

Abstract
Recent studies have demonstrated that DNA immunization is effective in eliciting antigen-specific antibody responses against a wide range of infectious disease targets. The polyclonal antibodies elicited by DNA vaccination exhibit high sensitivity to conformational epitopes and high avidity. However, there have been limited reports in literature on the production of monoclonal antibodies (mAb) by DNA immunization. Here, by using Clostridium difficile (C. diff) toxin A as a model antigen, we demonstrated that DNA immunization was effective in producing a panel of mAb that are protective against toxin A challenge and can also be used as sensitive reagents to detect toxin A from various testing samples. The immunoglobulin (Ig) gene usage for such mAb was also investigated. Further studies should be conducted to fully establish DNA immunization as a unique platform to produce mAb in various hosts.
Keywords: DNA immunization, monoclonal antibody, C. diff toxin, toxin A, immunoglobulin genes
Introduction
DNA immunization technology was discovered in the early 1990s. Significant amounts of work have been done in the past 20 y to demonstrate that DNA immunization is effective in eliciting antibody and T cell immune responses against various pathogens in almost every experimental animal model studied.1,2 More recently, promising immunogenicity results have been obtained in human studies when DNA vaccines were used to prime host immune responses as part of a heterologous prime-boost strategy3-5 or as part of an electroporation delivery method to deliver DNA vaccines.6,7 One hallmark of recent progress in DNA immunization is the confirmation that DNA immunization is effective in eliciting high quality polyclonal antibody responses in immunized animal or human sera, which show a high degree of conformation specificity and high avidity.3,8,9 Frequently, such antibodies are also highly functional in their expected biological activities, such as blocking the infection of highly virulent HIV-1, SARS-CoV, or influenza viruses to targeted cells.8,10,11 Parallel to such progress, DNA immunization has also been proposed as a useful technology to produce monoclonal antibodies (mAb)12-14 to decrease the need for protein and peptide antigens for immunization. This is significant progress to circumvent the need of production and purification of difficult and complex proteins while ensuring effective induction of antigen-specific antibody responses against native conformation. However, experience in using DNA immunization to produce mAb is limited and information on the detailed characterization of the quality of mAb elicited by DNA immunization is lacking. Furthermore, reports on the immunogenetic features of mAb elicited by DNA immunization are lacking in the current literature.
In the current study, we produced a group of mouse mAb against toxin A of Clostridium difficile (C. diff) by DNA immunization.15 C. diff is a top etiology for nosocomical infections among hospitalized patients in developed countries and toxin A is its key virulence factor.16 Toxin A-specific mAb elicited by DNA immunization demonstrated high biological functions in our study. Furthermore, the immunoglobulin genes from these mAb were also cloned and analyzed. Our data confirmed the utility of using DNA immunization to produce high quality mAb.
Results
In our recently published report on the immunogenicity of DNA vaccines expressing either toxin A or toxin B of C. diff,15 we tested two designs of inserts that encode the same 333aa segment of C-terminal toxin A (between 2378 to 2710) (Fig. 1A). Our data showed that the addition of a tPA leader (tPA-TcdA-C) did not further improve the high level antibody responses elicited by a similar insert without a putative signal peptide sequence (TcdA-C).15 In the current study, we used the DNA vaccine expressing TcdA-C insert to immunize BALB/c mice by electroporation. After four immunizations administered at Weeks 0, 2, 4, and 8, animals rested for 4 weeks, while the serum samples were collected and anti-toxin A antibody response levels were tested (Fig. 1B). Animals were then boosted twice with partially purified TcdA-C protein at Weeks 12 and 15, and after resting for 3 weeks, another boost was given 4 d before mouse splenocytes were collected.
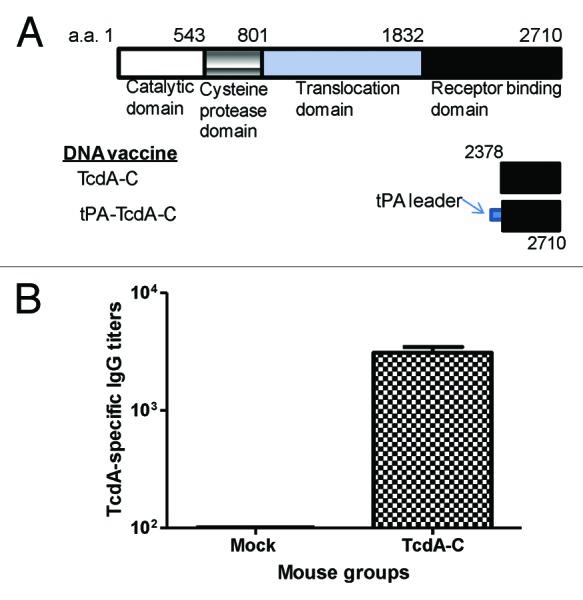
Figure 1. (A) Schematic designs of C. difficile toxin A (TcdA) DNA vaccines: TcdA-C (C-terminus of TcdA without leader sequence) and tPA-TcdA-C (TcdA-C with a tPA leader sequence). The amino acid positions for corresponding protein segments are indicated. (B) TcdA-C-specific antibody responses in mouse sera collected at one week after the 4th DNA immunization with either TcdA-C DNA vaccine or the empty vector (Mock) against TcdA-C protein expressed in supernatant of tPA-TcdA-C transfected 293T cells.
The conventional procedure for mouse hybridoma fusion was followed and those specific for toxin A were screened by ELISA. Commercially available C. diff toxin A was used to screen for positive hybridomas. After 5 rounds of screening, a total of 40 monoclonal positive hybridomas were identified. The top six monoclonal hybridomas for binding titers were shown in Figure 2. Supernatants of hybridomas at 1:2 dilution were used in ELISA against toxin A (Fig. 2A). Western blot analysis confirmed binding specificity and indicated that those mAb recognize linear epitopes (Fig. 2B). In this analysis, C-terminal toxin A segment was produced in transiently transfected 293T cells by tPA-TcdA-C DNA vaccine plasmid and was recognized by these six toxin A specific hybridomas.
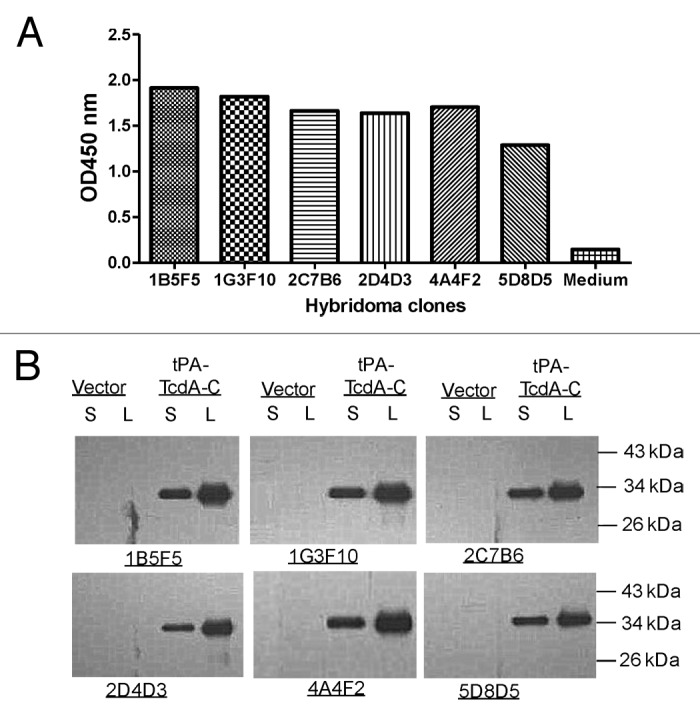
Figure 2. Immunological testing of TcdA-specific hybridoma clones generated from TcdA-C DNA vaccine immunized mice. (A) ELISA of culture supernatants (1:100 dilution) from selected hybridoma clones against TcdA-C. (B) Western blot analysis of mAb purified from hybridoma culture supernatants against TcdA-C protein expressed in transiently transfected 293T cell supernatant (S) and cell lysate (L). Supernatant (S) and cell lysate (L) of 293T cells transfected by the empty vector were included as negative control. The TcdA-C specific mAb used for Western blot analysis was at 1 µg/ml.
A sandwich ELISA was conducted to select the most sensitive toxin A-detecting mAb pairs (Fig. 3). Purified mAb from hybridoma cell cultures were used in this study. Each time, one mAb was used as the capture antibody in a regular ELISA plate while all six mAb, labeled with HRP, were tested individually as the detecting antibodies against captured toxin A. Using the OD value where the same mAb was used for both detection and capture as the baseline, 1G3 and 5D8 were identified as the most effective capture mAb to provide high OD values when the other four mAb were used as the detecting antibodies (Fig. 3A). The other four mAb showed lower OD values when they were used as capture antibodies.
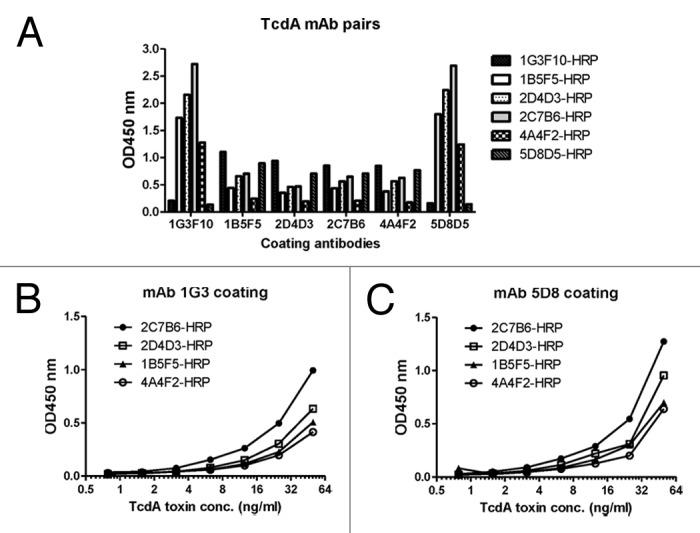
Figure 3. Identification of TcdA-specific mAb pairs to have the optimal detection of TcdA toxin by sandwich ELISA. (A) Screening of mAb pairs of six mAb, using one as the coating antibody (indicated under the columns) and the other as detecting antibody conjugated by HRP (indicated at the right side of the graph). Commercial TcdA toxin (0.05 µg/ml) was used as designated toxin in the ELISA assays. (B) Detection of toxin A with 4 mAb by sandwich ELISA using mAb 1G3 as the coating antibody. (C) Detection of toxin A with 4 mAb by sandwich ELISA using mAb 5G8 as the coating antibody.
More detailed analysis was conducted with mAb 1G3 and 5D8 as the capture mAb while the other four mAb, labeled with HRP, were used as the detecting mAb to test serially diluted toxin A. In both cases, 2C7 outperformed the other four detecting mAb while the 5D8–2C7 pair gave a slightly higher sensitivity over the 1G3–2C7 pair (Fig. 3B and C). Therefore, 5D8–2C7 and 1G3–2C7 were selected as two-paired mAb to detect C. diff toxin A from laboratory or clinical samples at a concentration as low as 2ng/ml.
One pilot study was conducted to detect toxin A from C. diff cultured samples. Coded samples were tested by either the sandwich ELISA using 5D8–2C7 mAb pair or a commercial C. diff toxin A detection kit X/pect by Remel. By using OD value > 0.1 as the cut off, each time a testing sample had a higher value than 0.1 in our mAb pair ELISA, the testing result was also positive by the commercial kit (Fig. 4A).
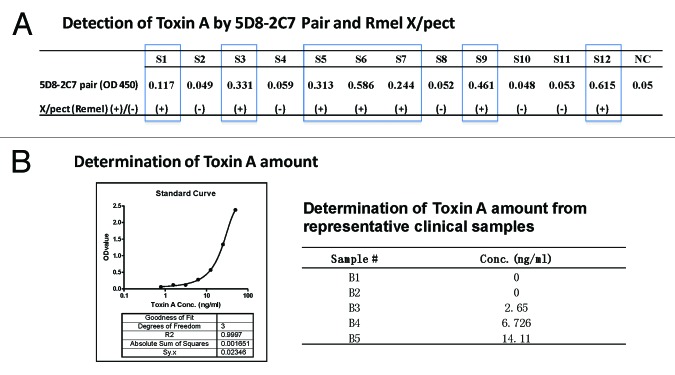
Figure 4. Detection of TcdA in clinical C. diff samples using TcdA-specific mAb 5D8/2C7-HRP pair by sandwich ELISA. (A) Detection of TcdA toxin in blinded clinical samples using mAb 5D8/2C7-HRP sandwich ELISA and commercial TcdA detection kit “Remel X/pect.” OD values, >0.1 in 5D8/2C7-HRP sandwich ELISA were considered as positive. The results “(+)” and “(-)“ indicate positive or negative for TcdA toxin in Remel X/pect assays, respectively. S1 – S12 were clinical samples, and NC was negative control using PBS. (B) Quantitative detection of TcdA toxin in clinical samples. The graph on the left is an example of the standard curve of commercial TcdA detected by mAb 5D8/2C7-HRP pair in a sandwich ELISA. The table on the right is a list of TcdA toxin amount in clinical samples B1-B5 as determined by mAb 5D8/2C7-HRP pair in sandwich ELISA.
In a separate study, known positive stool samples from C. diff-infected patients were tested. By using the 5D8–2C7 mAb pair-based ELISA, a quantitative determination of toxin A in a testing sample was feasible. The amount of toxin A in each sample was determined by using the resulting OD value against the standard curve, which was generated with the commercial toxin A using the same 5D8–2C7 mAb pair ELISA (Fig. 4B). So far no study about the correlation between clinical symptoms and toxin A amount in patients has been reported. The availability of a toxin A determination kit to quantify the amount of toxin in a testing sample may allow for such analysis.
The functional relevance of toxin A-specific mAb produced by DNA immunization was demonstrated in an in vitro protection study. Toxin A-treated CHO cells would demonstrate the effect of cytotoxicity in contrast to normal CHO cells without such treatment (Fig. 5A and B). Preincubating toxin A with each of the six toxin A-specific mAb was able to protect CHO (Fig. 5C–H). Similar protection effects with these mAb were observed when they were able to block the effects of toxin A on another target cell line, HT-29 (data not shown).

Figure 5. The neutralizing activities of TcdA-specific mAb measured by their protection against TcdA toxin in CHO cells. Light microscopic images of CHO cells with various treatments are shown: (A) “Cell control” lacks treatment by both TcdA toxin and mAb. (B) “Toxin control” is TcdA toxin without any antibody. (C–H) Cells were cultured with TcdA toxin with one TcdA mAb, as indicated under each panel. The mAb concentration was 15 µg/ml.
The in vivo protective effects of these novel mAb were further demonstrated in mice challenged with toxin A (Fig. 6). Individually, mAb 4A4 was the most protective (protecting 50% mice) while mAb 2C7 and 5D8 were not much different from isotype control or PBS (Fig. 6A). However, when 5D8 and 2C7 were used in combination with 4A4, a higher survival (90%) was achieved (Fig. 6B).
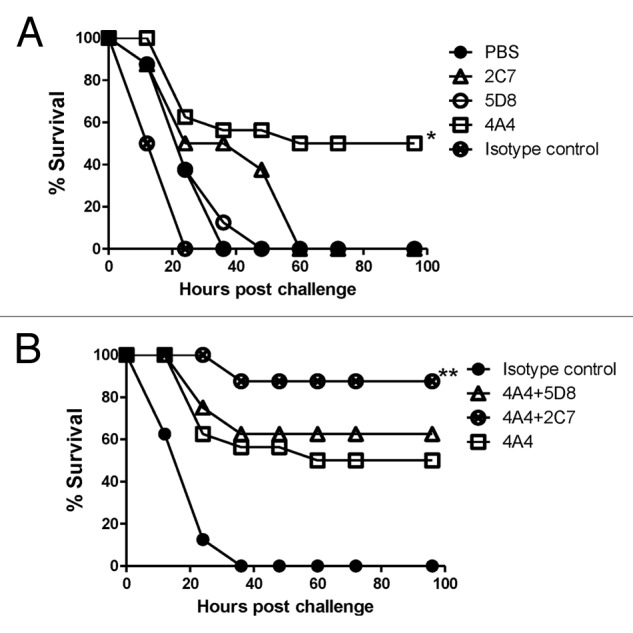
Figure 6. Passive mAb protection in mice against TcdA toxin challenge. (A) Passive protection experiments were conducted with individual mAb 2C7, 5D8 and 4A4 and negative controls PBS and mouse Ig isotype control matching mAb . * indicates a statistically significant difference (p < 0.05) compared with the negative controls (PBS or isotype Ig control groups). (B) Passive protection experiments were conducted with individual mAb 4A4 or a mixture of two mAb 4A+5D8 or 4A4+2C7, in addition to the Ig isotype control. ** indicates a statistically significant difference (p < 0.05) compared with the mAb 4A4 alone group. There were 24 Balb/C mice in 4A4 group and 8 mice/group in other groups.
The immunoglobulin (Ig) genes for the heavy and light chains of mAb 5D8 and 2C7 were PCR amplified and individually subcloned into the expression vector, pJW4303 (Fig. 7). Co-transfection of matched heavy chain and light chain expressing plasmids in 293F cells was able to produce a large quantity of recombinant mAb 5D8 and 2C7. The relative binding abilities against toxin A were compared between hybridoma-produced mAb and recombinant mAb produced by 293F cells. Both recombinant 5D8 and 2C7 mAb matched the high binding titers of the same mAb produced by hybridomas (Fig. 8).
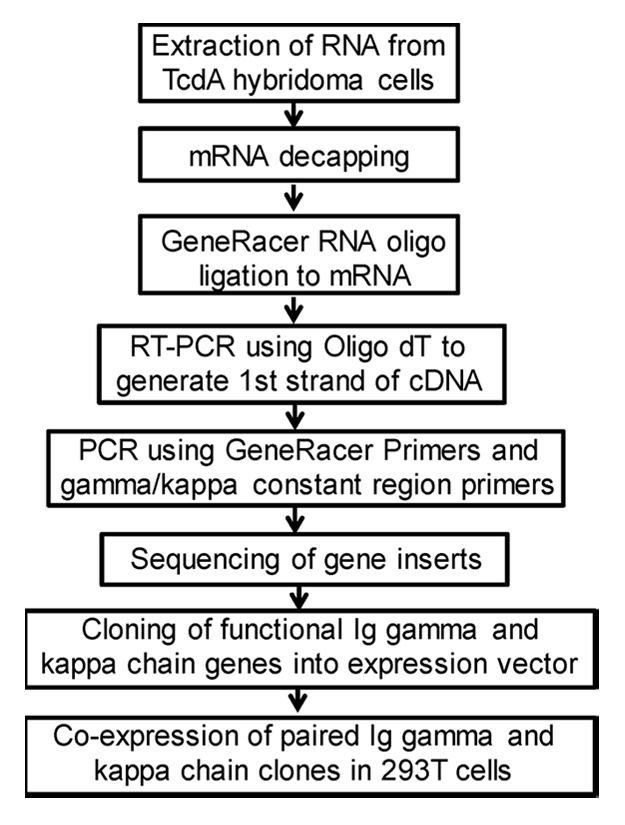
Figure 7. Flowchart of mouse mAb Ig gene cloning steps.
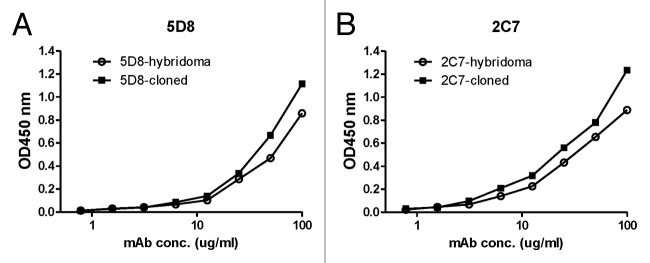
Figure 8. Comparison of TcdA toxin binding activities for purified mAb 5D8 (A) or 2C7 (B) produced either from hybridoma culture or from supernatant of 293T cells co-transfected with paired Ig gamma and kappa chain clones. ELISA curves labeled with solid squares indicate the mAb produced by hybridoma and open circles indicate mAb produced by cotransfection of paired Ig heavy and kappa chain clones. TcdA toxin was used as coating antigen (1 µg/ml).
Gene sequences of cloned mAb 5D8 and 2C7 were further analyzed by IMGT and results are shown in Table 1. Both mAb, 5D8 and 2C7, had similar levels of heavy (H) chain variable gene (VH) mutations while 5D8 had higher mutations than 2C7 in the heavy chain J gene (JH) region. On the other hand, 5D8 had a longer CDR3 than 2C7. 5D8 belongs to the 1–4*01 VH family and 2C7 is part of the VH family 9–2-1*01. Both 5D8 and 2C7 used kappa chain, which is consistent with the general dominance of kappa chain for mouse light (L) chain of immunoglobulin genes. They had the same level of kappa (K) chain variable gene mutation and the same length of complementarity-determining region 3 (CDR3) even though the kappa chain of 5D8 belongs to the 6–17*01 family and 2C7 belongs to the 3–2*01 family.
Table 1. TcdA mAb 5D8 and 2C7 lg gene germline and CDR3 analysis.
| Heavy Chain | ||||||
|---|---|---|---|---|---|---|
| mAb | VH family | VH mutation | D region | JH | JH mutation | CDR3 aa seq |
| 5D8 | 1–4*01 | 6% | 1–1*01 | 3*01 | 7% | CARIGDYYVSFAFW |
| 2C7 | 9–2-1*01 | 5% | 2–3*01 | 3*01 or 3*02 | 23% | CAAIHDENW |
| Kappa chain | ||||||
| mAb | VK family | VK mutation | JK | JK mutation | CDR3 aa seq | |
| 5D8 | 6–17*01 | 2% | 5*01 | 3% | CQQHFGTPLTF | |
| 2C7 | 3–2*01 | 2% | 1*01 | 0 | CQQTKEVPRTF | |
Discussion
While the use of DNA immunization to generate mAb was proposed almost at the same time when DNA immunization was discovered,12 there have been only limited reports on the use of this new immunization approach to elicit mAb against infectious disease targets in the last two decades.13,14,17-21 Three reports described the production of mAb against influenza HA antigens.17,18,21 These studies used DNA prime-inactivated virus or protein boost strategy as the immunization procedure. One study elicited eight mAb against H5 subtype hemaglutinin (HA) antigens and three of them exhibited hemagglutination inhibition (HI) activity. The other study generated two mAb with one targeting the stalk region of hemagglutinins of H1 subtype with broad neutralizing activities but not HI activity. The last study produced mAb against both HA1 and HA2 domains of H5 antigen collected from 1997 Hong Kong H5N1 outbreak and they do not cross react with HA antigens collected from 2003 H5N1 isolates. All three studies demonstrated the utility of DNA immunization to elicit antibodies against conformation sensitive epitopes. Similarly, DNA immunization followed by a live virus boost was able to produce conformation sensitive mAb against dengue NS1 protein and among 5 mAb produced, three were IgM.20 In a study to generate mAb against Rift Valley fever virus nucleocapsid protein, it was also shown that the DNA immunization strategy may have contributed to the proper exposure of surrogate determinant(s) mimicking the natural antigens.14 In this study, both DNA alone and DNA prime-protein boost methods were able to elicit mAb.
Two studies with mAb against non-viral pathogens were reported. Intramuscular injection of DNA vaccines produced mAb against H. Pylori vacuolating cytotoxin.19 The gene gun alone approach produced mAb against the 8 kDa Subunit of Echinococcus granulosus Antigen B (EgAgB8/2).13 But in both studies, only low affinity mAb were produced.
In the current study, we reported the use of DNA immunization to elicit mAb against a bacterial toxin. Electroporation (EP) was used as the DNA delivery approach and protein boosts were included in the immunization protocol. We do not know whether the protein boost is absolutely needed for the production of high quality mAb but using gene gun or intramuscular (IM) injection alone to deliver DNA vaccines were not successful in producing high quality mAb from our previous studies (unpublished observations). We have compared relative immunogenicity among IM needle injection, gene gun, and EP methods in generating polyclonal serum antibody responses and confirmed the Th1 bias for IM and EP and Th2 bias for gene gun approach.22 However, the impact of the DNA immunization method in the production of high quality mAb remains to be investigated. The current report attempted to include comprehensive analyses on mAb generated by the EP method, in combination with protein boost, and our results provide baseline information for more detailed and comprehensive studies.
Clostridium difficile infection represents a serious challenge to medical practice in many developed countries however, information from developing countries on this infection is generally lacking. One report on C. diff infection in China indicated that such infection was present but no systematic study was conducted to provide more complete epidemiology data.23 One reason is the lack of affordable diagnostic kits to be used for clinical or epidemiology studies.
In the current report, we describe the production of a panel of mAb against the toxin A of C. diff elicited by DNA immunization. Due to the large size of C. diff toxins and the toxicity of natural materials, it is desirable to use part of the toxin protein as the immunogen. DNA immunization is an ideal approach for such studies as various subdomains of the toxins can be designed and tested while there is no need for the production and purification of actual recombinant protein immunogens in vitro. Previously, we conducted a comprehensive analysis on candidate C. diff toxin antigens.15 Based on the results of this analysis, we selected the TcdA-C DNA vaccine to produce the related mAb, as described in the current report.
Toxin A-specific mAb identified in our study showed high antigen specificity and high antibody affinity. They have preserved functional activity as protective antibodies based on both in vitro cell protection and in vivo protection against a lethal challenge. More significantly, these mAb targeted different epitopes of toxin A, which allowed for the development of a “cocktail” formulation to improve the in vivo protein or a paired detection kit to measure the presence and levels of toxin A in testing samples, including clinical samples. Currently, most of the commercial toxin detection assays use one mAb to pair with polyclonal antibody. With our mAb pair, production costs and lot-to-lot variations can be significantly reduced. Recently, one study reported a significant correlation between toxin B protein concentrations and severities of clinical C. diff infection,24 however, no such correlation study has been reported for toxin A. By using the mAb pair we identified here, it will be feasible to measure toxin A with high sensitivity, making it a useful tool for such study in the future.
Finally, similar high quality toxin A-specific recombinant mAb were produced from the cloning of Ig genes and expressed in 293T cells. These reagents are very useful for the stable production and in-depth analysis of toxin A mAb. Furthermore, we have conducted the first immunogenetics analysis on mouse mAb generated by DNA immunization. It will be interesting to see if the method of immunization, including the delivery method of DNA vaccines, may impact immunoglobulin gene usage and affinity maturation. Again, our data provide useful baseline information for more in-depth analysis in future studies.
Given the recent rapid progress in mAb technology, including the direct cloning of Ig genes from immunized B cells from both animal and human sources,25-28 great opportunities to study and optimize the immunization approaches that can be effective in eliciting high titer and high affinity antibody responses in the hosts, which can then provide high quality B cells for Ig gene cloning, are becoming available. In combination with advanced deep sequencing technology, the entire process of antigen-specific B cell development can be monitored. DNA immunization offers an attractive approach to study the induction and production of high quality mAb and the current report confirmed such feasibility.
Materials and Methods
Construction of C. difficile toxin DNA vaccines
For pJW4303-TcdA-C and pJW4303-tPA-TcdA-C DNA vaccines, the codon optimized toxin A TcdA-C insert was cloned into the pJW4303, as previously reported.15 All of the DNA vaccine plasmids were produced from E. coli HB101 strain with Qiagen Plasmid Mega kit (QIAGEN, 12181) for in vitro transfection and in vivo animal immunization applications.
In vitro expression of target proteins
The expression of TcdA-C DNA vaccines was examined individually by transient transfection into 293T cells, as previously reported.15 western blot analysis was conducted to confirm the specific expression of expected C-terminal fragment of toxin A.
Animal immunization
BALB/c mice (6–8 weeks old) purchased from the Experimental Animal Study Center affiliated with Chinese Academy of Science were used for DNA immunizations. They were housed in the Animal Medicine Department of Nanjing Medical University in accordance with approved protocol by the institutional animal use committee. An electroporator (SCIENTZ-2C) from Scientz Co., Ltd. (SCIENTZ, 07–018), which has a non-penetrating caliper electrode, was used for the administration of DNA vaccines. Briefly, following intramuscular injection of plasmid (100μg per mouse divided at two different sites in the quadriceps muscle of the hindlimb), the caliper electrode was placed immediately adjacent to the skin of the injection sites; the injection sites were electroporated with the following parameters: 50V, 30ms and 30Hz. A total of four DNA immunizations were given at weeks 0, 2, 4 and 8. Sera were collected prior to the start of immunization and 2 weeks after each immunization. Then, two boost immunizations were conducted at Weeks 12 and 15 with partially purified TcdA-C fragment produced in 293T cells by IM after emulsified with incomplete Freund’s adjuvant (IFA). A final boost was given at Week 18, 4 d before splenocytes were collected.
Biotinylation of toxin A
Commercially available and lyophilized toxin A (List Biological Laboratories, 152C) was reconstituted to 0.1 mg/ml with water according to manufacturer’s instruction. Biotin Stock Solution was first prepared by dissolving 25 mg of NHS-PEG12-Biotin (EZ-link 21312, Thermo Scientific) in 82 µl of DMF (Thermo Scientific, 20673) to reach the final concentration of 250 mM. At the time of use, a working solution (250 µM) was produced by further diluting Biotin Stock Solution with DMF. To label 10 µg toxin A, 100 µl of toxin A (0.1 mg/ml in PBS) was mixed with 6.0µl of Biotin Working Solution for 30 min at room temperature with shaking. Slide-A-Lyzer dialysis Unit (Thermo scientific, 69558) was used to remove excess biotin for 30 min. Glycerol was added at 1:1 ratio by volume and the final product was stored at -20°C.
Enzyme-linked immunosorbent assay (ELISA)
The 96-well flat-bottom plates were coated with the supernatant from 293T cells transfected with the the TcdA-C DNA vaccine plasmid at 1:5 dilution or commercial purchased Toxin A at a concentration of 1 µg/ml dissolved in phosphate buffered saline (PBS). Detailed steps of ELISA were reported before.15
Generation of toxin A mAb
The last boost was performed using partial purified TcdA-C fragment produced in 293T cells 4 d before fusion. Splenocytes were fused with the SP2/0 myeloma cells using 50% polyethylene glycol (Roche, 10783641001). After HAT (Sigma, H0262) medium selection, culture supernatants were analyzed for antibody reactivity against TcdA whole toxin using capture ELISA. The specificity and sensitivity of mAb were assessed by ELISA and western blot analysis.
Capture ELISA for hybridoma screening
Capture antibody (Goat anti-mouse IgG-FC, 5µg/ml) (Thermo scientific, 31170) was prepared in PBS, 100 µl of this solution was then added to each well of a 96-well plate. Plates were incubated overnight at 4°C. Plates were then washed 2 times with washing buffer. A volume of 200 µL of blocking solution was pipetted into each well and incubated overnight at 4°C. Blocking solution was discarded and 100 µl of hybridoma supernatant (1:1 dilution with diluent) was transferred into the appropriate wells. Plates were incubated at 37°C for 1 h; plates were then washed twice with washing buffer. 100 µl of 1 µg/ml biotinylated toxin A was added into each well and plates were incubated at 37°C for 1 h. Plates were then washed 3 times with washing buffer. NeutrAvid-HRP (Thermo scientific, 31001) was added at 1:2000 dilution to each well. Plates were washed 3 times with washing buffer then 50 µl of TMB substrate solution (Thermo scientific, 34028) was added. Plates were allowed to develop and 50 µl stop solution (1M sulfuric acid) was then added. Absorbance was read at the appropriate wavelength in a microplate reader.
Western blot
The 293T cell transfection samples were subjected to denatured SDS-PAGE and blotted onto PVDF membrane (MILLIPORE, IPVH00010). The membranes were blocked with 5% dried skim milk. Specific animal immune sera raised against individual toxin segments were used as the detecting antibody at 1:500 dilution and incubated at 4°C overnight. The membranes were washed with blocking buffer and reacted with horseradish peroxidase (HRP)-conjugated goat-anti-mouse IgG (SouthernBiotech, GAR007) at 1:10,000 dilution and incubated at 37°C for 1 h. After the final wash, SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific Inc., 34077) was applied to the membranes for 5 min. Once they were dry, Kodak films were exposed to the membrane and developed in the dark room.
Preparation of bacteria culture filtrate
C. diff VPI 10463 was inoculated into a BacT/ALERT®N 259790 culture bottle (bioMérieux, 259790) and incubated at 37°C for 72 h. The culture medium was then centrifuged at 6000 g for 15 min at 4°C to remove cell debris. The culture medium was then filtered using 0.45 μm filter, and the filtrate was transferred into an Amicon Ultra-15 Centrifugal Filter Unit with Ultracel-100 Membrane (Millipore, UFC810008) and centrifuged at 5000 g for 30 min at 4°C. The final product, concentrated bacteria culture filtrate, was stored at -70°C before use.
In vitro protection study
CHO cells were seeded into a 96-well plate with a concentration of 1 × 104 cells per well and then cultured under 5% CO2 at 37°C for 24 h with Dulbecco’s Modified Eagle Medium (DMEM) with 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 10% inactivated fetal bovine serum. The medium was then discarded and the cells were washed with 200 μl pre-heated PBS. Samples were prepared as follows and added into triplicate wells: 150 µl of commercial purchased toxin A at a concentration of 1.2 µg/ml, which is the lowest cytoxic dose to cause all cells to round at 24 h,15 was mixed with 150 µl of 30 µg/ml individual mAb and incubated at 37°C for 1 h, the final concentration of toxin A was 0.6 µg/ml and mAb was 15 µg/ml, while the toxin A at the same concentration and DMEM medium only were added into control wells. After 24 h, the cell morphology was observed under a Nikon ECLIPSE TE2000-S microscope (Nikon). 100% inhibition of cell rounding was defined as protection function for mAb.
In vivo protection study
A pre-protection study was conducted first in BALB/c mice to determine 100 ng of Toxin A as the absolute lethal dose (LD100). Various mAb, either individual or in combination, were mixed with 100 ng of Toxin A and incubated at 37°C for 1 h before intraperitoneal (IP) injection, while PBS or irrelevant mAb with the same isotype were used in control groups. Each group contained 8 mice. Survival rates were observed every 12 h for two weeks.
PCR amplification of mAb genes from hybridomas
Total RNA was extracted from 107 hybridoma cells using a commercially available RNA isolation kit following the manufacturer's instructions (Qiagen, 74204). The purified RNA was quantitated spectrophotometrically. The first strand cDNA for the expressed H or L chain was synthesized using a commercially available kit (Invitrogen L1502-01). For the heavy chain, gamma 3′constant primer (IgG2a) was used for the reverse transcription reaction, and for the light chain, the corresponding primers were used for the κ and λ chains. The 20 μl reverse transcription reaction mixture consisted of 1 X PCR buffer II, 0.75 μM antisense primer, 10 mM dNTP mix, 5 mM MgCl2, 1U of RNase inhibitor, 50U of MuLV reverse transcriptase (Applied Biosystems), and 10 μg of total RNA. The reaction was performed at 42°C for 1 h. Reverse transcriptase was inactivated by incubating at 99°C for 5 min. For the first round PCR, 5 μl of unpurified cDNA was used as a template. The concentration of the components and cycling parameters were the same as described above for the genomic PCR except that PCR buffer I, AmpliTaq DNA polymerase, and J region primers were replaced by Pfu Ultra Hotstart buffer, Pfu Ultra Hotstart DNA polymerase (Clonetech, 639123), and C region primers, respectively. The Pfu DNA polymerase exhibits an average error rate of 1.3 × 10− 6 mutation frequency/bp/duplication.29 For experiments involving low copy templates (e.g., small numbers of B cells or single B cells), Taq DNA polymerase was preferred over Pfu DNA polymerase because of its higher amplification efficiency.
Acknowledgments
This study was supported in part by a grant (# BC2011096) from Science and Technology Department of Jiangsu Province, China under its Technology Innovation Funding Program for STE in Jiangsu Province. The authors would like to thank Dr Jill M Serrano for critical reading and editing of the manuscript.
Glossary
Abbreviations:
- TMB
3,3-,5,5-tetramethylbenzidine
- C. diff
Clostridium difficile
- TcdA
C. difficile toxin A
- TcdA-C
C. difficile toxin A C-terminus
- DMEM
Dulbecco's Modified Eagle Medium
- EP
electroporation
- ELISA
enzyme-linked immunosorbent assay
- HRP
horseradish peroxidase
- Ig
immunoglobulin
- IM
intramuscularly
- IP
intraperitoneally
- mAb
monoclonal antibodies
- PBS
phosphate buffered saline
- PCR
polymerase chain reaction
- tPA
tissue plasminogen activator
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/vaccines/article/25656
References
- 1.Liu MA. DNA vaccines: an historical perspective and view to the future. Immunol Rev. 2011;239:62–84. doi: 10.1111/j.1600-065X.2010.00980.x. [DOI] [PubMed] [Google Scholar]
- 2.Kutzler MA, Weiner DB. DNA vaccines: ready for prime time? Nat Rev Genet. 2008;9:776–88. doi: 10.1038/nrg2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang S, Parker C, Taaffe J, Solórzano A, García-Sastre A, Lu S. Heterologous HA DNA vaccine prime--inactivated influenza vaccine boost is more effective than using DNA or inactivated vaccine alone in eliciting antibody responses against H1 or H3 serotype influenza viruses. Vaccine. 2008;26:3626–33. doi: 10.1016/j.vaccine.2008.04.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ledgerwood JE, Wei CJ, Hu Z, Gordon IJ, Enama ME, Hendel CS, et al. VRC 306 Study Team DNA priming and influenza vaccine immunogenicity: two phase 1 open label randomised clinical trials. Lancet Infect Dis. 2011;11:916–24. doi: 10.1016/S1473-3099(11)70240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harari A, Bart PA, Stöhr W, Tapia G, Garcia M, Medjitna-Rais E, et al. An HIV-1 clade C DNA prime, NYVAC boost vaccine regimen induces reliable, polyfunctional, and long-lasting T cell responses. J Exp Med. 2008;205:63–77. doi: 10.1084/jem.20071331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan J, Corbitt N, Pankhong P, Shin T, Khan A, Sardesai NY, et al. Immunogenicity of a novel engineered HIV-1 clade C synthetic consensus-based envelope DNA vaccine. Vaccine. 2011;29:7173–81. doi: 10.1016/j.vaccine.2011.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pillet S, Kobasa D, Meunier I, Gray M, Laddy D, Weiner DB, et al. Cellular immune response in the presence of protective antibody levels correlates with protection against 1918 influenza in ferrets. Vaccine. 2011;29:6793–801. doi: 10.1016/j.vaccine.2010.12.059. [DOI] [PubMed] [Google Scholar]
- 8.Vaine M, Wang S, Crooks ET, Jiang P, Montefiori DC, Binley J, et al. Improved induction of antibodies against key neutralizing epitopes by human immunodeficiency virus type 1 gp120 DNA prime-protein boost vaccination compared to gp120 protein-only vaccination. J Virol. 2008;82:7369–78. doi: 10.1128/JVI.00562-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaine M, Wang S, Hackett A, Arthos J, Lu S. Antibody responses elicited through homologous or heterologous prime-boost DNA and protein vaccinations differ in functional activity and avidity. Vaccine. 2010;28:2999–3007. doi: 10.1016/j.vaccine.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang S, Chou TH, Sakhatskyy PV, Huang S, Lawrence JM, Cao H, et al. Identification of two neutralizing regions on the severe acute respiratory syndrome coronavirus spike glycoprotein produced from the mammalian expression system. J Virol. 2005;79:1906–10. doi: 10.1128/JVI.79.3.1906-1910.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang S, Taaffe J, Parker C, Solórzano A, Cao H, García-Sastre A, et al. Hemagglutinin (HA) proteins from H1 and H3 serotypes of influenza A viruses require different antigen designs for the induction of optimal protective antibody responses as studied by codon-optimized HA DNA vaccines. J Virol. 2006;80:11628–37. doi: 10.1128/JVI.01065-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barry MA, Barry ME, Johnston SA. Production of monoclonal antibodies by genetic immunization. Biotechniques. 1994;16:616–8, 620. [PubMed] [Google Scholar]
- 13.Al-Qaoud KM, Al-Omari MM, Al-Aghbar M, Abdel-Hafez SK. Production of monoclonal antibodies against the 8 kDa subunit of Echinococcus granulosus Antigen B (EgAgB8/2) using DNA immunization. Hybridoma (Larchmt) 2008;27:431–8. doi: 10.1089/hyb.2008.0039. [DOI] [PubMed] [Google Scholar]
- 14.Martín-Folgar R, Lorenzo G, Boshra H, Iglesias J, Mateos F, Borrego B, et al. Development and characterization of monoclonal antibodies against Rift Valley fever virus nucleocapsid protein generated by DNA immunization. MAbs. 2010;2:275–84. doi: 10.4161/mabs.2.3.11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin K, Wang S, Zhang C, Xiao Y, Lu S, Huang Z. Protective antibody responses against Clostridium difficile elicited by a DNA vaccine expressing the enzymatic domain of toxin B. Hum Vaccin Immunother. 2013;9:63–73. doi: 10.4161/hv.22434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin K, Wang S, Huang Z, Lu S. Clostridium difficile infections in China. J Biomed Res. 2010;24:411–6. doi: 10.1016/S1674-8301(10)60055-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei J, Yan B, Chen Z, Li T, Deng F, Wang H, et al. Production and characterization of monoclonal antibodies against the hemagglutinin of H5N1 and antigenic investigation of avian influenza H5N1 viruses isolated from China. Can J Microbiol. 2011;57:42–8. doi: 10.1139/W10-097. [DOI] [PubMed] [Google Scholar]
- 18.Tan GS, Krammer F, Eggink D, Kongchanagul A, Moran TM, Palese P. A pan-H1 anti-hemagglutinin monoclonal antibody with potent broad-spectrum efficacy in vivo. J Virol. 2012;86:6179–88. doi: 10.1128/JVI.00469-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ulivieri C, Burroni D, Telford JL, Baldari CT. Generation of a monoclonal antibody to a defined portion of the Helicobacter pylori vacuolating cytotoxin by DNA immunization. J Biotechnol. 1996;51:191–4. doi: 10.1016/0168-1656(96)01610-0. [DOI] [PubMed] [Google Scholar]
- 20.Puttikhunt C, Kasinrerk W, Srisa-ad S, Duangchinda T, Silakate W, Moonsom S, et al. Production of anti-dengue NS1 monoclonal antibodies by DNA immunization. J Virol Methods. 2003;109:55–61. doi: 10.1016/S0166-0934(03)00045-4. [DOI] [PubMed] [Google Scholar]
- 21.Horimoto T, Fukuda N, Iwatsuki-Horimoto K, Guan Y, Lim W, Peiris M, et al. Antigenic differences between H5N1 human influenza viruses isolated in 1997 and 2003. J Vet Med Sci. 2004;66:303–5. doi: 10.1292/jvms.66.303. [DOI] [PubMed] [Google Scholar]
- 22.Wang S, Zhang C, Zhang L, Li J, Huang Z, Lu S. The relative immunogenicity of DNA vaccines delivered by the intramuscular needle injection, electroporation and gene gun methods. Vaccine. 2008;26:2100–10. doi: 10.1016/j.vaccine.2008.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin K, Wang S, Huang Z, Lu S. Clostridium difficile infections in China. J Biomed Res. 2010;24:411–6. doi: 10.1016/S1674-8301(10)60055-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryder AB, Huang Y, Li H, Zheng M, Wang X, Stratton CW, et al. Assessment of Clostridium difficile infections by quantitative detection of tcdB toxin by use of a real-time cell analysis system. J Clin Microbiol. 2010;48:4129–34. doi: 10.1128/JCM.01104-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R, et al. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc. 2009;4:372–84. doi: 10.1038/nprot.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453:667–71. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao HX, Levesque MC, Nagel A, Dixon A, Zhang R, Walter E, et al. High-throughput isolation of immunoglobulin genes from single human B cells and expression as monoclonal antibodies. J Virol Methods. 2009;158:171–9. doi: 10.1016/j.jviromet.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lightwood DJ, Carrington B, Henry AJ, McKnight AJ, Crook K, Cromie K, et al. Antibody generation through B cell panning on antigen followed by in situ culture and direct RT-PCR on cells harvested en masse from antigen-positive wells. J Immunol Methods. 2006;316:133–43. doi: 10.1016/j.jim.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 29.Cline J, Braman JC, Hogrefe HH. PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 1996;24:3546–51. doi: 10.1093/nar/24.18.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]