

Members of the Bcl-2 protein family that contain only the Bcl-2 homology (BH) domain 3 are inducers of cell death when overexpressed.1 One of these BH3-only proteins, Bmf, was discovered because of its binding to the anti-apoptotic member Bcl-2, Bcl-XL, Bcl-w, and Mcl-1.2 It is expressed in the pancreas, liver, kidney, and hematopoietic tissues and in many cell lines of B and T lymphoid, myeloid, and fibroblastoid origin.2 Bmf has at least 2 isoforms translated from a single mRNA (BmfCUG and BmfS), due to an alternative start site from a common open reading frame.1 Bmf interacts with the actin-based myosin V motor complex via dynein light chain 2 (DLC2), which sequesters Bmf to the cytoskeleton under normal conditions. Certain cellular stressors, such as UV irradiation or loss of extracellular matrix adhesion, release Bmf from the cytoskeleton to localize to the mitochondria and elicit pro-apoptotic activity.2
Another BH3-only protein, Bim, which is found as 3 isoforms, BimEL, BimL, and BimS, has characteristics similar to Bmf. UV irradiation releases Bmf and Bim from the cytoskeleton, both proteins are required for anoikis in some cells,1 and both proteins are transcriptionally induced upon HDAC inhibitor treatment in diverse cancer cell lines.3 In fact, based on these similarities, the BMF and BIM genes are believed to have a common ancestral BH3-only gene, although their homology is restricted to the BH3 domain and the DLC-binding motif.1 Bim is inactivated in healthy cells by its interaction with the cytoskeleton by binding to the microtubule-based dynein motor complex through DLC1 (Fig. 1B). Upon activation by apoptotic stimuli, Bim is phosphorylated by JNK in its DLC binding motif, allowing Bim to dissociate from DLC1 and activate apoptosis.4
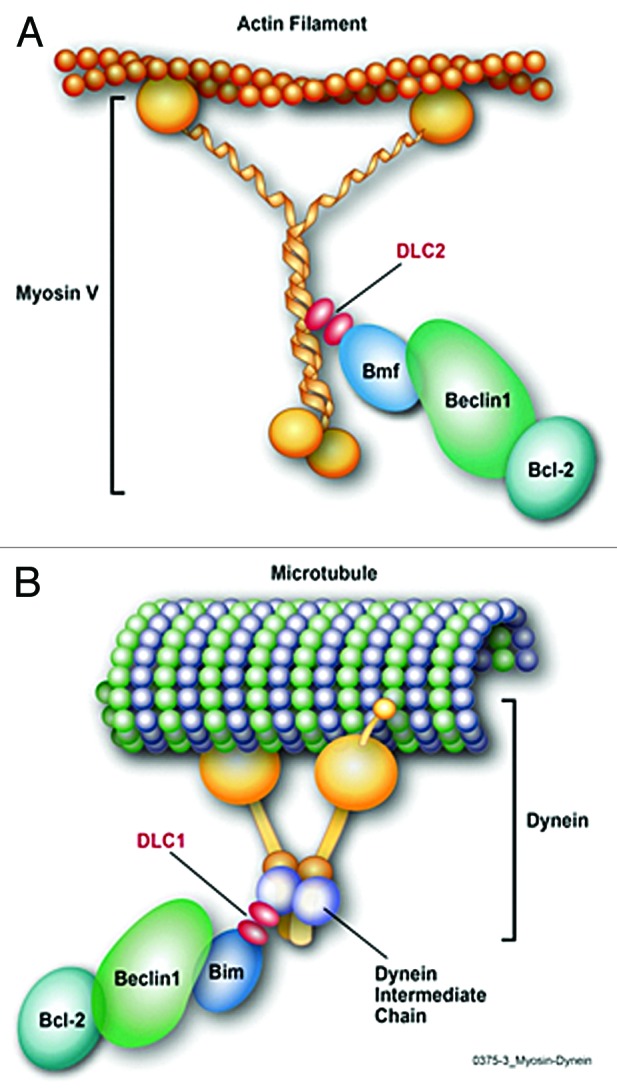
Figure 1. (A) Proposed model of Bmf tethering the Beclin1/Bcl-2 complex to the Myosin V by binding DLC2. In resting cells, Bmf binds to the Beclin1/Bcl-2 complex and maybe to DLC2, thereby linking the autophagy complex to myosin V, to inhibit autophagy initiation. (B) Bim tethers Beclin1 to the Dynein motor complex by binding DLC1. In resting cells, Bim binds to DLC1 and to Beclin1, and probably to the Beclin1/Bcl2 complex, and thereby links them to the dynein motor complex to inhibit autophagy initiation.
We reported that Bmf is a regulator of autophagy,5 a conserved process mediating degradation of bulk cytoplasm, long-lived proteins, and entire organelles to bridge stressful conditions, including nutrient deprivation and infection. Central to this pathway is the formation of the autophagosome, which contains a portion of the cytoplasm that fuses with the lysosome, where the sequestered cytoplasmic components are degraded.6 A major regulator of autophagy, Beclin1, forms a complex with Vps15, Vps34, and likely Ambra1,6 to activate a PI3P kinase to initiate the autophagosome formation. Initiation of autophagy is inhibited as long as the ER-localized Bcl-2 (Bcl-XL or Mcl-1) binds Beclin1, and some BH3-only proteins cause the release of Beclin1.6
We found that IFN-γ reduces Bmf levels in human or mouse airway epithelial cells and increases Beclin1 expression, while Bmf overexpression decreases Beclin1 levels.5 Primary airway epithelial cells or embryonic fibroblasts from bmf−/− compared with bmf+/+ mice present with more autophagosomes. Bmf co-immunoprecipitates with Beclin1 and Bcl-2, and Beclin1/Bcl-2 interaction is disrupted in the absence of Bmf, suggesting that Bmf stabilizes the inhibitory complex to avoid initiation of autophagy.5
Two different groups reported that the dynein motor complex-interacting proteins DLC1 and DLC2 can regulate autophagy by tethering Beclin1 to the cytoskeleton. These studies may provide clues to understand how Bmf negatively regulates autophagy. The first publication demonstrated that Bim sequesters Beclin1 to the dynein motor complex and thereby blocks initiation of autophagy.7 Upon starvation, Bim is phosphorylated by JNK, and Beclin1 is released to initiate autophagy.7 The second study showed that DLC1 and DLC2 bind the Ambra1/Beclin1/Vps34 autophagy initiation complex to the cytoskeleton via Ambra1.8 Induction of autophagy activates Ulk1 to phosphorylate Ambra1 and release the Ambra1/Beclin1/Vps34 complex from the dynein chain, allowing it to translocate to the ER and prime autophagosome formation.8 Curiously, in the second study, inhibition of JNK did not interfere with the dissociation process upon starvation,8 suggesting that Bim phosphorylation does not play a role in the association between DLC1/2 and Ambra1. Whether Bim/Beclin1 interaction7 is part of a pathway distinct from the Ulk1 activated Ambra1,8 or whether the complexes may differ depending on cell type, needs to be explored further.
Considering that Bmf and Bim have several characteristics in common, including the inhibition of autophagy and their binding to DLC proteins, it is reasonable to assume that these 2 BH3-only proteins tether Beclin1 to the dynein/myosin V motor complexes to inhibit autophagy. Based on our finding that Bmf interacts with Beclin1 and Bcl-2, and the studies showing Bim being involved in attaching Beclin1 to the cytoskeleton, we propose that Bmf, through DLC2, tethers the autophagy-initiation complex to the actin, as Bim tethers the complex to microtubule through DLC1 (Fig. 1A and B). Both Bmf and Bim are known substrates for kinases, supporting the idea that phosphorylation of these proteins could be involved in regulating the initiation of autophagy.
Collectively, the recent discoveries demonstrating that the pro-apoptotic BH3-only proteins either inhibit or induce autophagy by either stabilizing or inactivating the Beclin1-Bcl-2/Bcl-XL/Mcl-1 complex reveals the intricate connection between the apoptotic and autophagic pathways. Autophagy can inhibit or enhance apoptosis or induce a cell death independent of apoptosis, depending on the cell type and the type and duration of the stimulation.6 Accordingly, future studies will elucidate whether the net effect on autophagy, or cell death could be tissue- or cell type-specific and may result from conditions that favor the change in expression or posttranslational modifications of BH3-only proteins.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26696
References
- 1.Labi V, et al. J Exp Med. 2008;205:641–55. doi: 10.1084/jem.20071658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Puthalakath H, et al. Science. 2001;293:1829–32. doi: 10.1126/science.1062257. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, et al. Cell Death Differ. 2006;13:129–40. doi: 10.1038/sj.cdd.4401686. [DOI] [PubMed] [Google Scholar]
- 4.Lei K, et al. Proc Natl Acad Sci U S A. 2003;100:2432–7. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Contreras AU, et al. J Cell Biol. 2013;201:427–37. doi: 10.1083/jcb.201205064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kroemer G, et al. Mol Cell. 2010;40:280–93. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo S, et al. Mol Cell. 2012;47:359–70. doi: 10.1016/j.molcel.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Bartolomeo S, et al. J Cell Biol. 2010;191:155–68. doi: 10.1083/jcb.201002100. [DOI] [PMC free article] [PubMed] [Google Scholar]