

Epigenomic analyses are used for many applications, including discovering gene regulatory mechanisms, characterizing non-genetic transmission modes of adaptive and disease traits, identifying and detecting disease biomarkers, and for drug discovery, as epigenome modifying therapeutics are under development for therapeutic benefit. Epigenomic testing might also become as common in the clinic as genomic testing.
The most widely used methods for epigenomic analysis include chromatin immunoprecipitation (ChIP) and bisulfite sequencing (BS-seq), which respectively report the genomic locations of histone modifications and 5-methylcytosine (5mC). Both are powerful, yet have limitations. First, one epigenomic feature is typically assayed at a time. When multiple features are of interest, data sets are superimposed, and inferences are made about coincidence of those features. However, the existence of multiple cell populations in a sample limits the validity of such inferences. Experiments using serial ChIP or ChIP followed by BS-seq can circumvent this problem.1 Second, both sequencing-based approaches are costly and time consuming. These can be minimized by immunodetection methods that simply quantify the abundance of an epigenomic modification; however, such methods cannot quantify coincidence of multiple modifications. Mass spectrometry (MS) provides quantitative and coincidence data, but only for histone modifications that are closely spaced on the same histone peptide, unless MS is combined with ChIP.2 DNA and histone modifications cannot be simultaneously detected. MS is also costly, time consuming, and requires much technical expertise. Third, ChIP and MS typically require an abundance of materials, although low cell input studies have been reported.3 Methods that are quantitative, rapid, simple, and enable analyses of combinations of epigenomic features in small samples will leverage all efforts requiring epigenomic analyses.
Single-molecule analytical approaches we are developing provide tools for circumventing limitations of existing methods. SCAN (single chromatin molecule analysis at the nanoscale) is analogous to flow cytometry and fluorescence activated cell sorting, but rather than detecting, quantifying, and sorting single cells based on their binding to antibodies recognizing cell surface markers, SCAN performs these using single molecules of DNA or chromatin, and antibodies or probes recognizing epigenomic features.4-6 Use of nanoscale structures, fluorescent antibodies, and sensitive optics enables single-molecule analyses with analytes in low nanomolar range with throughputs of 6000 molecules per minute (Fig. 1).
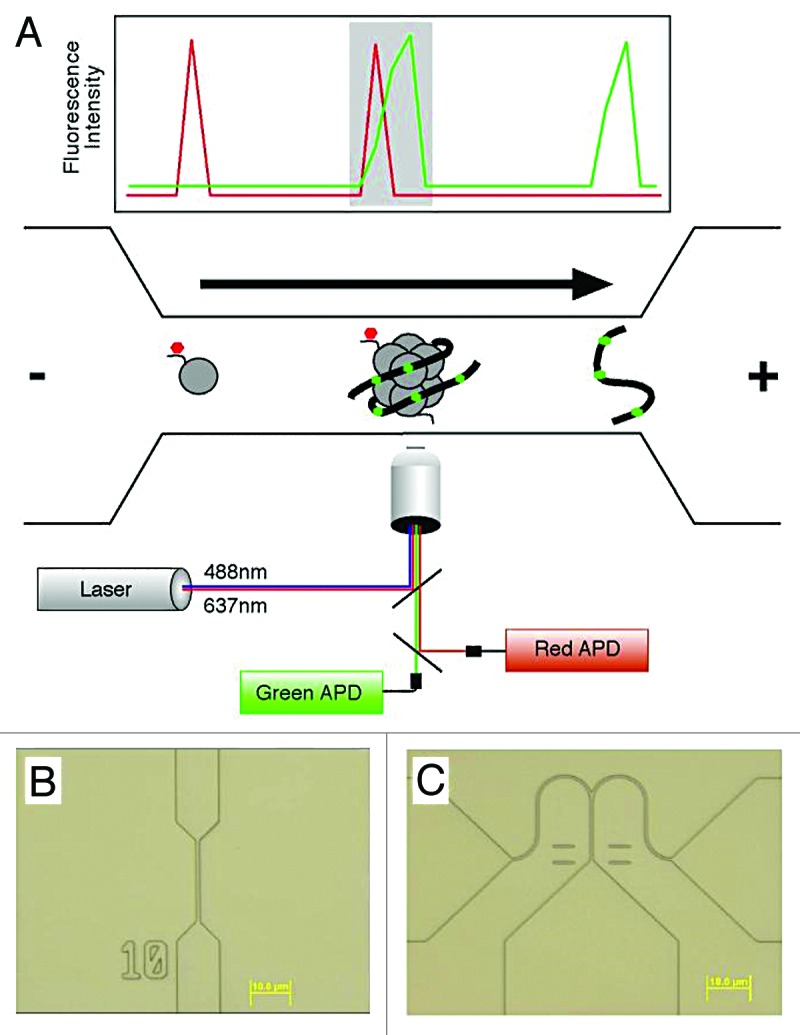
Figure 1. (A) A schematic diagram of a nanofluidic channel. The channel (bottom), fabricated in silica, consists of a microscale loading area (left), a 500 nm-wide constriction (middle) and a microscale outflow tract (right). Channel depth is 250 nm. An applied voltage drives DNA and chromatin through the channel toward the cathode as shown by the arrow. DNA is labeled with an intercalator (green), and chromatin modifications are detected by antibodies or other probes recognizing the modifications (red). Overlapping lasers illuminate the channel and define a 0.16 fL inspection volume. Light emitted by molecules occupying the volume is collected using a confocal microscope, and single photon detection is achieved using avalanche photodiodes (APDs). A graph representative of the collected data are shown above the channel. Single molecules are detected based on achieving a threshold of emitted photons relative to background. Intact chromatin fragments carrying epigenomic modifications of interest (shaded area) are identified by coincident detection of both a red and green single molecule. Recent changes in the optical design allow the use of 3 spectrally distinct lasers, enabling higher-order multiplexing and the detection of combinations of multiple epigenetic features. (B) A differential interference contrast optical micrograph of a typical nanofluidic channel used for enumerating epigenomic features. (C) Micrograph of a bifurcated nanofluidic channel designed to isolate individual molecules for subsequent analysis. The voltage bias is toggled between the 2 outflow tracts, controlled in real time based on the fluorescent properties of molecules as they pass through the inspection volume to achieve sorting.
In our efforts to develop SCAN, we first demonstrated that native chromatin could remain intact during voltage-driven movement through nanofluidic channels, and that 5mC could be specifically detected on DNA using MBD1 protein as the 5mC probe.4 These studies performed single-molecule counting only. Single-molecule sorting would enable recovery of materials for downstream sequencing. We therefore modified our counting platform to sort molecules based on their epigenomic features. In a proof-of-principle experiment, we demonstrated efficient and specific isolation of methylated DNAs from a mixture containing methylated and unmethylated molecules, based on their binding MBD1 protein.5 Most recently, we directly detected H3K9me3 and 5mC coincidence on the same chromatin molecules. Their coincidence was anticipated based on ChIP and BS studies, but never directly demonstrated. To extend this observation, and to demonstrate the utility of our methods, we explored the relationships between these 2 gene-silencing marks and a third silencing mark, H3K27me3. H3K27me3 is robustly antagonized by 5mC in mouse ES cells and primary fibroblasts, consistent with our observations using ChIP-seq.7 Intriguingly, upon immortalization or transformation of fibroblasts, the antagonism is lost; instead, H3K27me3 placement becomes dependent on 5mC. This reversal of H3K27me3 fate is also seen in human myeloleukemia cells. The results identify a point of epigenomic failure early in cancer development causing aberrant coordination between epigenomic marks.
There are many ways to extend and apply these methods. Our goals include sorting and sequencing of chromatin-bearing combinations of epigenomic features, increasing multiplexing to facilitate analyses of higher order combinations of epigenomic marks, increasing throughput for full genome coverage, and performing analyses with extremely low inputs of material. Use of quantum dots instead of organic dyes for reagent labeling will enable higher multiplexing because of the narrow emission spectra of quantum dots, and also permit higher single-molecule throughput because of their high emission intensity. Our analyses so far have used a single fluidic channel, whereas multiple channels will scale throughput proportionally. When analytes are in low abundance from rare cells or pathology sections, the exquisite sensitivity afforded by single-molecule approaches can enable analyses that are currently impossible. This will require non-standard tools for extracting chromatin or DNA from low cell inputs. One device we developed for single-cell extractions was designed to interface with the nanofluidic devices described above, providing a workflow for single-cell epigenomic analysis.8
Future epigenomic analyses will require new methods that provide higher information content than is now available and enable interrogation of few cells using a simple, fast, and inexpensive workflow. Single-molecule methods provide one pathway toward these goals.
Murphy PJ, et al. Proc Natl Acad Sci U S A. 2013;110:7772–7. doi: 10.1073/pnas.1218495110.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26694
References
- 1.Brinkman AB, et al. Genome Res. 2012;22:1128–38. doi: 10.1101/gr.133728.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Voigt P, et al. Cell. 2012;151:181–93. doi: 10.1016/j.cell.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adli M, et al. Nat Methods. 2010;7:615–8. doi: 10.1038/nmeth.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cipriany BR, et al. Anal Chem. 2010;82:2480–7. doi: 10.1021/ac9028642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cipriany BR, et al. Proc Natl Acad Sci U S A. 2012;109:8477–82. doi: 10.1073/pnas.1117549109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy PJ, et al. Proc Natl Acad Sci U S A. 2013;110:7772–7. doi: 10.1073/pnas.1218495110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hagarman JA, et al. PLoS One. 2013;8:e53880. doi: 10.1371/journal.pone.0053880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benítez JJ, et al. Lab Chip. 2012;12:4848–54. doi: 10.1039/c2lc40955k. [DOI] [PMC free article] [PubMed] [Google Scholar]