | <4d | –0.19 ± 0.06 (3) | <4d | –0.12 ± 0.082 (3) | <4d |
| R2e | 0.90 | 0.90 | 0.80 | 0.82 | ||||
Mean ± standard error of the mean (number of determinations).
Compound concentration 10 μM.
pIC50 (95% CI based on duplicate pIC50 measurements).
<25% inhibition at 100 μM.
Coefficient of determination based on a linear correlation between DSF Tm shift (abscissa) and AlphaScreen pIC50 (ordinate).
With an interesting series of inhibitors discovered through compound purchasing, additional analogues were designed and synthesized as shown in Scheme 1. Initial analogues focused on optimizing compounds 12 and 17 for CECR2 and CREBBP potency. Compound 12, although not the most potent analogue, had a preference for CECR2 and CREBBP by DSF Tm shift. Compound 17 was an attractive CREBBP lead due to its potency and synthetic accessibility. The fused methyl triazole ring was formed by substitution of 1,4-dichlorophthalazine 18 with acetyl hydrazide and in situ condensation. Suzuki coupling of the resulting aryl chloride with boronic ester 35 gave compound 13. Stepwise chloride displacement of compound 18 with hydrazine and subsequent condensation with carboxylic acids gave more elaborate substitution at the 2-position of the triazole in compounds 24–28 following Suzuki reaction. It was hoped that triazole substituents larger than methyl might compensate for the loss of activity on CECR2 and CREBBP seen between compounds 12 and 13 by displacing conserved waters in the bromodomain pocket (blue sphere in Figure 3). The hydrophobic ethyl and cyclopropyl groups of compounds 24 and 25, and hydrogen bonding groups of compounds 26 and 28 provided no additional binding beyond the methyl group of compound 13 and could not rescue the loss of the para-methyl group from compound 12 (Figure 4).17
Figure 4.

Synthetic inhibitors of multiple bromodomains.
The next group of analogues explored alternative additional substituents to the aryl sulfonamide. Compound 29 was made from the commercially available sulfonamide-containing boronic ester 36. To examine the effect of a chlorine in the 6-position of the phenyl ring, 2-chloro-5-nitrophenylboronic acid 37 was coupled to intermediate 19. Reduction and sulfonylation gave compound 30. Although compound 29 showed only weak binding to all bromodomains, compound 30 had modest binding to BRD4(1) and CREBBP.
As highlighted in Figure 3, CREBBP has an arginine (R1173) at the mouth of the acetylated peptide binding pocket and it was thought that the arylsulfonamide (calculated pKa 8.1)23 of compound 15 might be interacting with this charged arginine. Carboxylic acid-containing compounds were synthesized to try to exploit a potential salt bridge to R1173 and create CREBBP selective inhibitors. Compounds 31–33 were synthesized from compound 19 via Suzuki reaction with boronic acids 38–40 and hydrolysis of the methyl esters if required. Of the three, compound 33 with its extended acid was the most active against CREBBP but did not show selectivity over BRD4(1). Comparing compound 33 to compounds 12 and 13 showed that the acetic acid group could compensate to some extent for the loss of activity going from 12 to 13 and turn a CECR2 and CREBBP favoring inhibitor into a BRD4(1) and CREBBP inhibitor. Removing all substituents from the 6-phenyl group gave the simplest molecule described so far, compound 34, which, although it is only a modest inhibitor, has an intriguing selectivity for BRD9.24
The structure–activity relationship (SAR) established thus far had highlighted the importance of a group at the 4-position of the 6-phenyl substituent and the utility of a sulfonamide at the 3-position. The potent and lipophilic analogues in Figure 1, such as compounds 14 and 17, suffered from poor solubility. To identify more potent sulfonamides, without further loss in solubility, a polar replacement for the critical 4-methyl group was sought. As the docking of compound 17 in CREBBP (Figure 3) did not show a direct binding role of the 4-methyl group, its influence was hypothesized to be due to a positive conformational effect on the sulfonamide. N-Morpholino- and 4-methylpiperazinyl- groups were chosen as polar methyl substitutes, and analogues incorporating each were synthesized as shown in Scheme 2.
Scheme 2. The Synthesis of Compounds 49–57.

Reagents and conditions: (a) morpholine, iPrOH (100%); (b) 4-methylpiperazine, Et3N, iPrOH (100%); (c) (BOpin)2, Pd(dppf)Cl2, KOAc, p-dioxane/DMSO (47–72%); (d) 19, Pd(PPh3)4, K2CO3, p-dioxane/H2O (32–50%); (e) SnCl2, EtOH, reflux (26–100%); (f) RSO2Cl, Et3N, p-dioxane or RSO2Cl, pyridine, DCM (47–81%); (g) 22, Pd(PPh3)4, K2CO3, p-dioxane/H2O (62%); (h) (i) 23, Pd(PPh3)4, K2CO3, p-dioxane/H2O, (ii) HCl, EtOAc (60%).
In a route designed to allow synthesis of sulfonamide variants from a common intermediate, 2,5-dibromonitrobenzene 41 was selectively reacted with morpholine or 4-methylpiperazine to give compounds 42 and 43. After bromide to boronate substitution, Suzuki coupling with compound 19 gave compounds 45 and 47. Reduction with tin(II) chloride gave the anilines 46 and 48, which were capped with sulfonyl chlorides to give the inhibitors 49 – 55.
The resulting compounds 47–55 were tested by DSF in the panel of 17 bromodomains, and the results are summarized in Figure 5. The strategy to increase potency by replacing the para-methyl group with a cyclic amine was very successful; compound 50 was as potent as compound 15. However, increasing the size of the sulfonamide from methyl to para-tolyl to give compound 49 was detrimental to solubility and accurate Tm shift measurements were difficult to obtain. The chloro-substituted analogues of compound 49 were soluble, and compounds 51, 52, and 53 were potent and showed an apparent preference for CREBBP.
Figure 5.

Triazolophthalazines with para-aminophenyl substituents are potent bromodomain inhibitors.
Compound 53 is an interesting BRD4-, BRD9-, and CREBBP-favoring inhibitor targeting three different BRDs implicated in leukemia.12,25 More polar replacements for the morpholine and chlorophenyl substituents were incorporated in compound 55 in the form of 4-methylpiperazine and 4-methoxyphenylsulfonyl groups, respectively, which gave a preference for BRD9 over other bromodomains.
In a final attempt to displace two of the conserved waters in the bromodomain acetyl-lysine binding pocket (blue sphere in Figure 3), two analogues of the potent inhibitor 53 with triazole methyl group extensions, compounds 56 and 57, were synthesized from compound 42. But as in compounds 24–28, they both lost potency compared to the methyl analogue 53.
Four representative compounds (50, 51, 53, and 55) were chosen for confirmation screening and IC50 determination in AlphaScreen peptide displacement assays against five bromodomains (Table 2). In general, all compounds were inactive against TIF1α but the AlphaScreen assays showed less discrimination than the DSF assays for the remaining four bromodomains. This may be due to assay differences; DSF Tm shift is a measure of protein stability increased by ligand interaction, whereas AlphaScreen competition IC50 is also a function of the competing peptide’s affinity for the BRD. In addition, the AlphaScreen IC50 is consistently measured at 20–25 °C, whereas the DSF Tm shift is measured at the melting temperature of the protein which varies between proteins. Any compounds that bind with a large entropic contribution would be expected to show differences in potencies between assays run at different temperatures. Compound 50 was a promiscuous inhibitor and showed submicromolar IC50 values against BRD4(1), BRD9, CECR2, and CREBBP, whereas compound 51 was at least 100-fold selective for BRD4(1), BRD9, and CREBBP over CECR2. Compound 53 was similar in profile to compound 51, with less discrimination over CECR2. The only piperazine derivative, compound 55, had a slight preference for CECR2 over BRD4(1), but despite inclusion of polar substituents, poor solubility of this compound showed variable IC50 determinations for BRD9 and CREBBP as shown by the high standard errors.
Table 2. pIC50 of Representative Compounds As Measured by AlphaScreen and ΔTm As Measured by DSF.
|
50 |
51 |
53 |
55 |
|||||
|---|---|---|---|---|---|---|---|---|
| target | ΔTm °Ca,b,c | pIC50a,d | ΔTm °Ca,b,c | pIC50a,d | ΔTm °Ca,b,c | pIC50a,d | ΔTm °Ca,b,c | pIC50a,d |
| BRD4(1) | 2.6 ± 0.099 (6) | 6.6 ± 0.49 (2) | 4.4 ± 0.81 (11) | 6.8 ± 0.12 (2) | 4.4 ± 0.38 (9) | 6.0 ± 0.16 (2) | 2.5 ± 0.38 (8) | 5.3 ± 0.014 (2) |
| BRD9 | 4.9 ± 0.24 (4) | 6.5 ± 0.092 (3) | 6.6 ± 0.82 (6) | 6.7 ± 0.11 (3) | 6.2 ± 0.35 (5) | 6.0 ± 0.11 (2) | 7.3 ± 0.44 (3) | 6.2 ± 0.99 (2) |
| CECR2 | 3.7 ± 0.16 (3) | 6.8 ± 0.17 (2) | 1.62 ± 0.48 (4) | 4.6 ± 0.24 (2) | 2.0 ± 0.16 (3) | 4.7 ± 0.10 (2) | 2.0 ± 0.16 (3) | 6.0 ± 0.14 (2) |
| CREBBP | 3.9 ± 0.33 (4) | 6.2 ± 0.62 (2) | 7.6 ± 0.94 (7) | 6.7 ± 0.074 (2) | 7.9 ± 0.33 (6) | 6.2 ± 0.57 (2) | 6.7 ± 0.35 (6) | 6.1 ± 0.61 (2) |
Mean ± standard error of the mean (number of determinations).
Compound concentration 10 μM.
All compounds tested showed ΔTm < 0.2 °C for TIF1α.
All compounds tested showed <30% inhibition of TIF1α at 50 μM.
It is noteworthy that the compounds disclosed in this work are potent inhibitors of the CECR2, BRD4(1)/(2), CREBBP, BRD9, and TAF1L(2) bromodomains to varying degrees. The inhibited bromodomains do not cluster as expected from the sequence-based phylogenetic tree (Figure 1b). From the phylogenetic tree it would be expected that closely related BRDs such as PCAF (subfamily I shared with CECR2), PHIP(2) (subfamily III shared with CREBBP), BRPF3 and ATAD2 (subfamily IV shared with BRD9), and TAF1(1) (subfamily VII shared with TAF1L(2)) would also be affected by the inhibitors. It could be argued that the phylogenetic tree in Figure 1b described by Filippakopoulos et al.17 is based on a sequence alignment of the entire bromodomain, whereas only the residues in the ligand binding region are relevant to selectivity. A more ligand-focused phylogenetic tree has been described by Vidler et al. which clusters BRDs based on binding site residues.26 This refined analysis goes part of the way to explaining compound selectivity as it clusters PHIP(2) with PB1(5) (both not inhibited) and moves ATAD2 to its own branch, but it does not explain the remaining inconsistencies as PCAF still clusters with CECR2, BRPF3 with BRD9, and TAF1(1) with TAF1L(2). Calculated druggability is also insufficient to explain inhibitor preference as both PCAF and TAF1(1) have similar SiteMap D-scores to CECR2 and TAF1L(2).26
Compound 51 was chosen as a representative of this series, and co-crystallization was attempted with multiple bromodomains. High resolution structures were obtained with BRD4(1) and BRD9 (Figure 6). The overall pose of the compound with the two bromodomains was exactly as expected from the initial docking studies of the predecessor 17 in CREBBP (Figure 3). The triazole moiety formed hydrogen bonds to the conserved asparagine N140 in BRD4(1) and N100 in BRD9 and to a conserved pocket water molecule in both proteins. The phthalazine ring system was sandwiched securely between L94 and I146 in BRD4 and I53 and Y106 in BRD9. The pendant aryl ring allowed further interactions of the sulfonamide. In BRD4(1), the nitrogen of the sulfonamide appears to be deprotonated and acting as an H-bond acceptor to tryptophan-81 with a heavy atom distance of 2.1 Å. This unusual interaction has not been described for previous BET inhibitors, which rely on a hydrophobic interaction with W81 for potency. In BRD9, the sulfonamide is rotated to allow a hydrophobic interaction between the 2-chlorophenyl group and I53. This leaves the sulfonamide in a position to accept an H-bond from the phenol of Y106. The morpholine substituent does not make any direct interactions with the protein but keeps the sulfonamide conformation favorable for protein interactions. Although compound 51 was not crystallized in CECR2, it was hypothesized that the difference in selectivity could be rationalized by the large 2-chlorophenylsulfonamide being harder to accommodate in the smaller pocket of CECR2 than the smaller methylsulfonamide of the more promiscuous compound 50.
Figure 6.

(A) Compound 51 (green stick) in complex with BRD4(1) (PDB ID: 4NQM, blue ribbon and stick, top loop removed for clarity in first image) shows H-bonds (dashed lines) between the triazole moiety, the conserved asparagine (N140), and a pocket water (red sphere). The anionic sulfonamide forms an additional hydrogen bond to tryptophan (W81). (B) In complex with BRD9 (PDB ID: 4NQN, yellow ribbon and stick (the ZA loop was removed for clarity in first image)), compound 51 forms H-bonds to N100 and water but acts as an H-bond acceptor via a sulfonamide oxygen to Y106. The electron density map from the X-ray refinement is shown as a dark-blue mesh around the ligand.
The ability of inhibitors to displace the bromodomain of CREBBP from chromatin was assessed using fluorescence recovery after photobleaching (FRAP). A construct consisting of the multimerised bromodomain of CREBBP as well as a similar construct in which the conserved asparagine responsible for binding of acetylated lysine has been mutated to a phenylalanine was transfected into U2OS cells. Cells were treated with the histone deacetylase (HDAC) inhibitor SAHA in order to globally increase lysine acetylation, resulting in a better assay window (Figure 7A). Treatment of the cells with compounds 50, 51, 53, and 55 significantly decreased FRAP recovery times (Figures 7B), indicative of displacement of the BRD construct from hyper-acetylated chromatin. The piperazine derivative, compound 55, showed a slightly decreased recovery time, indicating a stronger binding to the CREBBP bromodomain.
Figure 7.

(A) Cells transfected with a trimerized CREBBP-BRD-GFP construct show rapid recovery of fluorescent intensity after photobleaching (FRAP) (black). Recovery time is increased by pretreating cells with 2.5 μM SAHA* (green) and restored by transfecting with incompetent mutant protein (N1168F, red). (B) Cells treated with SAHAa (2.5 μM) and compounds 50 (blue), 51 (yellow), 53 (purple), and 55 (red) (1 μM) show increased recovery rates. (C) Recovery half-lives of transfected (black), SAHA treated (green), and SAHA plus compound treated cells. (D) Fluorescent images of cells show rapid recovery of photobleached area (red circle) after compound treatment. aSAHA treated cells; bN1168F, mutation of N1168 to Phe.
Conclusions
A series of potent BRD inhibitor compounds has been developed. Initial SAR in this series shows the potential to develop selective inhibitors for individual bromodomains, with compounds showing some preference for the BRDs of CECR2, BRD4(1), CREBBP, BRD9, and TAF1L(2) over the likes of PCAF, PHIP(2), BRPF3, ATAD2, TIF1α, SP140, BAZ2A/B, TAF1(1), PB1(5), and SMARCA4. It is not clear from sequence- and structure-based clustering why these novel inhibitors have preference for some BRDs over others. Inhibitors with in vitro IC50 <1 μM have been identified for the previously untargeted BRDs of BRD9 and CECR2. A modular synthetic route allows diversification of multiple positions of the core and will be used to further explore this scaffold to find more selective molecules to probe the biological function of the less well studied members of this epigenetic reader family. Using a FRAP assay, selected compounds have been shown to be cell active. By adopting a chemical probe approach27 rather than a preselected target approach and characterizing compounds across the entire BRD family, compounds with intriguing polypharmacology have also been uncovered such as compound 53, which selectively inhibits three bromodomain-containing proteins implicated in leukemia (BRD4, CREBBP, and BRD9).
Experimental Section
General Experimental
Commercial reagents were used as received without further purification. Commercial anhydrous solvents were used in reactions, and HPLC grade solvents were employed for workup and chromatography. NMR spectra were recorded using a Varian Mercury 300 or 400 MHz for 1H and 75 or 101 MHz for 13C. The solvent was used as internal deuterium lock. Coupling constants (J) are quoted in Hz and are recorded to the nearest 0.5 Hz. Identical proton coupling constants are averaged in each spectrum and reported to the nearest 0.1 Hz. When peak multiplicities are reported, the following abbreviations are used: s = singlet, d = doublet, t = triplet, m = multiplet, br = broadened, dd = doublet of doublets, dt = doublet of triplets. LRMS employed an electrospray ionization source acquiring in positive and negative ionization mode. m/z values are reported in Daltons. Analytical HPLC was carried out on an Agilent 1100 equipped with photodiode array detector (DAD), quaternary gradient pump, and micro plate sampler (Agilent 220). Separation of the analytes was performed upon Centurysil C18-AQ + 5 μm, 50 mm × 4.6 mm (Johnson). The flow rate of the mobile phase was kept at 3.5 mL/min. Mobile phases B and C were acetonitrile with 0.35% CF3CO2H and water with 0.35% CF3CO2H, respectively. The gradient conditions were as follows: 0–0.5 min 1% B and 99% C, 3.7 min 90% B and 10% C, 5 min 99% B and 1% C. The injection volume was 10 μL. All compounds tested in biological assays were ≥95% pure by HPLC at 254 nm and by evaporative light scattering detection (ELSD).
Synthetic Procedure and Characterization of Compounds 13, 24–34, 49–57
6-Chloro-3-methyl-[1,2,4]triazolo[3,4-a]phthalazine (19)
1,4-Dichlorophthalazine 18 (5 g, 25.1 mmol) was mixed with n-butanol (100 mL) under argon, and acetic hydrazide (3.7 g, 50.2 mmol) was added. The reaction was stirred at reflux overnight. The mixture was cooled to room temperature, followed by filtration. The solid was washed with EtOAc and MeOH. The solid residue was purified by flash column chromatography (EtOAc:petroleum ether 1:3) to obtain title compound 19 (2.26 g, 41%). MS (ES+): m/z calcd for (C10H7ClN4 + H)+ 219.0, found 219.0. Purity (ELSD) >95%.
N-Benzyl-3-(3-methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)benzenesulfonamide (13)
A mixture of compound 19 (45 mg, 0.21 mmol), boronate 35 (50 mg, 0.17 mmol), Pd(PPh3)4 (20 mg), and K2CO3 (58 mg, 0.43 mmol) in dioxane and water was stirred under argon at 120 °C. The reaction was monitored by TLC. Upon completion, water was added and the aqueous layer was extracted with DCM. The organic layers were combined, washed with brine, and dried (Na2SO4). The solvents were removed in vacuo, and the residue was purified by flash column chromatography (DCM:MeOH 30:1) to give the title compound 13 (30 mg, 33%). MS (ESI): m/z calcd for (C23H19N5O2S + H)+ 430.1, found 429.9. 1H NMR (DMSO-d6) δ 8.60 (1H, d, J = 7.8), 8.11–8.02 (3H, m), 7.96–7.79 (3H, m), 7.66 (1H, d, J = 8.1), 7.24–7.11 (5H, m), 4.10 (2H, s), 2.71 (3H, s).
N-Benzyl-3-(3-ethyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)benzenesulfonamide (24)
1-Chloro-4-hydrazinylphthalazine28 (300 mg, 1.54 mmol) was dissolved in propanoic acid (3 mL), and the solution was heated to reflux. The reaction was monitored by TLC. Upon completion, the solvent was removed in vacuo, and the residue was purified by flash column chromatography (DCM:MeOH 20:1) to provide the intermediate 6-chloro-3-ethyl-[1,2,4]triazolo[3,4-a]phthalazine (20) (149 mg, 42%). A mixture of compound 20 (45 mg, 0.21 mmol), boronate 35 (50 mg, 0.17 mmol), Pd(PPh3)4 (20 mg), and K2CO3 (58 mg, 0.43 mmol) in dioxane and water was heated and stirred under argon at 120 °C. The reaction was monitored by TLC. Upon completion, water was added and the mixture was extracted with DCM. The organic layers were combined, washed with brine, and dried (Na2SO4). The solvents were removed in vacuo, and the residue was purified by flash column chromatography (DCM:MeOH 30:1) to give the title compound 24 (30 mg, 33%). MS (ESI): m/z calcd for (C24H21N5O2S + H)+ 444.1, found 443.9. 1H NMR (DMSO-d6) δ 8.62 (1H, d, J = 7.8), 8.12–7.80 (6H, m), 7.69 (1H, d, J = 8.4), 7.25–7.18 (5H, m), 4.12 (2H, s), 3.14 (2H, q, J = 7.5), 1.42 (3H, t, J = 7.8). HPLC retention time 3.088 min.
N-Benzyl-3-(3-cyclopropyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)benzenesulfonamide (25)
Following the same procedure as for 24, the title compound was obtained in 80% yield via compound 21. MS (ESI): m/z calcd for (C25H21N5O2S + H)+ 456.1, found 455.9. 1H NMR (DMSO-d6) δ 8.59 (1H, d, J = 7.2), 8.10–8.04 (3H, m), 7.95 (1H, d, J = 7.8), 7.91–7.80 (2H, m), 7.66 (1H, d, J = 7.8), 7.25–7.17 (5H, m), 4.11 (2H, s), 2.45 (1H, m), 1.21–1.15 (4H, m).
N-Benzyl-3-(3-(hydroxymethyl)-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)benzenesulfonamide (26)
Following the same procedure as for 24, title compound 26 was obtained in 37% yield via compound 22. MS (ESI): m/z calcd for (C23H19N5O3S + H)+ 446.1, found 446.1. 1H NMR (DMSO-d6) δ 8.65 (1H, d, J = 8.1), 8.14–8.04 (3H, m), 7.99–7.90 (2H, m), 7.83 (1H, t, J = 7.8), 7.69 (1H, d, J = 7.8), 7.25–7.18 (5H, m), 4.96 (2H, s), 4.11 (2H, s).
tert-Butyl ((6-(3-(N-Benzylsulfamoyl)phenyl)-[1,2,4]triazolo[3,4-a]phthalazin-3-yl)-methyl)carbamate (27)
1-Chloro-4-hydrazinylphthalazine28 (1.6 g, 8.24 mmol) and Boc-glycine (7.2 g, 40 mmol) were dissolved in THF (150 mL), and the mixture was stirred at reflux. The reaction was monitored by TLC. Upon completion, the mixture was concentrated in vacuo, followed by dilution with H2O (50 mL) and extraction with DCM (3 × 20 mL). The combined organic layers were dried (Na2SO4), concentrated in vacuo, and the residue was purified by flash column chromatography (DCM:MeOH 30:1) to provide the intermediate 23 (237 mg, 39%).
A mixture of tricyclic triazole 23 (169 mg, 0.51 mmol), boronate 35 (162 mg, 0.56 mmol), Pd(PPh3)4 (58 mg, 0.1 equiv), and K2CO3 (175 mg, 1.27 mmol) in dioxane (5 mL) and water (0.5 mL) was stirred and heated under argon at 120 °C. The reaction was monitored by TLC. Upon completion, water (20 mL) was added and the mixture was extracted with DCM (3 × 20 mL). The combined organic layers were dried (Na2SO4), concentrated in vacuo, and the residue was purified by flash column chromatography (DCM:MeOH 40:1) to give the title compound 27 (80 mg, 29%). MS (ESI): m/z calcd for (C28H28N6O4S + H)+ 545.2, found 545.1. 1H NMR (CDCl3): δ 8.72 (1H, d, J = 7.8), 8.17 (1H, m), 8.09 (1H, m), 7.95 (1H, m), 7.87 (1H, m), 7.76–7.72 (3H, m), 7.26–7.22 (5H, m), 5.47 (2H, br), 4.92 (2H, d, J = 5.7), 4.27 (2H, d, J = 6.3), 1.42 (9H, s).
3-(3-(Aminomethyl)-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)-N-benzylbenzenesulfonamide (28)
Compound 27 (42 mg, 0.08 mmol) was dissolved in a solution of HCl in EtOAc. The mixture was stirred at room temperature, and the reaction was monitored by TLC. Upon completion, the precipitate was filtered to afford the title compound (40 mg, 100%) as the HCl salt. MS (ESI): m/z calcd for (C23H20N6O2S + H)+ 445.1, found 445.1. 1H NMR (DMSO-d6): δ 8.70 (1H, d, J = 7.8), 8.17 (1H, m), 8.10–8.07 (2H, m), 8.01–7.98 (2H, m), 7.85 (1H, m), 7.75 (1H, d, J = 7.8), 7.27–7.22 (5H, m), 4.65 (2H, s), 4.12 (2H, s).
N-(3-Chloro-5-(3-methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)phenyl)benzenesulfonamide (29)
Benzenesulfonyl chloride (0.26 mL, 2 mmol) was added to a mixture of 3-bromo-5-chloroaniline (350 mg, 1.7 mmol) and pyridine (0.16 mL, 2 mmol) in THF (8 mL), and the resulting mixture was stirred at room temperature for 6 h. Water was added, and the aqueous layer was extracted with DCM. The organic layers were combined, washed with brine, and dried (Na2SO4). The solvent was removed in vacuo, and the residue was purified by flash column chromatography (EtOAc:petroleum ether 8:1) to give the intermediate N-(3-bromo-5-chlorophenyl)-benzene sulfonamide (380 mg).
A mixture of the sulfonamide from the preceding reaction (180 mg, 0.52 mmol), bis(pinacolato)diboron (145 mg, 0.57 mmol), KOAc (101 mg, 1.04 mmol), and Pd(dppf)Cl2 (11 mg, 0.016 mmol) in p-dioxane (3 mL) and DMSO (0.1 mL) was degassed with argon and heated at 85 °C for 26 h. The reaction was monitored by TLC. Upon completion, the solvents were removed in vacuo, aqueous NaOH (2 M, 20 mL) was added to the residue, and the mixture was stirred for 10 min at room temperature. The mixture was extracted with EtOAc, 6 M HCl was added to the aqueous layer to adjust the pH to 3–4, and it was then extracted with EtOAc. The organic layer was washed with brine, dried (Na2SO4), concentrated in vacuo, and purified by flash column chromatography to provide the compound 36 (126 mg).
A mixture of tricyclic triazole 19 (40 mg, 0.19 mmol), boronate 36 (70 mg, 0.18 mmol), Pd(PPh3)4 (20 mg), and K2CO3 (6 mg, 0.43 mmol) in dioxane and water was stirred and heated under argon at 120 °C. The reaction was monitored by TLC. Upon completion, water was added and the aqueous layer was extracted with DCM. The organic layers were combined, washed with brine, and dried (Na2SO4). The solvents were removed in vacuo, and the residue was purified by flash column chromatography (DCM:MeOH 30:1) to give title compound 29 (20 mg, 25%). MS (ESI): m/z calcd for (C22H16ClN5O2S + H)+ 450.1 (35Cl) and 452.1 (37Cl), found 449.8 (35Cl) and 451.9 (37Cl). 1H NMR (DMSO-d6) δ 8.57 (1H, d, J = 7.2), 8.06 (1H, t, J = 7.2), 7.81–7.77 (3H, m), 7.64–7.51 (5H, m), 7.34–7.27 (2H, m), 2.69 (3H, s).
N-(4-Chloro-3-(3-methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)phenyl)benzenesulfon-amide (30)
Step 1: 6-(2-Chloro-5-nitrophenyl)-3-methyl-[1,2,4]triazolo[3,4-a]phthalazine
A mixture of tricyclic triazole 19 (500 mg, 2.28 mmol), 2-chloro-5-nitrophenylboronic acid (690 mg, 3.43 mmol), Pd(PPh3)4 (264 mg, 10%), and K2CO3 (789 mg, 5.72 mmol) in dioxane and water was stirred and heated under argon at 120 °C. The reaction was monitored by TLC. Upon completion, water was added and the aqueous layer was extracted with DCM. The organic layers were combined, washed with brine, and dried (Na2SO4). The solvents were removed in vacuo, and the residue was purified by flash column chromatography (EtOAc:petroleum ether 10:1) to give the title compound (307 mg, 39%). MS (ESI): m/z calcd for (C16H10ClN5O2 + H)+ 340.0 (35Cl) and 342.0 (37Cl), found 339.9 (35Cl) and 341.9 (37Cl). 1H NMR (CDCl3) δ 8.79 (1H, d, J = 7.8), 8.46–8.42 (2H, m), 7.99 (1H, m), 7.82 (1H, d, J = 9.6), 7.74 (1H, m), 7.43 (1H, d, J = 8.1), 2.84 (3H, s).
Step 2: 4-Chloro-3-(3-methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)aniline
A mixture of tricyclic triazole from step 1 (115 mg, 0.34 mmol) and SnCl2 (381 mg, 1.69 mmol) in ethanol was stirred under reflux for 5 h. Water was added, the pH was adjusted to 7–8 using saturated aqueous NaHCO3, and the aqueous layers were extracted with DCM. The organic layers were combined, washed with brine, and dried (Na2SO4). The solvents were removed in vacuo, and the residue was purified by flash column chromatography (DCM:MeOH 20:1) to give the title compound (100 mg, 95%). MS (ESI): m/z calcd for (C16H12ClN5 + H)+ 310.0 (35Cl) and 312.0 (37Cl), found 309.9 (35Cl) and 311.9 (37Cl). 1H NMR (CDCl3) δ 8.71 (1H, m), 7.91 (1H, m), 7.70 (1H, m), 7.56 (1H, m), 7.33 (1H, d, J = 8.7), 6.86 (1H, dd, J = 8.4, 2.7), 6.79 (1H, d, J = 2.7), 3.63 (2H, br s), 2.83 (3H, s).
Step 3
Benzene sulfonyl chloride (0.03 mL, 0.24 mmol) was added to a solution of aniline from step 2 (50 mg, 0.16 mmol) in anhydrous THF (3 mL), followed by addition of pyridine (0.026 mL, 0.32 mmol). The resultant mixture was stirred at room temperature, and the reaction was monitored by TLC. Upon completion, water was added and the aqueous layer was extracted with DCM. The organic layers were combined, washed with brine, and dried (Na2SO4). The solvents were removed in vacuo, and the residue was purified by flash column chromatography (DCM:MeOH 30:1) to give title compound 30 (30 mg, 42%). MS (ESI): m/z calcd for (C22H16ClN5O2S + H)+ 450.1 (35Cl) and 452.1 (37Cl), found 449.9 (35Cl) and 451.9 (37Cl). 1H NMR (DMSO-d6) δ 8.58 (1H, d, J = 7.8), 8.07 (1H, t, J = 7.8), 7.85–7.79 (3H, m), 7.71–7.57 (4H, m), 7.39 (1H, dd, J = 8.7, 2.7), 7.32 (1H, d, J = 2.7), 7.19 (1H, d, J = 8.1), 2.69 (3H, s).
3-(3-Methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)benzoic Acid (31)
A mixture of tricyclic triazole 19 (24 mg, 0.11 mmol), boronic acid 38 (21 mg, 0.12 mmol), Pd(PPh3)4 (13 mg, 0.1 equiv), and K2CO3 (37 mg, 0.27 mmol) in dioxane and water was stirred and heated under argon at 120 °C. The reaction was monitored by TLC. Upon completion, water was added and the aqueous layers were extracted with DCM. The organic layers were combined and dried (Na2SO4). The solvents were removed in vacuo, and the residue was purified by flash column chromatography (DCM:MeOH 30:1) to give the Suzuki adduct methyl 3-(3-methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)benzoate (29 mg, 83%). The intermediate ester (29 mg, 0.09 mmol) was dissolved in methanol (14 mL), and KOH (99 mg, 1.76 mmol) was added. The reaction was monitored by TLC, and upon completion water (21 mL) was added. The mixture was adjusted to pH 3–4 with 6 M HCl and extracted with DCM. The combined organic layers were dried (Na2SO4), and the solvent was removed in vacuo to give the title compound 31 (26 mg, 95%). MS (ESI): m/z calcd for (C17H12N4O2 + H)+ 305.1, found 305.1. 1H NMR (DMSO-d6) δ 8.60 (1H, d, J = 7.8), 8.25–8.19 (2H, m), 8.08 (1H, m), 7.98 (1H, m), 7.88 (1H, m), 7.80–7.76 (2H, m), 2.72 (3H, s).
3-(3-Methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)-5-nitrobenzoic Acid (32)
Following the same procedure as for compound 31 step 1, the methyl ester hydrolyzed under the reaction conditions to yield title compound 32 directly (35%). MS (ESI): m/z calcd for (C17H11N5O4 + H)+ 350.1, found 350.0. 1H NMR (DMSO-d6) δ 8.84–8.79 (2H, m), 8.64–8.60 (2H, m), 8.09 (1H, m), 7.90–7.80 (2H, m), 2.73 (3H, s).
2-(3-(3-Methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)phenyl)acetic Acid (33)
Tricyclic triazole 19 and boronic acid 40 were coupled and hydrolyzed using the same procedures as for compound 31 give the desired compound (Suzuki, 53%; hydrolysis, 73%). MS (ESI): m/z calcd for (C18H14N4O2 + H)+ 319.1, found 319.1. 1H NMR (DMSO-d6) δ 8.58 (1H, d, J = 7.8), 8.21 (1H, m), 7.94–7.76 (2H, m), 7.69–7.46 (4H, m), 3.75 (2H, s), 2.72 (3H, s).
3-Methyl-6-phenyl-[1,2,4]triazolo[3,4-a]phthalazine (34)
1-Hydrazino-4-phenyl-phthalazine (300 mg, 1.27 mmol) was dissolved in acetic acid (3 mL) and refluxed for 2 h. The reaction mixture was concentrated in vacuo and the residue purified by flash column chromatography (EtOAc:petroleum ether 1:3) to give title compound 34 (269 mg, 81% yield). MS (ESI): m/z calcd for (C16H12N4 + H)+ 261.1, found 261.0. 1H NMR (CDCl3) δ 8.67 (1H, d, J = 6.0), 7.89–7.82 (2H, m), 7.64–7.49 (6H, m), 2.77 (3H, s).
4-(4-(3-Methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)-2-nitrophenyl)morpholine (47)
4-(4-Bromo-2-nitrophenyl)morpholine29 (1.03 g) was treated with bis(pinacolato)diboron (1 g, 3.94 mmol), KOAc (703 mg, 7.16 mmol), and Pd(dppf)Cl2 (89 mg) in p-dioxane (20 mL) and DMSO (0.5 mL) and heated to reflux under argon overnight. After removal of the solvents in vacuo, aqueous NaOH (2 M, 10 mL) was added to the residue and the mixture was stirred for 30 min at room temperature. The mixture was extracted with EtOAc, and 6 M HCl was added to the aqueous layer to adjust the pH to 3–4. The precipitate formed was collected by filtration and dried then purified by flash column chromatography (DCM:MeOH 50:1) to provide the corresponding boronate (860 mg, 72%) which was used directly.
A mixture of aryl chloride 19 (59 mg, 0.27 mmol), boronate from the previous reaction (100 mg, 0.30 mmol), K2CO3 (94 mg, 0.68 mmol), and Pd(PPh3)4 (31 mg) in p-dioxane (5 mL) and water (0.5 mL) was heated to reflux under argon. The reaction was monitored by TLC. Upon completion, the mixture was filtered, and the filtrate was evaporated to dryness. The residue was partitioned between EtOAc and water. The organic layer was washed with brine, dried (Na2SO4), concentrated in vacuo, and the crude residue was purified by flash column chromatography (petroleum ether:EtOAc 1:10) to give the title compound 47 (50 mg, 50%). MS (ESI): m/z calcd for (C20H18N6O3 + H)+ 391.1, found 390.9. 1H NMR (CDCl3) δ 8.81 (1H, d, J = 8.1), 8.19 (1H, d, J = 2.1), 8.02–7.77 (4H, m), 7.34 (1H, d, J = 8.7), 3.92 (4H, t, J = 4.5), 3.23 (4H, t, J = 4.5), 2.86 (3H, s).
5-(3-Methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)-2-morpholinoaniline (48)
A mixture of nitrophenyl 45 (50 mg, 0.13 mmol) and SnCl2 (144 mg, 0.64 mmol) in ethanol was stirred under reflux for 5 h. Water was added, and the pH was adjusted to 7–8 by adding saturated aqueous NaHCO3. The mixture was filtered to remove the precipitate, and the filtrate was extracted with DCM. The organic layers were combined, washed with brine, and dried (Na2SO4). The solvents were removed in vacuo, and the residue was purified by flash column chromatography (DCM:MeOH 60:1) to give title compound 48 (12 mg, 26%). MS (ESI): m/z calcd for (C20H20N6O + H)+ 361.1, found 361.1. 1H NMR (CDCl3) δ 8.72 (1H, d, J = 7.8), 7.99 (1H, d, J = 8.1), 7.91 (1H, t, J = 7.5), 7.71 (1H, t, J = 7.8), 7.17 (1H, m), 7.05–7.04 (2H, m), 3.92 (4H, t, J = 4.2), 3.04 (4H, t, J = 4.2), 2.83 (3H, s).
4-Methyl-N-(5-(3-methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)-2-morpholinophenyl)-benzenesulfonamide (49)
Et3N (36 μL, 0.249 mmol) was added to a solution of 48 (30 mg, 0.083 mmol) in p-dioxane (5 mL), followed by addition of p-toluenesulfonyl chloride (36 mg, 0.19 mmol). The reaction mixture was stirred at room temperature and monitored by TLC. Upon completion, water was added and the aqueous layers were extracted with DCM. The organic layers were combined, washed with brine, and dried (Na2SO4). The solvents were removed in vacuo, and the residue was purified by flash column chromatography (DCM:MeOH 60:1) to give title compound 49 (27 mg, 63%). MS (ESI): m/z calcd for (C27H26N6O3S + H)+ 515.1, found 514.9. 1H NMR (CDCl3) δ 8.78 (1H, d, J = 7.8), 8.01–7.70 (7H, m), 7.39–7.25 (3H, m), 3.86 (4H, t, J = 4.2), 2.84 (3H, s), 2.74 (4H,t, J = 4.2), 2.40 (3H, s).
N-(5-(3-Methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)-2-morpholinophenyl)methane-sulfonamide (50)
Methanesulfonyl chloride (14 mg, 0.12 mmol) was added to a solution of 48 (22 mg, 0.06 mmol) in DCM (1.6 mL), followed by addition of pyridine (15 μL, 0.03 mmol). The resulting mixture was stirred at room temperature, and the reaction was monitored by TLC. Upon completion, water was added and the aqueous layers were extracted with DCM. The organic layers were combined and dried (Na2SO4). The solvent was removed in vacuo, and the residue was purified by flash column chromatography (DCM:MeOH 30:1) to give compound 50 (16 mg, 60%). MS (ESI): m/z calcd for (C21H22N6O3S + H)+ 439.1, found 439.1. 1H NMR (CDCl3) δ 8.79 (1H, d, J = 7.8), 8.00–7.94 (2H, m), 7.90–7.85 (2H, m), 7.78 (1H, m), 7.48 (2H, m), 3.98–3.95 (4H, m), 3.20 (3H, s), 3.04–3.01 (4H, m), 2.86 (3H, s).
2-Chloro-N-(5-(3-methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)-2-morpholinophenyl)-benzenesulfonamide (51)
Following the same procedure as for compound 50 gave compound 51 (29 mg, 81%). MS (ESI): m/z calcd for (C26H23ClN6O3S + H)+ 535.1 for 35Cl and 537.1 for 37Cl, found 535.0 and 537.0. 1H NMR (CDCl3) δ 8.80 (2H, m), 8.13 (1H, d, J = 7.4), 7.96 (1H, m), 7.79–7.64 (3H, m), 7.56–7.45 (2H, m), 7.41–7.30 (3H, m), 3.98–3.87 (4H, m), 3.05–2.90 (4H, m), 2.80 (3H, s).
3-Chloro-N-[5-(3-methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)-2-morpholinophenyl]-benzenesulfonamide (52)
Following the same procedure as for compound 50 gave compound 52 (36 mg, 78%). MS (ESI): m/z calcd for (C26H23ClN6O3S + H)+ 535.1 for 35Cl and 537.1 for 37Cl, found 535.0 and 537.0. 1H NMR (CDCl3) δ 8.77 (1H, d, J = 8.0), 8.09 (1H, s), 7.96 (1H, m), 7.88–7.81 (2H, m), 7.81–7.70 (3H, m), 7.55 (1H, m), 7.40 (3H, m), 3.95–3.79 (4H, m), 2.82 (3H, s), 2.78–2.66 (4H, m).
4-Chloro-N-[5-(3-methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)-2-morpholinophenyl]-benzenesulfonamide (53)
Following the same procedure as for compound 50 gave compound 53 (37 mg, 64%). MS (ESI): m/z calcd for (C26H23ClN6O3S + H)+ 535.1 for 35Cl and 537.1 for 37Cl, found 535.1 and 537.0. 1H NMR (CDCl3) δ 8.83 (1H, d, J = 7.8), 8.06 (1H, s), 7.98 (1H, m), 7.85–7.69 (5H, m), 7.46–7.34 (4H, m), 3.93–3.81 (4H, m), 2.85 (3H, s), 2.81–2.70 (4H, m).
3,5-Dichloro-N-[5-(3-methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)-2-morpholinophenyl]-benzenesulfonamide (54)
Following the same procedure as for compound 50 gave compound 54 (33 mg, 64%). MS (ESI): m/z calcd for (C26H22Cl2N6O3S + H)+ 569.1 for 35Cl2 and 571.1 for 35Cl37Cl, found 569.0 and 571.0. 1H NMR (CDCl3) δ 8.76 (1H, d, J = 7.9), 8.17 (1H, br s), 7.95 (1H, m), 7.86–7.61 (5H, m), 7.54 (1H, m), 7.48–7.32 (2H, m), 3.90 (4H, m), 2.83 (3H, s), 2.77 (4H, m).
4-Methoxy-N-[5-(3-methyl-[1,2,4]triazolo[3,4-a]phthalazin-6-yl)-2-(4-methylpiperazin-1-yl)phenyl]benzenesulfonamide (55)
Starting from compound 41 and 4-methylpiperazine, compound 55 (16 mg) was synthesized in a manner analogous to compound 49. MS (ESI): m/z calcd for (C28H29N7O3S + H)+ 544.2, found 544.3. 1H NMR (300 MHz, CDCl3): δ 8.75 (1H, d, J = 7.2), 8.03 (1H, br s), 7.93 (1H, m), 7.86–7.80 (4H, m), 7.73 (1H, m), 7.36–7.35 (2H, m), 6.92 (2H, d, J = 9.0), 3.85 (3H, s), 2.84 (7H, m), 2.69 (4H, br s), 2.46 (3H, s).
4-Chloro-N-{5-[3-(hydroxymethyl)-[1,2,4]triazolo[3,4-a]phthalazin-6-yl]-2-morpholino-phenyl}benzenesulfonamide (56)
5-Bromo-2-morpholinoaniline2944 (640 mg, 2.49 mmol), bis(pinacolato)diboron (945 mg, 3.73 mmol), Pd(dppf)Cl2 (91 mg), and KOAc (610 mg, 6.23 mmol) were dissolved in dioxane (30 mL) and DMSO (1 mL) and heated to 85 °C under argon. The reaction was stirred and monitored by TLC; upon completion, the solvent was removed in vacuo and the residue was dissolved in 2 N NaOH (20 mL) and stirred for 10 min. The mixture was extracted with diethyl ether, and the combined organic layers were dried (Na2SO4). After removal of the solvent in vacuo, the residue was purified by flash column chromatography (petroleum ether:EtOAc 4:1) to give the intermediate boronate (960 mg, 100%).
4-Chlorobenzene-1-sulfonyl chloride (435 mg, 2.06 mmol) was added to a solution of the boronate from the previous reaction (314 mg, 1.03 mmol) in DCM (5 mL), followed by addition of pyridine (0.25 mL, 3.09 mmol). The mixture was stirred at room temperature, and the reaction was monitored by TLC. Upon completion, water was added and the aqueous layer was extracted with DCM. The combined organic layers were dried (Na2SO4), and the solvent was removed in vacuo. The residue was purified by flash column chromatography (petroleum ether:EtOAc 6:1) to give the sulfonamide (232 mg, 47%), which was coupled to the heteroaryl chloride 22 as in the preparation of compound 26 to give the title compound 56 (23 mg, 62%). MS (ESI): m/z calcd for (C26H23ClN6O4S + H)+ 551.1 for 35Cl and 553.1 for 37Cl, found 551.1 and 553.1. 1H NMR (300 MHz, CDCl3) δ 8.87 (1H, d, J = 7.8), 8.05 (2H, m), 7.89–7.81(5H, m), 7.49 (2H, d, J = 8.7), 7.44–7.37 (2H, m), 5.33 (2H, s), 3.91 (4H, m), 2.82 (4H, m).
N-{5-[3-(Aminomethyl)-[1,2,4]triazolo[3,4-a]phthalazin-6-yl]-2-morpholinophenyl}-4-chlorobenzenesulfonamide (57)
Following analogous procedures as for the preparation of compound 56 gave compound 57 from compound 44 (16 mg, 60%). MS (ESI): m/z calcd for (C26H24ClN7O3S + H)+ 550.1 for 35Cl and 552.1 for 37Cl, found 550.1 and 552.0. 1H NMR (300 MHz, CDCl3/CD3OD) δ 8.65 (1H, d, J = 8.1), 8.02 (1H, m), 7.84 (2H, d, J = 4.2), 7.64 (3H, m), 7.43 (1H, d, J = 8.1), 7.36–7.32 (3H, m), 4.64 (2H, s), 3.75 (4H, br s), 2.73 (4H, br s).
Biological Evaluation
Protein Expression and Purification
Proteins were cloned, expressed, and purified as previously described.7
Peptides
H4Ac4 peptide (BRD4 and CECR2 assays, H2N-YSGRGK(Ac)GGK(Ac)GLGK(Ac)-GGAK(Ac)RHRK-(Biotin)-CO2H), H3K56(Ac) peptide (CREBBP assay, H2N-ALREIRRYQK(Ac)-STELLIRKLK(Biotin)-CO2H), H2K9(Ac)K13(Ac)K15(Ac) peptide (BRD9 assay, H2N-YSGRGKQGGK(Ac)ARAK(Ac)AK(Ac)TRSSRA-biotin), H3K14(Ac) peptide (BAZ2B, PB1(5) and TIF1α assays, H2N-YQTARKSTGGK(Ac)APRKQLATKA-K(biotin)-CO2H) were synthesized by Tufts University Core Facility, Pepceuticals, or Alta Biosciences.
DSF Tm Shift Assay
Bromodomain DSF Tm shift assays were carried out as previously described.8a
AlphaScreen Peptide Displacement Assay
Bromodomain AlphaScreen assays were carried out as previously described.8a All experiments were carried out in duplicate on the same plate.
CREBBP Fluorescence Recovery After Photobleaching (FRAP) Assay
FRAP studies were performed using a protocol modified from previous studies.7,30,31 In brief, U2OS cells were transfected (Lipofectamine 2000, Life Technologies) with mammalian overexpression constructs encoding a GFP chimera with three tandem repeats of the CREBBP bromodomain (corresponding to amino acids 869–1341, with or without N1168F mutagenesis, of RefSeq CREBBP (NM_004380)) in pcDNA6.2/N-EmGFP-DEST (Life Technologies). SAHA was added 4 h post transfection and compounds as indicated 16 h post transfection The FRAP and imaging system consisted of a Zeiss LSM 710 scanhead (Zeiss GmbH, Jena, Germany) coupled to an inverted Zeiss Axio Observer.Z1 microscope equipped with a high-numerical-aperture (NA 1.3) 40× oil immersion objective (Zeiss GmbH, Jena, Germany) equipped with a heated chamber set at 37 °C. FRAP and GFP fluorescence imaging were carried out with an argon-ion laser (488 nm) and with a piezomultiplier tube (PMT) detector set to detect fluorescence between 500 and 550 nm. A 27.5 μm2 region of a GFP-positive nucleus was selected, and after 5 prescans, the region was bleached. A time-lapse series was then taken to record GFP recovery using 1% of the power used for bleaching with an interval time of ∼0.25 s. The image data sets and fluorescence recovery data were exported from ZEN 2009, the microscope control software, into Microsoft Excel. The average intensity at each imaging time point was measured for three regions of interest: the bleached region (It), the total cell nucleus (Tt), and a random region outside of the cell for background subtraction (BG). The relative fluorescence signal in the bleached region was calculated for each time point t, with the following equation:32
The baseline was normalized to zero and the prebleach to 1. Normalized data was imported into GraphPad Prism 6.0, and half times of recovery were calculated from individual single exponential curve fittings and presented as the mean. P Values were calculated using the unpaired t test.
X-ray Crystallography
Crystallization
Aliquots of the purified proteins were set up for crystallization using a mosquito crystallization robot (TTP Labtech, Royston UK). Coarse screens were typically set up onto Greiner 3-well plates using three different drop ratios of precipitant to protein per condition (100 + 50 nL, 75 + 75 nL, and 50 + 100 nL). Initial hits were optimized further by scaling up the drop sizes. All crystallizations were carried out using the sitting drop vapor diffusion method at 4 °C. Crystals of BRD4(1) with compound 51 were grown by mixing 200 nL of the protein (9.2 mg/mL and 5 mM final ligand concentration) with 100 nL of reservoir solution containing 0.1 M SPG pH 8.0 and 60% MPD. BRD9 crystals with compound 51 were grown by mixing 100 nL of protein (14.9 mg/mL and 5 mM final ligand concentration) with 200 nL of reservoir solution containing 0.2 M KSCN and 20% PEG3350. In both cases, diffraction quality crystals grew within a few days.
Data Collection and Structure Solution
BRD9 crystals were cryoprotected using the well solution supplemented with additional ethylene glycol and were flash-frozen in liquid nitrogen. BRD4 crystals were frozen without any additional cryoprotection. Data were collected in-house on a Rigaku FRE rotating anode system equipped with a RAXIS-IV detector at 1.52 Å. Indexing and integration was carried out using MOSFLM,33 and scaling was performed with SCALA.34 Initial phases were calculated by molecular replacement with PHASER35 using the known models of BRD4(1) (PDB ID 2OSS) and BRD9 (PDB ID 3HME). Initial models were built by ARP/wARP,36 followed by manual building in COOT.37 Refinement was carried out in REFMAC5.38 In all cases, thermal motions were analyzed using TLSMD39 and hydrogen atoms were included in late refinement cycles. Data collection and refinement statistics can be found in Table 3. The models and structure factors have been deposited with PDB accession codes: 4NQM (BRD4(1)/compound 51), 4NQN (BRD9/compound 51).
Table 3. Data Collection and Refinement Statistics for BRD4(1) and BRD9 Complexes.
| Data Collection | ||
|---|---|---|
| PDB ID | 4NQM | 4NQN |
| protein | BRD4(1) | BRD9 |
| ligand | compd 51 | compd 51 |
| space group | P212121 | P212121 |
| cell dimensions | ||
| a, b, c (Å) | 45.51, 46.57, 62.38 | 47.12, 48.42, 69.32 |
| α, β, γ (deg) | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 |
| resolution* (Å) | 1.58 (1.66–1.58) | 1.73 (1.82–1.73) |
| unique observations* | 18694 (2514) | 17147 (2438) |
| completeness* (%) | 98.7 (93.5) | 99.9 (99.8) |
| redundancy* | 3.9 (2.8) | 4.4 (4.1) |
| Rmerge* | 0.049 (0.439) | 0.060 (0.523) |
| I/σI* | 15.7 (2.0) | 12.6 (2.0) |
| Refinement | ||
|---|---|---|
| resolution (Å) | 1.58 | 1.73 |
| Rwork/Rfree (%) | 16.9/20.3 | 19.0/24.5 |
| no. of atoms (protein/other/water) | 1082/45/166 | 946/37/158 |
| B-factors (Å2) (protein/other/water) | 17.04/18.34/30.66 | 30.71/22.55/37.40 |
| rmsd bonds (Å) | 0.015 | 0.016 |
| rmsd angles (deg) | 1.666 | 1.587 |
| Ramachadran favored (%) | 99.17 | 98.11 |
| allowed (%) | 0.83 | 1.89 |
| disallowed (%) | 0.00 | 0.00 |
Values in parentheses correspond to the highest resolution shell.
Acknowledgments
We are grateful for support received by the SGC, a registered charity (no. 1097737) that receives funds from the Canadian Institutes for Health Research, the Canada Foundation for Innovation, Genome Canada, GlaxoSmithKline, Pfizer, Eli Lilly, Takeda, AbbVie, the Novartis Research Foundation, the Ontario Ministry of Research and Innovation, and the Wellcome Trust [(092809/Z/10/Z]). P.F. and S.P. are supported by a Wellcome Trust Career-Development Fellowship (095751/Z/11/Z).We also thank Yue Zhu and Xu Bai of Changchun Discovery Sciences for the synthesis of compounds 13, 24–34, and 49–57.
Glossary
Abbreviations Used
- HAT
histone acetyl transferase
- HDAC
histone deacetylase
- BET
bromodomain and extra terminal domain
- BRD
bromodomain-containing protein or bromodomain
- DSF
differential scanning fluorimetry
- BRD4(1)/(2)
first/second bromodomain of BRD4
- BRDT
bromodomain, testis-specific
- CREBBP
CREB (cyclic-AMP response element binding) binding protein
- BRD9
bromodomain-containing protein 9
- CECR2
cat eye syndrome chromosome region, candidate 2
- TAF1
TBP-associated factor RNA polymerase 1
- TAF1L
TAF1-like
- TIF1α
transcription intermediary factor 1-alpha
- ATAD2
ATPase family, AAA domain containing protein 2
- SMARCA4
SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 4
- P300 or EP300
E1A binding protein p300
- BAZ2A
bromodomain adjacent to zinc finger domain, 2A
- BAZ2B
bromodomain adjacent to zinc finger domain, 2B
- PB1
polybromo-1
- PCAF
P300/CBP-associated factor
- BRPF3
bromodomain and PHD finger containing protein 3
- PHIP
pleckstrin homology domain interacting protein
- FRAP
fluorescence recovery after photobleaching
Supporting Information Available
DSF Tm shifts with replicates and errors (XLSX). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Zentner G. E.; Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nature Struct. Mol. Biol. 2013, 203259–266. [DOI] [PubMed] [Google Scholar]
- Dawson M. A.; Kouzarides T. Cancer Epigenetics: From Mechanism to Therapy. Cell 2012, 150112–27. [DOI] [PubMed] [Google Scholar]
- Brennan P.; Filippakopoulos P.; Knapp S. The therapeutic potential of acetyl-lysine and methyl-lysine effector domains. Drug Discovery Today: Ther. Strategies 2012, 92–3e101–e110. [Google Scholar]
- Muller S.; Filippakopoulos P.; Knapp S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29/1–e29/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewings D. S.; Rooney T. P. C.; Jennings L. E.; Hay D. A.; Schofield C. J.; Brennan P. E.; Knapp S.; Conway S. J. Progress in the Development and Application of Small Molecule Inhibitors of Bromodomain–Acetyl-lysine Interactions. J. Med. Chem. 2012, 55229393–9413. [DOI] [PubMed] [Google Scholar]
- Müller S.; Lingard H.; Knapp S., Selective Inhibition of Acetyl-Lysine Effector Domains of the Bromodomain Family in Oncology. In Nuclear Signaling Pathways and Targeting Transcription in Cancer; Kumar R., Ed.; Springer: New York: 2014; pp 279–298. [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 46873271067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hewings D. S.; Wang M.; Philpott M.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Vuppusetty C.; Marsden B.; Knapp S.; Conway S. J.; Heightman T. D. 3,5-Dimethylisoxazoles Act As Acetyl-lysine-mimetic Bromodomain Ligands. J. Med. Chem. 2011, 54196761–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dawson M. A.; Prinjha R. K.; Dittmann A.; Giotopoulos G.; Bantscheff M.; Chan W.-I.; Robson S. C.; Chung C.-w.; Hopf C.; Savitski M. M.; Huthmacher C.; Gudgin E.; Lugo D.; Beinke S.; Chapman T. D.; Roberts E. J.; Soden P. E.; Auger K. R.; Mirguet O.; Doehner K.; Delwel R.; Burnett A. K.; Jeffrey P.; Drewes G.; Lee K.; Huntly B. J. P.; Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 4787370529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bamborough P.; Diallo H.; Goodacre J. D.; Gordon L.; Lewis A.; Seal J. T.; Wilson D. M.; Woodrow M. D.; Chung C.-w. Fragment-Based Discovery of Bromodomain Inhibitors Part 2: Optimization of Phenylisoxazole Sulfonamides. J. Med. Chem. 2011, 552587–596. [DOI] [PubMed] [Google Scholar]; d Hay D.; Fedorov O.; Filippakopoulos P.; Martin S.; Philpott M.; Picaud S.; Hewings D. S.; Uttakar S.; Heightman T. D.; Conway S. J.; Knapp S.; Brennan P. E. The design and synthesis of 5- and 6-isoxazolylbenzimidazoles as selective inhibitors of the BET bromodomains. MedChemComm 2013, 41140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Hewings D. S.; Fedorov O.; Filippakopoulos P.; Martin S.; Picaud S.; Tumber A.; Wells C.; Olcina M. M.; Freeman K.; Gill A.; Ritchie A. J.; Sheppard D. W.; Russell A. J.; Hammond E. M.; Knapp S.; Brennan P. E.; Conway S. J. Optimization of 3,5-Dimethylisoxazole Derivatives as Potent Bromodomain Ligands. J. Med. Chem. 2013, 5683217–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Albrecht B. K.; Audia J. E.; Cote A.; Gehling V. S.; Harmange J.-C.; Hewitt M. C.; Leblanc Y.; Naveschuk C. G.; Taylor A. M.; Vaswani R. G.. Preparation of compounds containing azepine-based ring systems as bromodomain-containing protein inhibitors and therapeutic uses thereof. WO2012075383A2, 2012.
- Picaud S.; Da Costa D.; Thanasopoulou A.; Filippakopoulos P.; Fish P. V.; Philpott M.; Fedorov O.; Brennan P.; Bunnage M. E.; Owen D. R.; Bradner J. E.; Taniere P.; O’Sullivan B.; Müller S.; Schwaller J.; Stankovic T.; Knapp S. PFI-1, a Highly Selective Protein Interaction Inhibitor, Targeting BET Bromodomains. Cancer Res. 2013, 73113336–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C.-w.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 46873271119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirguet O.; Lamotte Y.; Donche F.; Toum J.; Gellibert F.; Bouillot A.; Gosmini R.; Nguyen V.-L.; Delannée D.; Seal J.; Blandel F.; Boullay A.-B.; Boursier E.; Martin S.; Brusq J.-M.; Krysa G.; Riou A.; Tellier R.; Costaz A.; Huet P.; Dudit Y.; Trottet L.; Kirilovsky J.; Nicodeme E. From ApoA1 upregulation to BET family bromodomain inhibition: Discovery of I-BET151. Bioorg. Med. Chem. Lett. 2012, 2282963–2967. [DOI] [PubMed] [Google Scholar]
- Zuber J.; Shi J.; Wang E.; Rappaport A. R.; Herrmann H.; Sison E. A.; Magoon D.; Qi J.; Blatt K.; Wunderlich M.; Taylor M. J.; Johns C.; Chicas A.; Mulloy J. C.; Kogan S. C.; Brown P.; Valent P.; Bradner J. E.; Lowe S. W.; Vakoc C. R. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 4787370524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.-Y.; Lee A. Y.; Lai H.-T.; Zhang H.; Chiang C.-M. Phospho Switch Triggers Brd4 Chromatin Binding and Activator Recruitment for Gene-Specific Targeting. Mol. Cell 2013, 495843–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Banerjee C.; Archin N.; Michaels D.; Belkina A. C.; Denis G. V.; Bradner J.; Sebastiani P.; Margolis D. M.; Montano M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukocyte Biol. 2012, 9261147–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li Z.; Guo J.; Wu Y.; Zhou Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 411277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C.-W.; Nicodeme E.. Quinoline, azoloquinoline, triazolobenzodiazepine derivatives as bromodomain inhibitors for treating autoimmune and inflammatory diseases and their preparation. WO2011054843A1, 2011.
- Albrecht B. K.; Harmange J.-C.; Cote A.; Taylor A. M.. Bromodomain inhibitors for cancer therapy. WO2012174487A2, 2012.
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J.-P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Müller S.; Pawson T.; Gingras A.-C.; Arrowsmith; Cheryl H.; Knapp S. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 1491214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Supporting Information for complete DSF Tm shifts with replicates and errors.
- Compounds 5–12 and 14–17 were purchased from Interbioscreen (http://www.ibscreen.com).
- Chung C.-w.; Dean T. W.; Woolven J. M.; Bamborough P. Fragment-based discovery of bromodomain inhibitors part 1: inhibitor binding modes and implications for lead discovery. J. Med. Chem. 2011, 552576–586. [DOI] [PubMed] [Google Scholar]
- Totrov M.; Abagyan R. Flexible protein–ligand docking by global energy optimization in internal coordinates. Proteins 1997, Suppl 1, 215–220. [DOI] [PubMed] [Google Scholar]
- Philpott M.; Yang J.; Tumber T.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Keates T.; Felletar I.; Ciulli A.; Knapp S.; Heightman T. D. Bromodomain–peptide displacement assays for interactome mapping and inhibitor discovery. Mol. BioSyst. 2011, 7102899–2908. [DOI] [PubMed] [Google Scholar]
- Calculated using the free ACD/I-Lab calculator at http://ilab.cds.rsc.org.
- Compound 34 was synthesized from the commercialy available 1-hydrazinyl-4-phenylphthalazine in a manner analogous to compound 20.
- Mullighan C. G.; Zhang J.; Kasper L. H.; Lerach S.; Payne-Turner D.; Phillips L. A.; Heatley S. L.; Holmfeldt L.; Collins-Underwood J. R.; Ma J.; Buetow K. H.; Pui C.-H.; Baker S. D.; Brindle P. K.; Downing J. R. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011, 4717337235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidler L. R.; Brown N.; Knapp S.; Hoelder S. Druggability Analysis and Structural Classification of Bromodomain Acetyl-lysine Binding Sites. J. Med. Chem. 2012, 55177346–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards A. M.; Bountra C.; Kerr D. J.; Willson T. M. Open access chemical and clinical probes to support drug discovery. Nature Chem. Biol. 2009, 57436–440. [DOI] [PubMed] [Google Scholar]
- Sternfeld F.; Carling R. W.; Jelley R. A.; Ladduwahetty T.; Merchant K. J.; Moore K. W.; Reeve A. J.; Street L. J.; O’Connor D.; Sohal B.; Atack J. R.; Cook S.; Seabrook G.; Wafford K.; Tattersall F. D.; Collinson N.; Dawson G. R.; Castro J. L.; MacLeod A. M. Selective, Orally Active γ-Aminobutyric AcidA α5 Receptor Inverse Agonists as Cognition Enhancers. J. Med. Chem. 2004, 4792176–2179. [DOI] [PubMed] [Google Scholar]
- Garino C.; Tomita T.; Pietrancosta N.; Laras Y.; Rosas R.; Herbette G.; Maigret B.; Quéléver G.; Iwatsubo T.; Kraus J.-L. Naphthyl and Coumarinyl Biarylpiperazine Derivatives as Highly Potent Human β-Secretase Inhibitors. Design, Synthesis, and Enzymatic BACE-1 and Cell Assays. J. Med. Chem. 2006, 49144275–4285. [DOI] [PubMed] [Google Scholar]
- French C. A.; Ramirez C. L.; Kolmakova J.; Hickman T. T.; Cameron M. J.; Thyne M. E.; Kutok J. L.; Toretsky J. A.; Tadavarthy A. K.; Kees U. R.; Fletcher J. A.; Aster J. C. BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene 2007, 27152237–2242. [DOI] [PubMed] [Google Scholar]
- Kedersha N.; Tisdale S.; Hickman T.; Anderson P. Real-time and quantitative imaging of mammalian stress granules and processing bodies. Methods Enzymol. 2008, 448, 521–552. [DOI] [PubMed] [Google Scholar]
- Phair R. D.; Gorski S. A.; Misteli T. Measurement of dynamic protein binding to chromatin in vivo, using photobleaching microscopy. Methods Enzymol. 2004, 375, 393–414. [DOI] [PubMed] [Google Scholar]
- Leslie A. G. W.; Powell H.. MOSFLM, 7.01; MRC Laboratory of Molecular Biology: Cambridge, 2007.
- Evans P.SCALA—scale Together Multiple Observations of Reflections, 3.3.0; MRC Laboratory of Molecular Biology: Cambridge, 2007.
- McCoy A. J.; Grosse-Kunstleve R. W.; Storoni L. C.; Read R. J. Likelihood-enhanced fast translation functions. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2005, 61, 458–464. [DOI] [PubMed] [Google Scholar]
- Perrakis A.; Morris R.; Lamzin V. S. Automated protein model building combined with iterative structure refinement. Nature Struct. Biol. 1999, 65458–463. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Painter J.; Merritt E. A. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 439–450. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.