

Abstract
The immune system responds to tumor cells. The challenge has been how to effectively use these responses to treat or protect against cancer. Toward the goal of developing a cancer vaccine, we are pursuing methodologies for the discovery and testing of useful antigens. We present an array-based approach for discovering these B cell antigens by directly screening for specific host-sera reactivity to lysates from tumor-derived cDNA expression libraries. Several cancer-specific antigens were identified, and these are currently being validated as potential candidates.
Keywords: cancer, tumor, vaccine, immunogen, cDNA, library, sera, antibody
Introduction
The need for better approaches against cancer
Cancer is the second leading cause of death worldwide.1 Conventional treatments for cancer such as surgery, chemotherapy, and radiation, have harsh side effects and usually slow progression but do not cure disease. A newer class of treatments comprised of monoclonal antibodies such as Herceptin, which recognizes the breast tumor-associated antigen HER2/neu, and Rituximab which binds the lymphoma associated antigen CD20. These immunotherapies have the significant benefit of being far more specific than conventional approaches, but the disadvantage of their high cost and requirement for continuous re-administration.
An even less conventional, and recently emerging approach to treating cancer is vaccination. Vaccines are typically considered for battling infectious pathogens, not one’s own cells. However, at least in theory the immune system could be capable of learning to recognize the patterns that set tumor cells apart from normal self cells. Not only does a cancer vaccine theoretically make sense, but cancer vaccines have been demonstrated to work in animal models. For example, vaccination of rats with a tumor-associated antigen formulation significantly delayed growth of chemically induced tumors.2 Injecting rats with lysates from whole tumor cells was also able to provide some level of tumor growth protection.3 Human self tumor mixtures in which tumor lysates are prepared from patients’ own tumor are also being tested in clinical trials.4 While self tumor mixtures hold potential, preparing these tumor mixtures from tumor-bearing patients is often not feasible relative to both disease timeframes and costs. In addition, effectiveness is compromised by the very fact that disease must be sufficiently progressed such that a tumor can be detected and extracted. A more attractive cancer vaccine that is economically and technically more practical might consist of a subunit vaccine containing tumor-specific antigens to be administered prior to tumor development. For this approach, prior knowledge of tumor antigens is needed.
Methods for discovering tumor antigens
Identifying tumor-specific antigens can be tackled using a variety of methods. One could sequence the DNA of cancer and normal cells to search for genomic differences.5 Alternatively, a researcher could examine the RNA transcripts.6 Another method is to actually use the immune system of the patient. Researchers have demonstrated that both T and B cell based immune responses are generated against tumors.7,8 The presence of lymphocytes in a tumor is associated with a better patient outcome.9-12 While much of these immune responses are cellular based, there are humoral responses as well. Antibodies produced against tumor proteins make good biomarkers since antibodies can persist in the body longer than a tumor protein biomarker, and antibodies are continuously amplified in the presence of continued immunogen. Tumor specific antibodies can even be elicited before disease symptoms are apparent.
Many experimental methods to probe tumor proteins with antibodies have been developed. Before the advent of high throughput methods, researchers could only probe a few antigens at a time using assays such as 1D SDS/PAGE and ELISA. Later on in 1995, the serological analysis of recombinant cDNA expression (SEREX) libraries was developed.13 Briefly, the method involved producing a tumor cDNA expression library, lysing clones on a nitrocellulose membrane filter, and then probing this membrane with autologous sera. Using this approach a large number of antibody and recombinant tumor protein interactions can be assayed at one time as compared with the single antigen assays. Nevertheless the SEREX method does have some disadvantages such as the fact the proteins must be capable of being expressed in bacteria, proteins with the most transcripts in the tumor cell may be detected most frequently while tumor associated antigens of low abundance may be missed, and there will be antibodies to non-tumor associated antigens. Most vexing has been the poor reproducibility of the process and the time-consuming and labor-intensive protocol.
Microarrays overcome some of the limitations of SEREX. With a microarray, recombinant proteins are arrayed onto a small slide rather than spread out across a large membrane. This arrangement allows for a small sample amount to be used. There can be two types of microarray approaches: biased and unbiased. The biased approach contains known proteins. For example, Invitrogen manufactures a Proto-Array containing more than 9,000 proteins which researchers can probe with sera. The disadvantage of this method is that new immunogens, such as undiscovered mutated proteins that may not be present in current databases, cannot be discovered using these microarrays. The unbiased approach spots recombinant phage or cDNA library cell lysate onto a slide, and this method allows for the discovery of previously unknown proteins since any protein encoded in the original tumor could be in the library. The benefits of this microarray approach include its high-throughput nature, reproducibility, small reaction surface along with even sera distribution across the surface, high dynamic range of signal intensities, and the quantity of data that one experiment can produce.14 The disadvantages currently involve the cost, and the fact that so much data are produced that more sophisticated data analysis software must be used to interpret the results.
Serological proteome analysis (SERPA) is another method for probing tumor proteins with antibody containing sera.15 In this method, 2D electrophoresis is first performed to fractionate the lysate’s tumor proteins. A western blot is then performed to detect binding events between sera from the tumor-bearing host and the tumor proteins. Tumor proteins of interest are then extracted from the gel and the identity of the protein is determined by mass spectrometry. This method does not require the construction of a cDNA library. Post-translational modifications can potentially be detected and heterologous expression is not needed since the tumor proteins are directly assessed. Unfortunately, this method suffers from limiting amounts of material, poor reproducibility, and the low resolution of 2D electrophoresis.
Known cancer immunogens
The experimental methods described above have discovered > 2500 tumor proteins, some of which may be immunogenic. Many of these proteins are stored in the Cancer Immunome Database.16 An analysis of these proteins reveals that there are many different categories of non-normal proteins recognized by the immune system of a tumor-bearing host. Well-studied tumor antigens include cancer testis (CT) antigens, heat shock proteins,17 and oncoproteins such as HER-2/Neu and p53. These have been identified as normal cell antigens that are overexpressed in tumor cells.18 Viral antigens such as antigens from the human papillomavirus,19 and cell differentiation antigens such as RAB38, and NY-BR-120 have also been identified. Other categories of tumor antigens are the result of frameshift mutations due to microsatellite regions in the genome such as the CDX2 antigen in colon cancer.21
Ideal cancer vaccine
Although many cancer immunogens have been discovered, other criteria need to be satisfied if they are to be useful components of a cancer vaccine. Even if tumor-specific antigens do exist in each individual tumor, these antigens would not prove very useful targets for a vaccine unless they occurred with a relatively high prevalence within the human population. If every tumor-specific antigen was unique to each tumor, then a separate vaccine would need to be discovered and developed for each patient. If antigens could be identified that were commonly found among cancer patients, then vaccines could be economically and effectively developed to treat them. Supporting this possibility, common mutations in oncogenes p5322 and Ras have been found.23 In addition, the ideal cancer vaccine should be comprised of multiple antigens so as to prevent immune escape that can occur as tumors continually evolve to evade the detection of the immune system. Note that the challenges of immune escape are mitigated in earlier stages of cancer and for prophylactic vaccines since there will be fewer tumor cells at these stages. Even though many tumor antigens have been discovered, the requirements for using these to treat cancer are high, and the current list may not be sufficient for the development of an effective vaccine. There may still be many critical proteins left to discover which will aid in the understanding of cancer as well as the development of a cancer vaccine.
Screening pooled tumor cDNA library lysates
In the presented research, a microarray of pooled tumor cDNA library lysates was used to screen tumor antigens. A tumor cDNA library was constructed and 3,000 lysate pools each containing approximately 1,000 original transformants were contact printed onto a nitrocellulose slide. This approach allowed three million library components to be quickly and simultaneously screened on one platform. One high binding sublibrary lysate-pool was then selected and re-arrayed onto another slide, as 3,000 individual features. Thus ~1,000 unique constituents would be represented at approximately one per spotted position since the colonies were diluted to contain one colony per well volume. High binding lysate spots from this array were then used to construct an array containing exactly one clone for a final round of screening. This multi-round pooled lysate approach allowed for the screening of many clones at one time in a very high-throughput manner. This high-throughput approach would not have been practical at the time when some tumor antigen screening methods such as SEREX were first developed since this new approach makes use of automated equipment for handling large numbers of PCR plates, small nitrocellulose slides, high resolution scanners, and accurate printing machinery. The high-throughput tumor lysate screening for tumor antigens approach described in this paper may be the type of method that could lead to the discovery of important tumor immunogens that aid in the understanding and treatment of cancer.
Results
Complexity of cDNA library
From the 15 colonies that were sequenced after the construction of the tumor cDNA library, an estimate of the complexity of the transcripts represented in the library was calculated. Several of the sequences were very short products (three sequences), and nine of the remaining 12 insert sequences were oriented in the forward orientation relative to the Lac promoter of the bacterial expression plasmid. One of these nine sequences was a duplicate, such that eight from this sampling of 15 library components were found to be unique probes for screening the binding reactivites of test and control sera.
Slide quality assessment
Before arriving at the final protocol used for the tumor cDNA library lysate slides, many different slide surfaces, incubation conditions, and reagent concentrations were tested. The final protocol optimized tumor lysate spot morphology without smearing one lysate pool into another (Fig. 1). We determined that a test antibody can recognize its cognate antigen when diluted up to 1000 fold into control lysate with a fold change of 1.66 and a p-value of 0.01. An experiment in which anti-SMC1Afs antibody was applied to the three million component tumor library array demonstrated that the SMC1Afs-containing lysate pools were selectively recognized (Fig. 2).
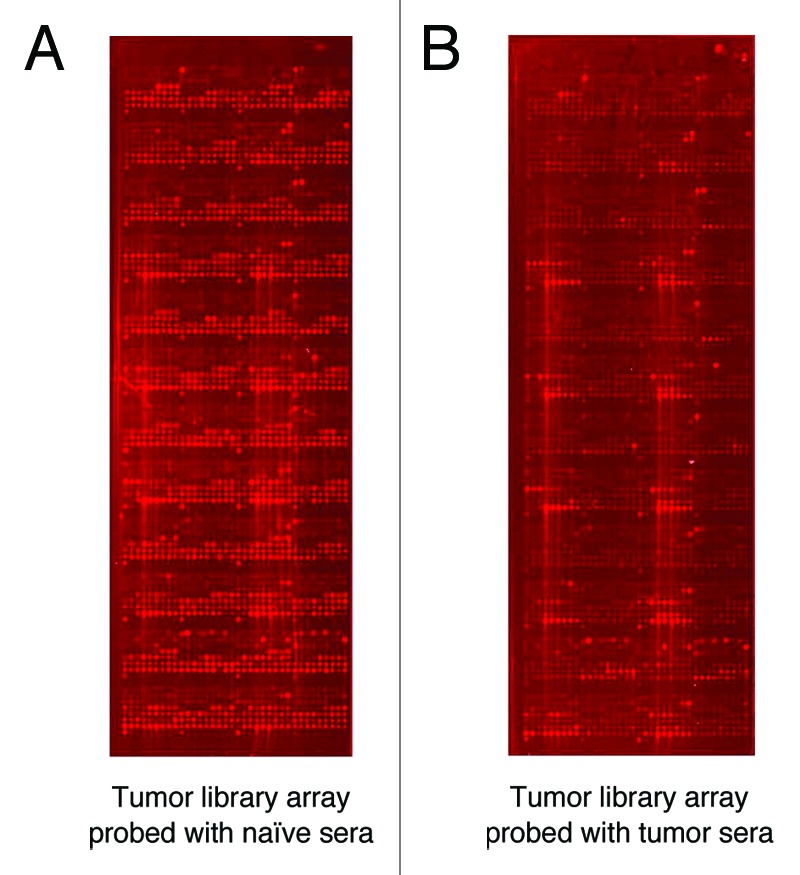
Figure 1. Array scan of slide surface. Lysates from a 4T1 tumor cDNA expression library printed onto nitrocellulose slides. (A) Library probed with naïve mouse serum. (B) Library probed with serum from 4T1 tumor-bearing mouse. Nitrocellulose slides were spotted with 3,000 pools of bacterially expressed tumor cDNA library clones, probed with the indicated sera. Specific lysate binding was detected with an anti-mouse IgG secondary antibody conjugated to AF647 and then scanned (Tecan Power Scanner) at 647 nm. An image of one slide with naive sera and one slide with tumor sera is presented. Each one of the 3,000 pools is comprised of 1,000 original 4T1 tumor cDNA library transformants.
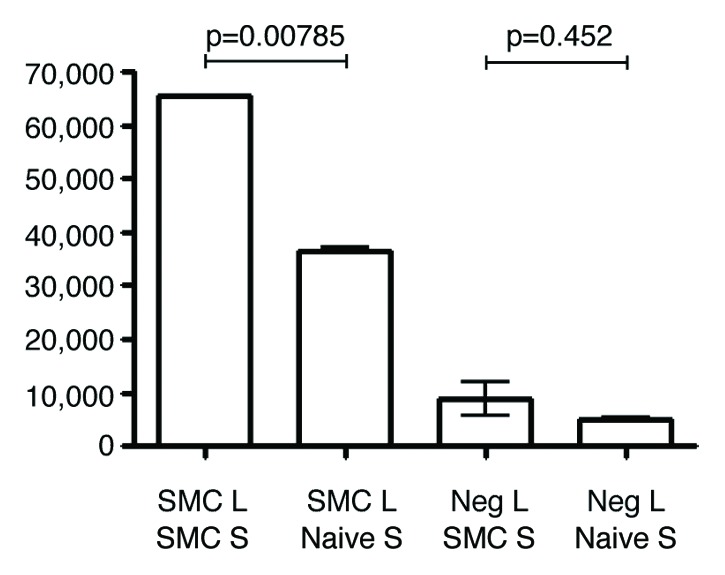
Figure 2. Bar graph of detected intensity of controls. Intensity of lysate containing either SMC1Afs peptide or negative control β-galactosidase protein. The intensities were detected after either sera against SMC1Afs or sera from a naïve mouse were applied to the 3 million component tumor lysate library. SMC, SMC1Afs; Neg, negative control β-galactosidase; S, sera; and L, lysate.
Tumor lysate screening
Tumor library screen
Pooled tumor lysates were subjected to several rounds of screening as described in the Materials and Methods section. Table 1 lists the array-feature selection criteria used through the screening protocol, which is presented in Figure 3A‒D. In the first round of screening, the nitrocellulose slide contained 3,000 pools each consisting of approximately 1,000 unique bacteria clones. Several pools from this round of experiments were selected for reduction and further characterization. The five tumor library lysate pools displaying the most significant increases in reactivity between normal and tumor sera samples were selected (p-values ranged from 2.04E-5 to 4.27E-3; fdr q-values ranged from 3.78E-3 to 1.86E-2). The five pools with the most significant decreases in reactivity were also selected (p-values ranged from 2.40E-6 to 2.04E-5; q-values ranged from 1.35E-3 to 3.78E-3). The five pools with the least significant p-values were selected as negative controls (p-value of 1; fdr q-value of 0.5). These fifteen selected pools were printed onto a new array for further characterization.
Table 1. Feature Performance Categories. The categories of feature subsets displayed in scatterplots (Fig. 3) are described relative to their performance against disease vs. normal sera.
| Symbol | Description | Stage of Screen |
|---|---|---|
| Significant p-valuea low tumorb | Three million component libraryc | |
| Significant p-value high tumor | Three million component library | |
| Pool selected from three million component library for further partitioning | Three million component library | |
| Greatest fold changee | 3,000 component sublibraryd | |
| Greatest tumor intensityf | 3,000 component sublibrary | |
| Significant p-value | 3,000 component sublibrary |
a Tumor sera or normal sera followed by AF647-labeled anti-mouse IgG antibody were applied to nitrocellulose slides that had been printed with lysate pools, in replicate. The slides were scanned at 632 nm and fluorescent intensities were measured for each feature. The intensity values obtained for each pool probed with tumor sera were compared with those obtained when probed with naïve sera. Calculated p-values were ranked by significance. b“Low tumor” indicates that the intensity of a pool was lower with tumor sera than naïve sera and “high tumor” indicates the opposite. cThe three million component library was made up of 3,000 pools (spots) each containing approximately 1,000 unique transformants. dThe 3,000 component sublibrary was partitioned from one of the pools in the three million component library. In this sublibrary there were 3,000 pools each containing approximately one unique transformant per spot. eThese pools had the greatest fold change in intensity that was higher with tumor sera than naïve sera fThese pools had the greatest intensity with tumor sera than any of the other pools with tumor sera.

Figure 3. Scatterplots of Tumor Library Lysate Screens. Naive and tumor sera was applied to the tumor library and sublibrary features. Values of features from tumor to control are displayed for the screen of the tumor library (A). At this stage of the screen, there are 1,000 unique transformants per spot. In (B) the validation of performance of selected lysate pools from the tumor library screen is presented. In this validation experiment to confirm that selected pools exhibited reproducible behavior in a new sera screen, there are still 1,000 unique transformants per spot. The most significant pool was partitioned onto a new array in the pool reduction screen presented in (C) with approximately one unique transformant per spot. A total of 9 lysate pools were selected from this screen and partitioned onto a new array for the single clone array screen presented in (D) containing exactly one single unique transformant per spot. The categories of features are described in Table 1. In (D), each lysate is labeled with the wild type transcript which the cloned sequence aligned with.
A scatterplot of the 3,000 lysate pools is presented in Figure 3A. The base two log of the fold change is represented on the x-axis so that values with a decreasing fold change from naïve to tumor are on the left side of the y-axis and values with an increasing fold change from naïve to tumor are on the right side of the y-axis. The negative base ten log of the p-value is represented on the y-axis so that values with the most significant p-values are present at the top of the graph. Note that false discovery rates are useful for making assessments about many p-values from an experiment. An FDR adjusted p-value, or q-value, of 3.78E-3 implies that 0.378% of all tests with a q-value less than or equal to 3.78E-3 are false positives. This provides a metric to use across the entire experiment rather than to use only for each individual lysate pool. In support of the validity of the array-based library screening method, the pool with the most significant p-value within the set of increased reactivities in one screening experiment was also in the top three of the same category in a second screening experiment (data not shown).
Validation of performance of selected lysate pools from tumor library screen
A validation array containing only the ten lysate pools from the three million clone library selected as reacting significantly different to the tumor vs. control sera, and the five negative control pools was printed and tested. The results support the outcomes obtained from the original full screen. The least significant p-value lysates in the tumor library screen had the least significant p-value in the validation screen with p-values and fold changes that would place them near the origin of the p-value vs. fold change plots. The lysate pool within the increased intensity set with the most significant p-value (2.93E-18) was selected to partition and re-array on a nitrocellulose slide as distinct features for further testing and reduction of pool complexity (Table 1 and Fig. 3B).
Pool reduction screen
Since the number of original transformants being expressed within each pool was estimated to be 1,000, the identification of an individual antigen requires further testing. The sublibrary pool with the most significant p-value and increasing tumor intensity was partitioned and regrown in 3,000 microtiter wells. The 3-fold redundancy is to facilitate capturing a complete sublibrary representation, and each new sample is estimated to be comprised of approximately one to three unique clones. Slides were printed and tumor test and naïve control sera were applied. Analysis of fluorescent reactivity readouts was used to select several samples for further characterization. Three lysate samples were selected based on the best p-value for increase-reactivities (2.05E-6, 2.05E-6, and 3.80E-6; q-values of 5.55E-4, 5.55E-4, and 5.84E-4), three lysate samples were selected based on the highest fold change in reactivity relative to control sera (1.81, 1.82, and 1.84), and three lysate samples were selected based on the highest measured reactivity levels (10,052; 9,205; and 9,598) (Fig. 3C).
Single clone array screen
A final set of lysate slides were constructed that contained single clones derived from these nine lysate pools, which were selected based on their p-value, fold change, or high intensity. This was done by sequencing ten single colonies from each of the nine complexity-reduced pools. These were determined to contain from one to three unique clones. Therefore, the final lysate array consisted of three lysates derived from the high p-value lysate pools, five lysates derived from high fold change lysate pools, and four lysates from high intensity lysate pools. Lysates from a bacterium producing no library-derived clone was included as a negative control. A scatterplot of the fold changes in reactivity vs. p-values between test and control demonstrates that one of the clone-lysates from each of the three characteristic-category displayed a significant increase in reactivity to tumor sera relative to naïve sera (Fig. 3D). Note that in Figure 3D, the category of each single clone feature represents the category of the 3,000 component sublibrary feature from which the single clone feature was derived from. In other words, the Eif1a feature did not have one of the most significant p-values in the single clone array screen, but the Eif1a feature was derived from a feature pool which did have the most significant p-value in the 3,000 component sublibrary screen.
Sequence Information
Sequence information was obtained for the single clones tested as lysates on the final nitrocellulose arrays. Most of these sequences matched the 3′ end of wild-type mouse RNA transcripts but were truncated at the 5′ end. Some of the cDNAs inserted into the plasmid out of frame relative to the wild type protein (5/9) or in reverse orientation (1/9). Several of the sequences also contained point mutations relative to the murine database. Table 2 summarizes some of the characteristics of these sequences. Note that the CcoI sequence is an unusual sequence since it contains the 3′ end of the CcoI mouse mitochondrial gene transcript upstream of the tRNA-Ser sequence and a poly A tail. For the reverse-oriented Hiatl1, a ribosome translating this would slip along the poly T region present in the sequence and all three reading frames would be expressed which is why multiple lengths to the stop codon are listed in the table for this transcript. The Wfdc17 sequence almost spans the full wild-type sequence; there are only 12 bp missing from the 5′ end.
Table 2. Information for sequences in clones. Information for the sequences present in the tumor cDNA library clones in the final round of screening with tumor and naïve. mouse sera.
| Sequencea | Length (bp)b | Length to SC (bp)c | Forward Orientation?d |
5′ truncated?e | Frame-shift?f | No. of PMg | Extra Seq (bp)h |
|---|---|---|---|---|---|---|---|
| Rps8 | 443 | 9 | Yes | Yes | Yes | 0 | 0 |
| Eif1a | 288 | 51 | Yes | Yes | No | 0 | 0 |
| Rnf130 | 467 | 276 | Yes | Yes | No | 2 | 17 |
| Cbx3 | 483 | 27 | Yes | Yes | Yes | 0 | 0 |
| Hiatl1 | 676 | 336 or 42 or 12 | No | Yes | Yes | 2 | 0 |
| Sec61b | 278 | 177 | Yes | Yes | Yes | 0 | 0 |
| Wfdc17 | 458 | 111 | Yes; only 12 bp missing | Yes | 0 | 0 | |
| Mxd1 | 101 | 20 | Yes | Yes | No | 0 | 0 |
| Cco1 | 447 | 18 | Yes | Yes | No | 3 | 0 |
a The wild type mouse transcript which the cloned sequence aligned with. bThe length of the inserted sequence.
c The length to the stop codon from the insertion site. dIndicates whether the sequence inserted into the plasmid in the forward orientation relative to the Lac promoter of the bacterial expression plasmid eIndicates whether the sequence in the clone is missing the normal 5′ end of the corresponding wild type transcript. fIndicates whether the sequence expressed from the Lac promoter in the bacterial plasmid is frame-shifted relative to the frame of the wild-type transcript. gIndicates the number of point mutations in the sequence. hIndicates how many additional base pairs are present in the sequence which are not present in the wild type transcript sequence.
Discussion
Summary
The goal of this research was to explore methods for screening tumor immunogens efficiently, and much knowledge has been acquired throughout this process. The basic process utilizes several technologies which were not widely available when the SEREX method was first developed. Such technologies include automated equipment to handle large numbers of 96-well plates, accurate printing machinery, and high resolution scanners. The process of screening multiple rounds of pooled lysates also allows the researcher to hone in on a particular clone of interest beginning from clones that cover the entire transcriptome. Several potential tumor antigens have also been identified, but more verification will be required before any strong conclusions can be made about any of these candidates.
Library transcript representation
The complexity of the sequences contained in the constructed cDNA library indicated that 53.3% of the sequences were not truncated products and were correctly oriented for expression. While informative, this number is likely an overestimate caused by the small sampling size. Namely, more than half of the clones in the sampled set were in the correct orientation (11/15 = 73% in correct orientation). Unless there is some fortuitous bias that we are unaware, 50% should be forward oriented and 50% should be reverse oriented). If more clones were sequenced, the calculated expressed-clone complexity would presumably approach the expected. This can be approximated as (1/2)*11/15 = 37% since 11/15 of the transcripts were unique non-truncated sequences, and approximately 50% of these sequences would be expected to insert into the plasmid in the correct orientation. Of these 37% only 33% would be anticipated to be in the same frame as the original transcript and therefore approximately 12% of the clones in the library would be expected to express products faithful to the original tumor RNA products.
The nitrocellulose slides are printed with E. coli cell lysate from this tumor cDNA library. There are 3,000 features total. Each feature represents proteins translated from approximately 1,000 original transformants. The total number of components present on a single microarray is ~3 million. This number includes transcripts that were inserted backward or out-of-frame. After taking these artifacts into account, we estimate a total of 3.6E5 unique transcripts are properly translated and presented on the microarray.
There are between 20,000 and 50,000 transcripts in a mammalian cell, depending on how one defines a unique transcript, and not taking into account post-translational modifications or rare splice variants. 94% of the mammalian transcripts occur between one and five times,24 and about 25% are present in one or fewer copies per cell.25 Given 3,000,000 clones, each mammalian transcript would be represented in our library 3E6/50,000 = 60 times on average. However, since only 12% of the library would correspond to the original RNA transcript sequence, each transcript would be expected to be represented approximately 60*0.12 = 7.2 times on average. We expect that there are certain transcripts for which this would not be true, given that some transcripts are extremely high copy and would appear more than 7.2 times presenting an opportunity to create a noise threshold. However, given a 7.2-fold minimum representation for low-copy transcripts, we expect sufficient sensitivity to detect single-copy events. A spike-in dilution experiment indicated our sensitivity is greater than one copy per 1000 clone pool (feature).
Experiment conditions
Different slide surfaces, incubation conditions, and reagent concentrations were tested during the development of the protocol. Although the data for all of these different conditions is not presented in this paper, a brief description will be provided. Many slide surfaces were used before consistently using the nitrocellulose slide surfaces used for the screens presented in the Materials and Methods and Results section. In hindsight, the nitrocellulose slides were highly likely to work since other protein assays such as a Western Blot assay make use of a nitrocellulose surface. However, cell lysate was printed onto Nexterion Slide A+ Aminosilane Coated Substrate slides from Schott, custom made polyethylenimine slides,26 and CodeLink Amine-binding slides from Surmodics. All of these slide surfaces resulted in smearing of cell lysate across the cell surface whereas printing lysate onto nitrocellulose slides resulted in no smearing and good spot morphology.
Other conditions such as blocking buffer, incubation time, and primary and secondary antibody concentrations also affect the resulting data. The blocking buffer used with the nitrocellulose affects the ability to detect antibody-protein interactions. Blocking with Super G blocking buffer as opposed to BSA results in lower overall signal intensity, but also results in an increased ability to detect a particular protein above background in a dilution series. Incubation condition tests also demonstrated that incubating the sera with the slide at 23 C for 16 h results in better detection of antibody-protein interactions than incubating the sera with the slide at 37 C for 1 h. A secondary antibody titration experiment was also performed to determine the optimal concentration of secondary to use, and a 1 nM concentration was then chosen for many experiments. These conditions were then used to demonstrate that anti-SMC1Afs antibody could detect the SMC1Afs protein in lysate from clones producing this protein as presented in the Results section.
Note that there are groups of features showing similar intensities. These were obtained in the same bacterial production batch, and this effect can be seen from the patterns of bright and faint spots on the nitrocellulose slide (Fig. 1). This bias in intensity which is introduced by technical experiment variation rather than a biological phenomenon does not affect the data analysis because each lysate feature intensity obtained with naïve sera is compared with the same lysate feature intensity obtained with tumor sera. In other words, the lysates compared are identical and are of the same batch, and lysates from different batches were not compared against one another.
No strong conclusions can be made from the current tumor cDNA library lysate screening results. However, there are some candidate antigens worthy of further investigation. There are several lysates which bound to tumor sera more than naïve sera at the last stage of screening. Note that the last round of screening resulted in lower p-values than was obtained in the previous rounds of screening. This last round of screening was repeated several times, and p-values comparable to the previous p-values were never obtained (data not shown). A likely reason for these less significant p-values is that the protein and lysate production for this batch of slides was not of the same quality as the protein and lysate production of the previous experiments. Nevertheless, p-values less than 0.05 were still often obtained. Note that several primary sera dilutions were also tried for this single clone screen and some counterintuitive results were obtained. The greatest fold change values between naïve and tumor with higher intensity for the tumor sera were obtained at the lower dilutions used. Perhaps this reflects greater affinity demanded for successful binding at the lower antibody concentrations, thereby reducing off target binding. Dilutions of 100 fold, 500 fold, 1,000 fold, and 2000 fold were tested, and the results from the 1,000 fold dilution are presented in this paper (Fig. 3D). A general observation from the experiments performed is that the Rps8, Wfdc17, Cbx3, Rnf130, and Sec61b clones often bound to sera better than other clones such as Mxd1 and the poly T clones. Some of the clones at this final stage of screening came from the same pool in a previous round of screening, but this does not imply that only one of the sequences is tumor specific. On the contrary, a lysate pool may have bound to antibodies in the tumor sera more than naïve because it contained lysate from more than one tumor specific sequence (positive clone). With lysate from a better batch of protein expression there is a possibility that all of the sequences may demonstrate higher binding reactivity to tumor than normal sera.
Transcript sequences and cancer
Do any of these sequences encode for proteins that have functions which relate to cancer in any way? Wfdc17, CcoI, Rps8, and Cbx3 produced the best p-values in a screen with tumor vs control sera. The protein encoded by Wfdc17 acts as a counter-regulator of proinflammatory responses.27 No papers associating this sequence with cancer were found. CcoI is a part of a complex in the mitochondia electron transport chain.28 This protein has been found to have altered expression levels in prostate cancer.29 The Rps8 gene encodes a ribosomal protein that is a component of the 40S subunit.30 Increased expression of this gene and other ribsomal proteins has been observed in some cancers such as colorectal cancer.31 The protein encoded by Cbx3 can act as a transcriptional regulator and is a component of heterochromatin.32 This sequence has previously been associated with neoplastic transformation and progression.33 Rps and Cbx sequences are currently present in the Cancer Immunome Database, but Wfdc and Cco proteins are not.16
The other sequences had a p-value less than 0.05 in the screen displayed in Figure 3D. Rnf130 encodes a protein that is a zinc finger protein structural domain, and these proteins are often involved in the ubiquitin degradation pathway.34 RNF130 interacts with miRNAs which are overexpressed in tumors.35 The protein encoded by Sec61b is a component of the machinery necessary to translocate proteins across the endoplasmic reticulum.36 Sec61b has been found to be one of the top 20 genes downregulated by miR-133a which is a tumor-suppressive microRNA.37 Mxd1 is a dimerization protein that competes with MYC for binding to MAX to form a DNA binding complex,38 and a mutated version of Myc is found in many cancers.39,40 Eif1a is an RNA binding protein essential for translation initiation.41 The EIF1A locus has been determined to be hypermethylated in human ovarian carcinoma CP70 cells.42 Hiatl1 belongs to the major facilitator superfamily and is involved in facilitating transport across membranes.43 The expression level of Hiatl1 was found to aid in the classification of gastric cancer.44 Sec, Rnf, and Eif sequences are currently present in the Cancer Immunome Database, but Hiatl, and Mxd sequences are not.16
Analysis of the sequences in the clones reveals that most of the sequences were 3′ truncated. The library was constructed using poly dT primers which would bind to the 3′ poly A tail of RNA transcripts. Although the reverse transcriptase used, polymerase used, and the SMART (Switching mechanism at 5′ end of RNA template) mechanism should have resulted in mostly full-length transcripts, many 3′ truncated transcripts were present. There could be important information upstream of the 3′ region in the original RNA transcript which was not detected in the library. Such information could include certain mutations as well as possible gene fusions which could cause expression of a frameshifted string of amino acids. A number of the sequences of the clones in the library were frameshifted relative to wild type murine databases. Some of these frameshifts may correspond to genomic events; others may be at the transcript level such as alternative splicing or trans-splicing events. Further experiments would be required to determine if there are any unusual transcripts in the RNA pool which correspond to the truncated 3′ transcripts detected.
Conclusion
This research outlines a platform for screening tumor cDNA library lysate for tumor-specific antigens. The platform involves using automated equipment to handle large numbers of PCR plates, small nitrocellulose slides, high resolution scanners, and the screening of several rounds of pools. The process is currently being repeated using random pentadecamers to construct a new tumor cDNA library rather than poly dT primers which may result in many 3′ truncated products as was the case for this cDNA library. As researchers use high throughput methods such as the method demonstrated in this paper to discover new tumor immunogens, we will better understand how the immune system responds to cancer and thereby be able to select optimal components to include in a preventative cancer vaccine.
Materials and Methods
Procedures with BALB/c mice
Fifteen four week old female BALB/c mice were purchased from Jackson laboratories and divided into three groups: a non-treated group, a tumor challenged group, and an SMC1Afs protein immunized group. Sera was collected weekly from each group starting at five weeks of age. 7,000 4T1 cells in 100 µL PBS were injected s.c into the flank of the mice when they were six weeks old. The tumor volume was measured daily after the tumor was palpable, and mice were euthanized when the tumor volume exceeded 2,000 mm^3 as calculated by the formula L^2*W/2. Splenocytes and tumor tissue were collected after euthanasia in accordance with the protocols outlined in Current Protocols in Immunology.45,46 Splenocytes were used to construct a phage library which is work not presented in this paper. Tumor tissues were used to construct a tumor cDNA library. Murine experiments were conducted under a protocol approved by the Arizona State University Institutional Animal Care and Use Committee
Mice in the SMC1Afs designated group were immunized with SMC1Afs DNA and protein constructs. These sera were used as a positive control against the SMC1Afs protein for following experiments. Five female six week old mice were genetically immunized with 1 µg SMC1-pCMVi plasmid and 1 µg CpG2395 adjuvant using a gene gun at day zero, day one, and day 15. A protein boost was performed with 5 µg 17 amino acid SMC1Afs fused to GST along with Alum and 5 µg SMC1Afs 17 amino acid peptide at day 34 and day 57 respectively. All of the mice were euthanized at day 73 and sera and splenocytes were collected.
Construction of tumor cDNA library
A tumor cDNA library was constructed from the tumor tissue from the tumor bearing BALB/c mice. RNA was extracted from 100–1000 mg of tumor tissue from mice using trizol (Cat No 15596–018) according to the manufacturer’s instructions.
The In-Fusion SMARTer cDNA Library Construction Kit (Cat No of 634929) from Clontech was used to construct the cDNA library from the obtained RNA. The 1st strand cDNA synthesis was performed according to the manual instructions. Briefly the initial reaction consisted of 4 µg of RNA mixed with the poly T 3′ SMART CDS Primer IIA oligo and ddH20, incubated at 72 C 3 min, and then incubated at 42 C 2 min. The following components were added to the mixture and incubated 42 C 90 min: 5X First-Strand buffer, DTT, dNTP, SMARTer II A Oligonucleotide, RNase Inhibitor, and SMARTscribe Reverse Transcriptase. For the 2nd strand synthesis several different numbers of cycles were used in different reactions. The reaction consisted of first-strand cDNA, ddH20, 10X Advantage 2 PCR buffer, 50X dNTP mix, 5′ PCR Primer IIA, and 50X Advantage 2 Polymerase Mix. The PCR reaction was performed with the following conditions: 95 C 1 min, (95 C 15 sec, 65 C 30 sec, 68 C 6 min)Xcycles. The sample which yielded a moderately strong smear from about 0.1 to 4 kb with a few prominent bands for abundant transcripts was selected for further reactions, and this sample corresponded with a cycle number of 21. The ds cDNA was purified to remove many small side reaction products. The purification was performed with CHROMA SPIN DEPC-1000 Columns from Clontech which exclude molecules larger than the pore size from the resin so that they pass through the column quickly. An ethanol precipitation was performed to purify the DNA further.
An In-Fusion reaction was performed to clone the cDNA products into the pSMART2IF vector. An In-Fusion reaction fuses DNA fragments that contain the same 15 base pairs at their end using the In-Fusion enzyme. In this library the sequence of overlapping 15 bp was identical at both sides of the insertion site into the plasmid. Therefore, sequences from the library could insert into the plasmid in the forward or reverse orientation. Additionally, translation of the sequence to protein in the bacterial clone starts from the Lac promoter in the plasmid, and therefore the frame of the tumor library sequence expressed may be different than the wild type frame. The In-Fusion reaction was performed by combining 5X In-Fusion reaction buffer, pSMART2IF linearized vector, cDNA, In-Fusion enzyme, and ddH20 and then incubating at 15 min 37 C followed by 15 min 50 C. This In-Fusion product was ethanol precipitated and electroporated into DH010B T1 electrocompetent cells from Invitrogen (Cat No 12033-015). These transformed cells were then induced to produce protein corresponding to the cDNA sequence inserted into their plasmid. Note that the final protein lysate that was used for screening was produced in several different batches.
Transformed colonies on numerous plates were counted using ImageJ software from the NIH.47 Images of the plates were obtained using the Universal Hood II from BIO-RAD Laboratories. The colony counting was performed by adjusting the brightness and contrast, setting the image to an 8 bit image type, applying a threshold so that only the colonies remained on the image, and using the analyze particles function to obtain a cell count.
Protein production and lysate printing
The proteins of the tumor library sequences were produced in pooled cultures in 96 well plates. There were three libraries handled: a library containing 3,000 lysate pools representing three million clones, a library containing 3,000 lysate pools representing 3,000 clones, and a library containing 12 lysates representing 12 clones. For the libraries with about 3,000 lysate features, 32 96-well plates containing bacteria cultures were used. The transfer of liquid cultures and reagents for these plates was performed with the Biomek FX Laboratory Automation Workstation from Beckman Coulter. Briefly, the transformed cultures were incubated overnight at 37 C at 250 rpm. These plates were incubated in stacks in a HiGro shaker from DIGILAB to make handling the numerous plates more convenient. After the overnight incubation, the culture was diluted 1 to 100 in fresh LB with Carbenicillin and grown to an OD600 of 0.5. IPTG was added to a final concentration of 1 mM to each well in the 96 well plates to induce the production of protein. The cultures were incubated at 37 C for 4 h. Cultures were harvested by centrifuging at about 4000 rcf for 10 min, discarding the supernatant, and resuspending in 100 µL of lysis buffer. The lysis buffer consisted of PBS, 1% Triton X-100 detergent, 1 mM PMSF protease inhibitor, and Complete Protease Inhibitor Cocktail from Roche (1 tablet per 10 mL solution). Lysozyme was added to the culture at a final concentration of 0.05 ug/uL. Plates were incubated for 15 min at room temperature with moderate shaking. The plates were then subjected to three freeze/thaw cycles to break up the cell membrane. DNase and MgCl2 were added to the solutions for a final concentration of 0.02 ug/mL and 1.1 mM MgCl2 respectively. Plates were incubated for 1 h at 4 C with moderate shaking. Soluble protein was isolated by centrifuging at 4,000 rcf for 20 min, and the supernatant was transferred to a new plate. EDTA was added at a final concentration of 1 mM to prevent contaminating growth, and glycerol was added at a final concentration of 2% to help maintain the integrity of the protein throughout future freeze/thaw cycles. A portion of the solution in each well was transferred from a 96 well plate to a 384 well plate to allow for printing.
Positive control lysate pools were included on the 1st library array containing three million clones. These positive control lysates were from clones containing the pGEX-SMC1fs-27mer plasmid, and these clones produced 17 amino acids of frameshift SMC1Afs sequence and 10 amino acids of wild-type SMC1A sequence fused to GST. Antibodies from mice immunized with this protein were later tested against this protein. Negative control lysates from transformants containing the PUC19 plasmid were also included in the library.
The soluble protein lysate was printed onto Oncyte SuperNova Nitrocellulose Film-Slides slides from Grace Biolabs (Cat No GBL705177) with the Nanoprint 60 microarray printer from ArrayIt Technologies with 500 µm spacing between spots. For the data presented in this paper, four print runs were performed: 1) a three million clone library containing 3,000 lysate pools (actually 3,072 lysate pools to be precise) each containing approximately 1,000 unique clones each, 2) a validation of performance print of 15 lysate pools selected based on p-values and intensity values from the three million clone library, 3) a pool reduction screen print consisting of a 3,000 feature sublibrary (specifically 3,072) with approximately one unique clone per feature, and 4) a single clone array screen print consisting of 12 lysates deconvoluted from features in the 3,000 feature sublibrary.
Another print run was also performed which contained lysate pools with a dilution series of 10-fold, 100 fold, and 1,000 fold SMC1Afs lysate into lysate from transformants containing the negative control PUC19 plasmid with three replicates for each dilution. This slide was later screened with anti-SMC1Afs sera, and this array was printed to demonstrate that a protein can be detected at low concentrations in a lysate pool.
Any printed slides were used for an experiment with sera one day after the print run to allow for enough time for the lysates to completely dry, but not allow for enough time for the proteins to degrade significantly. The lysate plates were stored at -80 C when not in use.
Application of sera to tumor library lysates
Numerous print runs and experiments with sera were performed, and the data from four of these experiments is presented (Fig. 3). The same protocol was followed for all four of these experiments with slight variations as knowledge was gained throughout the screening process.
Tumor Library screen
The nitrocellulose slides were first locked into the Tecan HS 4800 Pro Microarray Hybridization Station from Tecan. Two slides were used for each condition: tumor sera, naïve sera, and sera from SMC1Afs immunized mice. The slides were first washed for 30 sec with TBST, blocked with Super G blocking buffer, incubated at 23 C for 1 h, and washed for 30 sec with TBST. Primary sera at a 500 fold dilution in a 200 µL volume of incubation buffer consisting of BSA, Tween 20, and PBS was then applied to the nitrocellulose slides. This sera was from the tumor bearing, naïve, or SMC1Afs immunized mice described in section I. The slides were incubated at 23 C for 16 h, washed for 30 sec with TBST, and then 5 nM of AF647 goat anti-mouse IgG H+L antibody from Bethyl was added. The slides were incubated at 23 C 1 h, washed 30 sec with TBST, washed 30 sec with ddH20, and then dried with nitrogen for 5 min. The slides were scanned in the Tecan Power Scanner at 10 µm resolution to produce 16 bit images with intensity values for each pixel ranging from 0–65,535. The analysis of the data are described in section III. After the p-value was determined by comparing the tumor and naïve samples, the five most significant p-value lysate pools with high intensity with tumor sera, the most significant p-value lysate pools with low intensity with tumor sera, and the five least significant p-value lysate pools were used in a validation screen.
Validation of performance of selected lysate pools from the tumor library clone screen
Selected lysate pools (15 total) were printed onto a set of new nitrocellulose slides. Each lysate pool was present on the array with ten replicates. Sera was applied as before at a 500 fold dilution to two slides for tumor sera and two slides for naïve sera. This was followed by 1 nM secondary antibody. The lysate pool with the most significant p-value calculated by comparing the tumor and naïve sample was used to construct a new library containing 3,000 lysate pools with 1,000 unique clones per pool.
Pool reduction screen
Sera was applied at a 500 fold dilution to four nitrocellulose slide replicates per group containing lysate representing 3,000 unique clones. This was followed by 5 nM secondary antibody. Several lysate pools were chosen for further screening. These lysates corresponded to the three lysate pools with the most significant p-value, the greatest fold change, and the highest detected intensity with tumor sera.
Single clone array screen
Individual unique clones derived from the selected pools in the 3,000 clone screen were printed onto new nitrocellulose slides with three replicates per group and sera was applied at a 1,000 fold dilution. This was followed by 1 nM secondary antibody. The p-value and fold change of the lysates between tumor and naïve samples was then analyzed.
Data analysis
High intensity spots in the images of the scanned slides were correlated with the identity of specific lysate pools or lysates containing single clones by aligning the spots with labeled circles in a gal file. This process was performed with GenePix software. The resulting intensity values for each lysate were then analyzed in Microsoft Excel to perform T tests to determine the p-value for a lysate with tumor sera or naïve sera. The fold change between naïve and tumor was also determined. The QVALUE program for the R statistics software was also used to determine the false discovery rate (fdr) distribution of the intensities from a tumor library lysate screen.48 All of the default settings were used to calculate the false discovery rates. Graphs and figures were created using GraphPad Prism 4 for Windows (GraphPad Software, www.graphpad.com) and Inkscape 0.48 for Windows (www.inkscape.org).
Acknowledgments
Glossary
Abbreviations:
- BSA
bovine serum albumin
- PMSF
phenylmethylsulfonyl fluoride
- s.c
subcutaneously
- SMC1Afs
structural maintenance chromosome 1A frameshift
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/vaccines/article/25634
Stephen Johnston for experiment-plan suggestions; John Lainson for printing the tumor cDNA library lysate arrays; Maran Montgomery for assistance in cDNA library making, printing, and screening ; Krupa Navlkar for technical suggestions; Luhui Shen for technical advice and sharing SMC1Afs reagents; Phillip Stafford for experiment-plan suggestions and statistical advice.
References
- 1.Yach D, Hawkes C, Gould CL, Hofman KJ. The global burden of chronic diseases: overcoming impediments to prevention and control. JAMA. United States, 2004:2616-22. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Hur H, Kossoy G, Sandler B, Zusman I. Vaccination with soluble low-molecular weight tumor-associated proteins suppresses chemically-induced mammary tumorigenesis in rats. In Vivo. 2000;14:551–4. [PubMed] [Google Scholar]
- 3.Ward S, Casey D, Labarthe MC, Whelan M, Dalgleish A, Pandha H, et al. Immunotherapeutic potential of whole tumour cells. Cancer Immunol Immunother. 2002;51:351–7. doi: 10.1007/s00262-002-0286-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biragyn A, Tani K, Grimm MC, Weeks S, Kwak LW. Genetic fusion of chemokines to a self tumor antigen induces protective, T-cell dependent antitumor immunity. Nat Biotechnol. 1999;17:253–8. doi: 10.1038/6995. [DOI] [PubMed] [Google Scholar]
- 5.Kallioniemi OP, Kallioniemi A, Piper J, Isola J, Waldman FM, Gray JW, et al. Optimizing comparative genomic hybridization for analysis of DNA sequence copy number changes in solid tumors. Genes Chromosomes Cancer. 1994;10:231–43. doi: 10.1002/gcc.2870100403. [DOI] [PubMed] [Google Scholar]
- 6.Alon U, Barkai N, Notterman DA, Gish K, Ybarra S, Mack D, et al. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci U S A. 1999;96:6745–50. doi: 10.1073/pnas.96.12.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casiano CA, Mediavilla-Varela M, Tan EM. Tumor-associated antigen arrays for the serological diagnosis of cancer. Mol Cell Proteomics. United States, 2006:1745-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson KS, LaBaer J. The sentinel within: exploiting the immune system for cancer biomarkers. J Proteome Res. 2005;4:1123–33. doi: 10.1021/pr0500814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58:3491–4. [PubMed] [Google Scholar]
- 10.Nakano O, Sato M, Naito Y, Suzuki K, Orikasa S, Aizawa M, et al. Proliferative activity of intratumoral CD8(+) T-lymphocytes as a prognostic factor in human renal cell carcinoma: clinicopathologic demonstration of antitumor immunity. Cancer Res. 2001;61:5132–6. [PubMed] [Google Scholar]
- 11.Schumacher K, Haensch W, Röefzaad C, Schlag PM. Prognostic significance of activated CD8(+) T cell infiltrations within esophageal carcinomas. Cancer Res. 2001;61:3932–6. [PubMed] [Google Scholar]
- 12.Eerola AK, Soini Y, Pääkkö P. A high number of tumor-infiltrating lymphocytes are associated with a small tumor size, low tumor stage, and a favorable prognosis in operated small cell lung carcinoma. Clin Cancer Res. 2000;6:1875–81. [PubMed] [Google Scholar]
- 13.Sahin U, Türeci O, Schmitt H, Cochlovius B, Johannes T, Schmits R, et al. Human neoplasms elicit multiple specific immune responses in the autologous host. Proc Natl Acad Sci U S A. 1995;92:11810–3. doi: 10.1073/pnas.92.25.11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stempfer R, Syed P, Vierlinger K, Pichler R, Meese E, Leidinger P, et al. Tumour auto-antibody screening: performance of protein microarrays using SEREX derived antigens. BMC Cancer. England, 2010:627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki A, Iizuka A, Komiyama M, Takikawa M, Kume A, Tai S, et al. Identification of melanoma antigens using a Serological Proteome Approach (SERPA). Cancer Genomics Proteomics. Greece, 2010:17-23. [PubMed] [Google Scholar]
- 16.Jongeneel V. Towards a cancer immunome database. Cancer Immun. United States, 2001:3. [PubMed] [Google Scholar]
- 17.Uemura M, Nouso K, Kobayashi Y, Tanaka H, Nakamura S, Higashi T, et al. Identification of the antigens predominantly reacted with serum from patients with hepatocellular carcinoma. Cancer. 2003;97:2474–9. doi: 10.1002/cncr.11374. [DOI] [PubMed] [Google Scholar]
- 18.Scanlan MJ, Gout I, Gordon CM, Williamson B, Stockert E, Gure AO, et al. Humoral immunity to human breast cancer: antigen definition and quantitative analysis of mRNA expression. Cancer Immun. United States, 2001:4. [PubMed] [Google Scholar]
- 19.Jochmus I, Osen W, Altmann A, Buck G, Hofmann B, Schneider A, et al. Specificity of human cytotoxic T lymphocytes induced by a human papillomavirus type 16 E7-derived peptide. J Gen Virol. 1997;78:1689–95. doi: 10.1099/0022-1317-78-7-1689. [DOI] [PubMed] [Google Scholar]
- 20.Jäger D, Stockert E, Güre AO, Scanlan MJ, Karbach J, Jäger E, et al. Identification of a tissue-specific putative transcription factor in breast tissue by serological screening of a breast cancer library. Cancer Res. 2001;61:2055–61. [PubMed] [Google Scholar]
- 21.Ishikawa T, Fujita T, Suzuki Y, Okabe S, Yuasa Y, Iwai T, et al. Tumor-specific immunological recognition of frameshift-mutated peptides in colon cancer with microsatellite instability. Cancer Res. 2003;63:5564–72. [PubMed] [Google Scholar]
- 22.Ang HC, Joerger AC, Mayer S, Fersht AR. Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J Biol Chem. United States, 2006:21934-41. [DOI] [PubMed] [Google Scholar]
- 23.Andreyev HJ, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, et al. Kirsten ras mutations in patients with colorectal cancer: the 'RASCAL II' study. Br J Cancer. Scotland: 2001 Cancer Research Campaign., 2001:692-6. [Google Scholar]
- 24.Scott H. What transcripts are found in a human cell? Genome Biology 2000, 1:reports031. http://genomebiology.com/2000/1/1/reports/031
- 25.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. Landscape of transcription in human cells. Nature. 2012;489:101–8. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Domenyuk V, Loskutov A, Johnston SA, Diehnelt CW. A technology for developing synbodies with antibacterial activity. PLoS One. 2013;8:e54162. doi: 10.1371/journal.pone.0054162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karlstetter M, Walczak Y, Weigelt K, Ebert S, Van den Brulle J, Schwer H, et al. The novel activated microglia/macrophage WAP domain protein, AMWAP, acts as a counter-regulator of proinflammatory response. J Immunol. United States, 2010:3379-90. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Park JS, Deng JH, Bai Y. Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex. J Bioenerg Biomembr. 2006;38:283–91. doi: 10.1007/s10863-006-9052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herrmann PC, Gillespie JW, Charboneau L, Bichsel VE, Paweletz CP, Calvert VS, et al. Mitochondrial proteome: altered cytochrome c oxidase subunit levels in prostate cancer. Proteomics. 2003;3:1801–10. doi: 10.1002/pmic.200300461. [DOI] [PubMed] [Google Scholar]
- 30.Davies B, Fried M. The structure of the human intron-containing S8 ribosomal protein gene and determination of its chromosomal location at 1p32-p34.1. Genomics. United States, 1993:68-75. [DOI] [PubMed] [Google Scholar]
- 31.Pogue-Geile K, Geiser JR, Shu M, Miller C, Wool IG, Meisler AI, et al. Ribosomal protein genes are overexpressed in colorectal cancer: isolation of a cDNA clone encoding the human S3 ribosomal protein. Mol Cell Biol. 1991;11:3842–9. doi: 10.1128/mcb.11.8.3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye Q, Worman HJ. Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. J Biol Chem. 1996;271:14653–6. doi: 10.1074/jbc.271.25.14653. [DOI] [PubMed] [Google Scholar]
- 33.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci U S A. United States, 2004:9309-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lovering R, Hanson IM, Borden KL, Martin S, O’Reilly NJ, Evan GI, et al. Identification and preliminary characterization of a protein motif related to the zinc finger. Proc Natl Acad Sci U S A. 1993;90:2112–6. doi: 10.1073/pnas.90.6.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee KH, Goan YG, Hsiao M, Lee CH, Jian SH, Lin JT, et al. MicroRNA-373 (miR-373) post-transcriptionally regulates large tumor suppressor, homolog 2 (LATS2) and stimulates proliferation in human esophageal cancer. Exp Cell Res. United States, 2009:2529-38. [DOI] [PubMed] [Google Scholar]
- 36.Hartmann E, Sommer T, Prehn S, Görlich D, Jentsch S, Rapoport TA. Evolutionary conservation of components of the protein translocation complex. Nature. 1994;367:654–7. doi: 10.1038/367654a0. [DOI] [PubMed] [Google Scholar]
- 37.Uchida Y, Chiyomaru T, Enokida H, Kawakami K, Tatarano S, Kawahara K, et al. MiR-133a induces apoptosis through direct regulation of GSTP1 in bladder cancer cell lines. Urol Oncol. 2013;31:115–23. doi: 10.1016/j.urolonc.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 38.Ayer DE, Lawrence QA, Eisenman RN. Mad-Max transcriptional repression is mediated by ternary complex formation with mammalian homologs of yeast repressor Sin3. Cell. United States, 1995:767-76. [DOI] [PubMed] [Google Scholar]
- 39.Arends MJ, McGregor AH, Toft NJ, Brown EJ, Wyllie AH. Susceptibility to apoptosis is differentially regulated by c-myc and mutated Ha-ras oncogenes and is associated with endonuclease availability. Br J Cancer. 1993;68:1127–33. doi: 10.1038/bjc.1993.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niklinski J, Furman M. Clinical tumour markers in lung cancer. Eur J Cancer Prev. 1995;4:129–38. doi: 10.1097/00008469-199504000-00002. [DOI] [PubMed] [Google Scholar]
- 41.Battiste JL, Pestova TV, Hellen CU, Wagner G. The eIF1A solution structure reveals a large RNA-binding surface important for scanning function. Mol Cell. United States, 2000:109-19. [DOI] [PubMed] [Google Scholar]
- 42.Shi H, Wei SH, Leu YW, Rahmatpanah F, Liu JC, Yan PS, et al. Triple analysis of the cancer epigenome: an integrated microarray system for assessing gene expression, DNA methylation, and histone acetylation. Cancer Res. 2003;63:2164–71. [PubMed] [Google Scholar]
- 43.Law CJ, Maloney PC, Wang DN. Ins and outs of major facilitator superfamily antiporters. Annu Rev Microbiol. 2008;62:289–305. doi: 10.1146/annurev.micro.61.080706.093329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cui J, Li F, Wang G, Fang X, Puett JD, Xu Y. Gene-expression signatures can distinguish gastric cancer grades and stages. PLoS One. 2011;6:e17819. doi: 10.1371/journal.pone.0017819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pulaski BA, Ostrand-Rosenberg S. Mouse 4T1 breast tumor model. Curr Protoc Immunol 2001; Chapter 20:Unit 20 2. [DOI] [PubMed] [Google Scholar]
- 46.Reeves JP, Reeves PA, Chin LT. Survival surgery: removal of the spleen or thymus. Curr Protoc Immunol 2001; Chapter 1:Unit 1 10. [DOI] [PubMed] [Google Scholar]
- 47.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Storey JD. A direct approach to false discovery rates. J R Stat Soc, B. 2002;64:479–98. doi: 10.1111/1467-9868.00346. [DOI] [Google Scholar]