

Abstract
A convergent and efficient strategy was developed for the synthesis of lipomannan (LM), useful for vaccine development. Thioglycosides were employed as glycosyl donors to construct two key pseudotrisaccharide and tetramannose intermediates through pre-activation-based glycosylation strategy. These building blocks were then successfully coupled to form the LM core, which was lapidated, phospholipidated, and finally globally deprotected to afford the target molecule. The intermediate LM core involved in this synthesis contained orthogonal protections, which would facilitate its variable modifications for the preparation of other complex LM derivatives and for the synthesis of LM conjugates as LM-based vaccines.
Introduction
Mycobacterium tuberculosis (Mtb) is the causative pathogen of tuberculosis (TB), one of the most detrimental diseases worldwide, which causes more than two million deaths each year. A major virulent factor of Mtb is its cell envelope glycolipids, especially the phosphatidylinositol-anchored lipoglycans, such as lipoarabinomannans (LAMs) and lipomannans (LMs).1,2 LMs have exhibited a variety of bioactivities, such as stimulating proinflammatory cytokine secretion through the toll-like receptor 2/CD14-dependent pathway and inducing macrophage apoptosis and IL-12 expression.3–6 Due to the structural complexity and the intriguing immunoregulatory activities, LMs and related molecules have been the targets of a number of chemical total syntheses.7–18
In an effort to explore LM-derived vaccines, we have developed a highly convergent and efficient strategy for LM synthesis via pre-activation-based iterative one-pot glycosylation using thioglycosides as glycosyl donors.19,20 Many syntheses have proved that this strategy can save time and improve efficiency by abolishing multiple experimental preparation and intermediate separation steps. Our synthetic plan (Scheme 1) was to assemble the target molecule 1 from pseudotrisaccharide 2, tetramannose 3, and phosphoglycerolipid 4. The key intermediate 3 was constructed from two monosaccharide building blocks 6 and 8 via iterative one-pot glycosylation, whereas the orthogonal allyl (All), tert-butyldimethylsilyl (TBS) and para-methoxybenzyl (PMB) protecting groups in 2 allowed for regioselective glycosylation, lipidation, and phospholipidation. Furthermore, the 2-O-positions of 6, 7, 8, and 9 were protected as acetyl esters to safeguard α-selective glycosylation resulting from neighboring group participation.
Scheme 1.

Retrosynthesis of the target molecule 1
Results and Discussion
Mannosyl donors and acceptors 6, 7, 8, and 9 were prepared from 1021 and 1222 according to the procedures shown in Scheme 2. Stannylene acetal-directed benzylation of 10 was selective for the 3-O-position, which was followed by 2-O-acetylation and then regioselective benzylidene ring-opening in the presence of borane tetrahydrofuran complex (BH3·THF) and trimethylsilyl trifluoromethanesulfonate (TMSOTf) to afford 6. Thereafter, the free hydroxyl group in 6 was silylated with TBSCl under the influence of imidazole to obtain 7. Compound 8 was prepared from orthoester 12 following perbenzylation and subsequent thioglycosylation using SnCl4 as the promoter. En route to 9, the 6-O-position of 12 was regioselectively protected with TBS first, followed by benzylation of the remaining free hydroxyl groups to produce 14. Consecutively, the 6-O-TBS group was swapped for an allyl group to distinguish from 7, via sequential desilylation mediated by tetrabutylammonium fluoride (TBAF) and reaction with allyl bromide and sodium hydride. Finally, reaction of 15 with p-thiocresol and SnCl4 gave 9 which was derived from 12 in five steps and a 47% overall yield, involving only two column purification operations.
Scheme 2.

Synthesis of mannose building blocks 6–9
The assembly of pseudotrisaccharide 2 (Scheme 3), which had two mannose residues linked to the inositol 2-O- and 6-O-positions while the 1-O-position was uniquely protected to facilitate subsequently selective deprotection and phospholipidation, commenced from an optically pure 1,2,6-O-differentiated myo-inositol derivative 5 obtained from methyl α-D-glucopyranoside according to a reported procedure.23 Glycosylation of 5 with 7 under the influence of p-toluenesulfenyl triflate (p-TolSOTf) generated in situ from the reaction of p-toluenesulfenyl chloride (p-TolSCl) and silver triflate (AgOTf)19,20 gave α-linked pseudodisaccharide 16 (81%) stereospecifically, as a result of neighboring acetyl group participation in the reaction. The allyl group on the inositol 6-O-position was thereafter removed by Iridium complex-catalyzed olefin rearrangement and then Hg(II)-catalyzed hydrolysis24 to form 17. Mannosylation of 17 with 9 was again promoted by p-TolSOTf to give the desired 2 in an 83% yield. The anomeric C-H coupling constants (1JC,H = 175 and 177 Hz) of 2 confirmed the α-stereochemistry of its glycosidic linkages.25 Cleavage of the allyl protection by the Ir-complex/Hg(II) method mentioned above finally afforded 18, which was ready for further elongation of the glycan to get the target molecule or other LM derivatives.
Scheme 3.

Synthesis of the key pseudotrisaccharide 2
The synthesis of tetramannose 3 via pre-activation-based iterative one-pot glycosylation is outlined in Scheme 4. Pre-activation of the thioglycosyl donors was achieved at −78 °C with p-TolSOTf as the promoter. Glycosylation reactions were furnished using a sterically hindered base, 2,4,6-tri-tert-butylpyrimidine (TTBP), as a scavenger for trifluoromethanesulfonic acid generated from the reactions. Each glycosylation was kept at room temperature for ca. 20 min to endorse complete reaction as shown by TLC. It is noteworthy that 1.0 eq. of p-TolSCl and 0.9 eq. of 6 (relative to the donors) were used in the reactions to further assure complete consumption of the glycosyl acceptor in each step so as to minimize any potential interference with the reactions followed. Clearly, iterative one-pot glycosylation for oligosaccharide synthesis could improve the synthetic efficiency by obviating some time-consuming purification processes.26–28 Thus, after three sequential glycosylation steps, 3 was obtained in 6 h and a 39% overall yield, giving an average of more than 73% yield for each glycosylation step. All of the reactions were proved α-specific (anomeric 1JC,H values of 3 were between 169 and 176 Hz). Although 3 could be directly utilized as a glycosyl donor for the glycosylation of 18, the reactions employing p-TolSCl/AgOTf/TTBP or NIS/AgOTf/TTBP as the promoters gave relatively low yields of the desired product with an orthoester as the main byproduct. Due to neighboring group participation, glycosylation reactions using 2-O-acylated donors, such as 3, usually involve orthoesters as the reaction intermediates, which can be transformed into the desired glycosides in the presence of strong Lewis acids. However, 3 and 18 were both complex and sterically hindered, and the reaction condition was almost neutral, thus the reaction might have stopped at the orthoester stage. To avoid this problem, 3 was converted to trichloroacetimidate 19 via hydrolysis of the thioglycoside in the presence of NIS/AgOTf/TTBP and then reaction of the resultant hemiacetal with trichloroacetonitrile and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU).
Scheme 4.
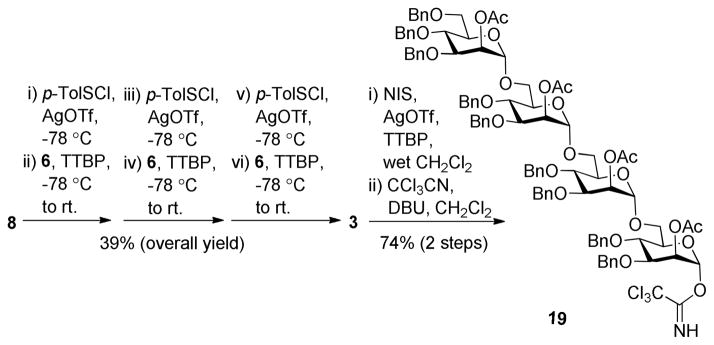
Pre-activation-based iterative one-pot synthesis of 3 and 19
Glycosylation of 18 with 19 in the presence of TMSOTf was α-specific to afford 20 in a 77% yield (Scheme 5). The 13C NMR spectrum of 20 displayed six discrete carbon signals at δ 98.6, 98.3, 98.2, 98.1, 98.0 and 97.9 with 1JC,H values between 172 and 176 Hz. Upon O-deacetylation and benzylation, the acetyl groups in 20 were replaced with benzyl groups to provide 22, which was ready for the installation of lipids. The purpose for the protecting group exchange was to avoid using bases for global deprotection later on, as the target molecule contained ester linkages that were rather sensitive to basic conditions. Treatment of 22 with Et3N·3HF to remove TBS was followed by acylation of the exposed hydroxyl group with stearic acid under the influence of N,N′-dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) and thereafter cleavage of the PMB ether with 10% TFA in CH2Cl2 to generate 25. Compound 25 was smoothly phospholipidated using a two-step one-pot protocol, including reaction with freshly prepared phosphoramidite 4 in the presence of 1H-tetrazole and oxidation of resultant phosphite intermediate with meta-chloroperoxybenzoic acid (m-CPBA) to afford 26 (72%) as a 1:3 diastereomeric mixture, originating from the stereogenic phosphorus atom. Global debenzylation of 26 was achieved in a mixture of chloroform, methanol, and water (3:3:1) under a H2 atmosphere with 10% Pd/C as the catalyst to eventually yield the synthetic target 1, which was characterized with 1H and 31P-NMR spectroscopy and MALDI-TOF MS.
Scheme 5.

Final assembly of the synthetic target 1
Conclusions
In summary, an efficient and convergent strategy was developed for the synthesis of LMs. It is highlighted by the construction of tetramannose 3 through pre-activation-based iterative one-pot glycosylation, which has markedly reduced the number of synthetic and purification steps as compared to the reported syntheses7–18 and thus improved the synthetic efficiency. Furthermore, taking advantage of neighboring group participation, all of the glycosylation reactions involved in the synthesis were stereoselective to form α-glycosidic linkages. The synthetic strategy reported here can be generally applicable to the preparation of various LM derivatives and conjugates. For example, intermediate 22, which contained orthogonal TBS and PMB protecting groups, can be selectively deprotected to facilitate regioselective introduction of linkers for the conjugation with proteins or other carrier molecules to formulate LM-based vaccines or introduction of various lipids or phospholipids at these positions to obtain different LM derivatives. Furthermore, using differently protected monosaccharides from 8 as starting materials for Scheme 4, intermediates suitable for further elongation of the carbohydrate chain of the LM skeleton can be obtained for the synthesis of more complex LM molecules.
Experimental Section
General Experimental Methods
Chemicals and materials were obtained from commercial sources, and were used as received without further purification unless otherwise noted. MS 4Å was flame-dried under high vacuum and used immediately after cooling under a N2 atmosphere. Analytical TLC was carried out on Silica Gel 60Å F254 plates with detection by a UV detector and/or by charring with 15% (v/v) H2SO4 in EtOH. NMR spectra were recorded on a 400, 500 or 600 MHz machine with chemical shifts reported in ppm (δ) downfield from tetramethylsilane (TMS), which was used as an internal reference.
p-Tolyl 2-O-acetyl-3-O-benzyl-4,6-O-benzylidene-1-thio-α-D-mannopyranoside (11):29
The mixture of diol 10 (2.0 g, 5.35 mmol) and Bu2SnO (1.6 g, 6.42 mmol) in toluene (80 mL) was refluxed under a N2 atmosphere for 6 h. After the reaction was cooled to room temperature, toluene was removed under vacuum. The residue was dissolved in 20 mL of anhydrous DMF and mixed with CsF (2.4 g, 16.05 mmol) and BnBr (960 μL, 8.03 mmol). The mixture was stirred at room temperature for 24 h, at the end of which time TLC indicated the complete reaction. The solution was diluted with EtOAc (300 mL) and washed with saturated aq. NaCl. The organic layer was dried with Na2SO4 and evaporated. The residue was dried under high-vacuum for 1 h and then dissolved in 15 mL of pyridine and 2 mL of acetic anhydride. After 1 h of reaction, the mixture was co-evaporated with toluene and the residue was finally purified by silica gel column chromatography with a 1:7 mixture of EtOAc and hexane as eluent to give 11 (2.2 g, 81% for two steps) as syrup. The following NMR data agreed well with that of the reported.29 1H NMR (600 MHz, CDCl3) δ: 7.52-7.49 (m, 2H, Ph), 7.40-7.24 (m, 10H, Ph), 7.12 (d, J = 8.4 Hz, 2H, Ph), 5.63 (s, 1H, PhCH), 5.61 (dd, J = 3.3, 1.0 Hz, 1H, H-2), 5.37 (d, J = 1.0 Hz, 1H, H-1), 4.70 (m, 2H, Bn), 4.36 (dt, J = 10.2, 4.8 Hz, 1H, H-5), 4.22 (dd, J = 10.2, 4.8 Hz, 1H, H-6), 4.12 (t, J = 10.2, 1H, H-4), 4.01 (dd, J = 10.2, 3.3 Hz, 1H, H-3), 3.85 (t, J = 10.2 Hz, 1H, H-6′), 2.32 (s, 3H, Tol), 2.14 (s, 3H, Ac). 13C NMR (150 MHz, CDCl3) δ 170.0, 138.3, 137.7, 137.4, 132.7, 129.9, 128.9, 128.3, 128.1, 127.7, 126.1, 101.6, 87.5 (C-1), 78.5, 74.1, 72.3, 71.3, 68.4, 65.1, 21.1, 21.0. MS (ESI-TOF) m/z: calcd for C29H30O6SNa [M + Na]+ 529.1; found, 528.8.
p-Tolyl 2-O-acetyl-3,4-di-O-benzyl-1-thio-α-D-mannopyranoside (6)
To a solution of 11 (2.0 g, 3.95 mmol) and MS 4Å in 30 mL of anhydrous THF at −40 °C was added BH3·THF (19.75 mL, 19.75 mmol) under a N2 atmosphere. Fifteen minutes later, TMSOTf (924 μL, 5.14 mmol) was added dropwise to this solution. The mixture was stirred under this condition for 1 h, and then stirred at room temperature for another 24 h. The reaction was quenched with saturated aq. NaHCO3 at 0 °C. The solution was diluted with CH2Cl2 (400 mL) and washed with saturated aq. NaHCO3 and NaCl solutions. The organic layer was dried with Na2SO4 and concentrated; the residue was purified by silica gel column chromatography with a mixture of EtOAc and hexane (1:4) as the eluent to give 6 (76%, 1.53 g) as syrup. 1H NMR (400 MHz, CDCl3) δ 7.40-7.29 (m, 12H, Ph), 7.13 (d, J = 8.0 Hz, 2H, Ph), 5.62 (dd, J = 3.2, 1.6 Hz, 1H, H-2), 5.40 (d, J = 1.6 Hz, 1H, H-1), 4.95 (d, J = 11.2 Hz, 1H, Bn), 4.75 (d, J = 11.7 Hz, 1H, Bn), 4.66 (d, J = 10.7 Hz, 1H, Bn), 4.59 (d, J = 11.2 Hz, 1H, Bn), 4.22 (dt, J = 9.6, 3.4 Hz, 1H, H-5), 3.98 (dd, J = 9.4, 3.2 Hz, 1H, H-3), 3.90 (t, J = 9.2 Hz, 1H, H-4), 3.80-3.85 (m, 2H, H-6,6′), 2.33 (s, 3H, Tol), 2.14 (s, 3H, Ac). 13C NMR (100 MHz, CDCl3) δ 170.2, 138.3, 138.2, 137.6, 132.8, 129.9, 129.4, 128.5, 128.2, 128.0, 127.9, 127.8, 86.6 (C-1), 78.3, 75.3, 74.3, 72.9, 71.9, 70.3, 70.2, 62.0, 21.1. HR MS (ESI-TOF) m/z: calcd for C29H32O6SNa [M + Na]+ 531.1817; found, 531.1824. MS (ESI-TOF): found, 530.9.
p-Tolyl 2-O-acetyl-3,4-di-O-benzyl-6-O-tert-butydimethylsilyl-1-thio-α-D-mannopyranoside (7)
To a solution of 6 (388 mg, 0.764 mmol) in anhydrous DMF (10 mL) was added imidazole (104 mg, 1.53 mmol) and TBSCl (192 mg, 1.15 mmol) at 0 °C. The mixture was stirred at room temperature for 2 h and then diluted with EtOAc (150 mL). The organic phase, after being washed with saturated aq. NaCl solution, was dried over Na2SO4, concentrated in vacuo and the residue was purified by silica gel column chromatography with EtOAc and hexane (1:14) as the eluent to give 7 (427 mg, 90%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 7.34-7.27 (m, 12H, Ph), 7.08 (d, J = 7.8 Hz, 2H, Ph), 5.54 (s, 1H, H-2), 5.37 (s, 1H, H-1), 4.90 (d, J = 10.8 Hz, 1H, Bn), 4.71 (d, J = 11.2 Hz, 1H, Bn), 4.64 (d, J = 10.8 Hz, 1H, Bn), 4.56 (d, J = 11.2 Hz, 1H, Bn), 4.10-4.12 (m, 1H, H-5), 3.95-3.91 (m, 3H, H-3,4,6), 3.81 (d, J = 11.4 Hz, 1H, H-6′), 2.30 (s, 3H, Tol), 2.09 (s, 3H, Ac), 0.89 (s, 9H, tBu), 0.03 (s, 3H, SiMe), 0.04 (s, 3H, SiMe). 13C NMR (150 MHz, CDCl3) δ 170.2, 138.6, 137.7, 137.6, 132.1, 130.3, 129.7, 128.4, 128.3, 128.1, 127.9, 127.8, 127.6, 86.5 (C-1), 78.3, 75.2, 74.4, 73.8, 71.9, 70.6, 62.2, 60.0, 25.9, 21.0, 20.9, 18.3, −5.2, −5.4. HR MS (ESI-TOF) m/z: calcd for C35H46O6SSiNa [M + Na]+ 645.2682; found, 645.2689. MS (ESI-TOF): found, 644.9.
p-Tolyl 2-O-acetyl-3,4,6-tri-O-benzyl-1-thio-α-D-mannopyranoside (8):30
To a solution of 12 (410 mg, 1.74 mmol) in anhydrous DMF were added NaH (208 mg, 8.69 mmol) and BnBr (1.04 mL, 8.69 mmol) at 0 °C under N2. Half an hour later, the reaction was quenched with saturated aq. NaCl solution, and the mixture was diluted with EtOAc (150 mL). The organic phase, after being washed with saturated aq. NaCl, was dried over Na2SO4, concentrated in vacuo and the residue was purified by silica gel column with EtOAc and hexane (1:8) as the eluent to give 13 as a pale yellow solid. Compound 13, p-thiocresol (248 mg, 2.0 mmol), and activated 4Å MS were mixed in 20 ml of anhydrous CH2Cl2. Then, a catalytic amount of SnCl4 (300 μL, 0.3 mmol, 1M in CH2Cl2) was added at 0 °C under N2. The mixture was stirred at room temperature for 30 min, and TLC indicated the completion of the reaction. After the reaction was quenched with triethylamine, the solvent was evaporated in vacuum to give the crude oil, which was purified by silica gel column chromatography with EtOAc and hexane (1:10) as the eluent to give 8 (717 mg, 69% for 2 steps) as a white solid. The following NMR data agreed well with that of the reported.30 1H NMR (600 MHz, CDCl3) δ 7.38-7.25 (m, 15H, Ph), 7.20 (d, J = 7.2 Hz, 2H, Ph), 7.05 (d, J = 7.2 Hz, 2H, Ph), 5.60 (d, J = 1.8 Hz, 1H, H-2), 5.46 (s, 1H, H-1), 4.89 (d, J = 10.8 Hz, 1H, Bn), 4.73 (d, J = 10.8 Hz, 1H, Bn), 4.66 (d, J = 12.0 Hz, 1H, Bn), 4.57 (d, J = 11.4 Hz, 1H, Bn), 4.51 (d, J = 10.8 Hz, 1H, Bn), 4.46 (d, J = 12.0 Hz, 1H, Bn), 4.34 (m, 1H, H-5), 3.96-3.93 (m, 2H, H-3,4), 3.85 (ddd, J = 10.8, 4.8, 1.8 Hz, 1H, H-6), 3.73 (d, J = 10.8 Hz, 1H, H-6′), 2.30 (s, 3H), 2.13 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 170.3, 138.3, 138.2, 137.8, 137.6, 132.3, 129.9, 129.8, 128.4, 128.3, 128.2, 128.1, 127.8, 127.7, 127.6, 127.5, 86.5 (C-1), 78.5, 75.2, 74.6, 73.3, 72.4, 71.9, 70.3, 68.9, 21.07, 21.06. MS (ESI-TOF) m/z: calcd for C36H38O6SNa [M + Na]+ 621.2; found, 620.9.
3,4-Di-O-benzyl-6-O-allyl-1,2-O-methoxyethylidene-β-D-mannopyranose (15)
To a solution of 12 (330 mg, 1.40 mmol) in anhydrous DMF (20 mL) was added imidazole (190 mg, 2.80 mmol) and TBSCl (252 mg, 1.68 mmol) at 0 °C. The mixture was stirred at room temperature for 2 h and then diluted with EtOAc (200 mL). The organic phase, after being washed with saturated aq. NaCl solution, was dried over Na2SO4 and concentrated in vacuo to afford a residue, which was dissolved in anhydrous DMF (10 mL) and then mixed with NaH (101 mg, 4.2 mmol) and BnBr (670 μL, 5.6 mmol) at 0 °C under N2. Half an hour later, the reaction was quenched with methanol, diluted with EtOAc (200 mL) and washed with saturated aq. NaCl solution. The organic phase was dried over Na2SO4 and concentrated in vacuo to give the crude 14. To a solution of crude 14 in THF (8 mL) was added TBAF (4.2 mL, 4.2 mmol, 1M in THF). The mixture was stirred at room temperature for 3 h, at which time TLC indicated the completion of the reaction, and then diluted with EtOAc (200 mL). The organic phase, after being washed with saturated aq. NaCl solution, was dried over Na2SO4 and concentrated in vacuo to afford a residue, which was dissolved in anhydrous DMF (10 mL) and mixed with NaH (67 mg, 2.8 mmol) and allyl bromide (242 μL, 2.8 mmol) at 0 °C under N2. An hour later, the reaction was quenched with methanol, diluted with EtOAc (100 mL) and then washed with saturated aq. NaCl solution. The organic phase was dried over Na2SO4, concentrated in vacuo and the product was purified by silica gel column chromatography with EtOAc and hexane (1:8) as the eluent to give 15 (408 mg, 64% for 4 steps) as a white solid. 1H NMR (600 MHz, CDCl3) δ 7.39-7.28 (m, 10H, Ph), 5.87 (m, 1H, All), 5.33 (d, J = 2.4 Hz, 1H, H-1), 5.26 (dd, J = 17.2, 1.8 Hz, 1H, All), 5.14 (dd, J = 10.2, 1.2 Hz, 1H, All), 4.92 (d, J = 10.8 Hz, 1H, Bn), 4.80-4.75 (m, 2H, Bn), 4.66 (d, J = 10.8 Hz, 1H, Bn), 4.38 (dd, J = 3.6, 2.4 Hz, 1H, H-2), 4.04 (dd, J = 13.2, 5.4 Hz, 1H, All), 3.99 (dd, J = 13.2, 5.4 Hz, 1H, All), 3.89 (t, J = 9.6 Hz, 1H, H-4), 3.72-3.63 (m, 3H, H-3,4,6′), 3.39-3.36 (m, 1H, H-5), 3.27 (s, 3H, OMe), 1.73 (s, 3H, Me). 13C NMR (150 MHz, CDCl3) δ 138.3, 137.8, 134.6, 128.5, 128.4, 128.0, 127.9, 127.7, 123.9, 116.6, 97.5 (C-1), 79.0, 77.1, 75.2, 74.2, 74.1, 72.4, 72.3, 68.9, 49.7, 24.3. HR MS (ESI-TOF) m/z: calcd for C26H32O7Na [M + Na]+ 479.2046; found, 479.2052. MS (ESI-TOF): found, 478.9.
p-Tolyl 2-O-acetyl-3,4-di-O-benzyl-6-O-ally-1-thio-α-D-mannopyranoside (9)
To a solution of 15 (400 mg, 0.877 mmol), p-thiocresol (131 mg, 1.05 mmol) and activated MS 4Å in 15 mL of anhydrous CH2Cl2 was added SnCl4 (176 μL, 0.176 mmol, 1M in CH2Cl2) at 0 °C under N2. The reaction mixture was stirred at room temperature for 30 min, at which time TLC indicated the completion of the reaction. The reaction was quenched with triethylamine, and the solvent was evaporated in vacuo. The residue was purified by silica gel column chromatography EtOAc and hexane (1:8) as the eluent to give 9 (344 mg, 74%) as colorless syrup. 1H NMR (400 MHz, CDCl3) δ 7.38-7.30 (m, 12H, Ph), 7.11 (d, J = 8.0 Hz, 2H, Ph), 5.98-5.88 (m, 1H, All), 5.61 (s, 1H, H-2), 5.47 (s, 1H, H-1), 5.30 (dd, J = 17.2, 1.2 Hz, 1H, All), 5.18 (d, J = 10.4 Hz, 1H, All), 4.95 (d, J = 10.8 Hz, 1H, Bn), 4.75 (d, J = 11.2 Hz, 1H, Bn), 4.63 (d, J = 10.8 Hz, 1H, Bn), 4.58 (d, J = 11.2 Hz, 1H, Bn), 4.32 (m, 1H, H-5), 4.11 (dd, J = 12.8, 5.2 Hz, 1H, All), 4.01-3.94 (m, 3H, H-3,4,All), 3.82 (dd, J = 10.8, 4.4 Hz, 1H, H-6), 3.69 (d, J = 10.8 Hz, 1H, H-6′), 2.33 (s, 3H, Tol), 2.16 (s, 3H, Ac). 13C NMR (100 MHz, CDCl3) δ 170.3, 138.4, 137.8, 137.7, 134.7, 132.2, 129.9, 129.8, 128.5, 128.4, 128.2, 127.9, 127.7, 116.9, 86.6 (C-1), 78.5, 75.3, 74.6, 72.36, 72.34, 71.9, 70.4, 68.9, 21.1. HR MS (ESI-TOF) m/z: calcd for C32H36O6SNa [M + Na]+ 571.2130; found, 571.2137. MS (ESI-TOF): found, 570.9.
Pre-activation-based glycosylation (General Procedure A)
After a solution of glycosyl donor (1.1 eq.) and acceptor (1.0 eq.) in anhydrous CH2Cl2 mixed with TTBP (1.0 eq.) and MS 4Å was stirred at room temperature for 40 min, it was cooled to −78 °C. Then, a solution of AgOTf (3.3 eq.) in acetonitrile was added. Fifteen minutes later, p-TolSCl (1.1 eq.) was added dropwise. The reaction mixture was warmed to room temperature slowly within 1 h and stirred for another 20 min. The reaction was quenched with Et3N, and the mixture was diluted with CH2Cl2 and filtered. The filtrate was concentrated in vacuum, and the resultant residue was purified by flash silica gel column chromatography with a mixture of EtOAc and toluene as eluent to afford the desired product.
Ir-complex/Hg(II)-catalyzed removal of the allyl group (General Procedure B)
The solution of [Ir(COD)(PMePh2)2]PF6 (0.15 eq.) in anhydrous THF was stirred under a H2 atmosphere at room temperature until the red color turned to pale yellow (in ca. 15 min). Then, H2 was exchanged with Argon for three times, before a solution of the allyl protected intermediate (1.0 eq.) in anhydrous THF was added slowly. The reaction mixture was stirred at room temperature for 40 min, at which point TLC showed the complete reaction. The reaction mixture was concentrated in vacuum, and after the residue was dissolved in acetone and water (9:1, V/V), the solution was treated with HgCl2 (5.0 eq.) and HgO (0.15 eq.). Ten minutes later, the solution was concentrated and the residue was purified by silica gel column chromatography to give the desired product.
(2-O-Acetyl-3,4-di-O-benzyl-6-O-tert-butydimethylsilyl-α-D-mannopyranosyl)-(1→2)-1-O-(para-methoxybenzyl)-3,4,5-tri-O-benzyl-6-O-allyl-D-myo-inositol (16)
General Procedure A was used to prepare 16 (279 mg, 81%) from 7 (213 mg, 0.342 mmol) and 5 (190 mg, 0.311 mmol). 1H NMR (600 MHz, CDCl3) δ 7.35-7.19 (m, 27H), 6.86 (d, J = 8.4 Hz, 2H), 5.99-5.93 (m, 1H, All), 5.42 (s, 1H), 5.28 (dd, J = 17.4, 1.8 Hz, 1H, All), 5.20 (d, J = 1.2 Hz, 1H, Man H-1), 5.17 (dd, J = 10.8, 1.2 Hz, 1H, All), 4.86-4.53 (m, 12H), 4.38 (dd, J = 12.0, 6.0 Hz, 1H), 4.32 (dd, J = 12.0, 6.0 Hz, 1H), 4.27 (s, 1H), 3.98-3.92 (m, 3H), 3.81-3.76 (m, 5H), 3.67 (dd, J = 12.0, 2.4 Hz, 1H), 3.44 (d, J = 10.8 Hz, 1H), 3.37 (t, J = 9.6 Hz, 1H), 3.27 (m, 2H), 2.05 (s, 3H), 0.89 (s, 9H), 0.04 (s, 3H), 0.007 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 169.9, 159.1, 139.1, 138.7, 138.6, 138.1, 138.0, 135.4, 130.3, 129.0, 128.3, 128.2, 128.1, 128.0, 127.9, 127.6, 127.4, 127.3, 127.1, 116.7, 113.7, 98.5 (Man C-1, JCH = 177 Hz), 83.4, 81.1, 81.0, 80.3, 78.9, 77.6, 76.1, 75.7, 75.0, 74.6, 73.9, 72.37, 72.35, 72.0, 71.8, 68.8, 61.8, 55.2, 25.9, 20.9, 18.3, −5.1, −5.4. HR MS (ESI-TOF) m/z: calcd for C66H80O13SiNa [M + Na]+ 1131.5266; found, 1131.5243. MS (MALDI-TOF): found, 1132.4.
(2-O-Acetyl-3,4-di-O-benzyl-6-O-tert-butydimethylsilyl-α-D-mannopyranosyl)-(1→2)-1-O-(para-methoxybenzyl)-3,4,5-tri-O-benzyl-D-myo-inositol (17)
General Procedure B was used to prepare 17 (197 mg, 89%) from 16 (230 mg, 0.208 mmol). 1H NMR (400 MHz, CDCl3) δ 7.38-7.20 (m, 27H), 6.87 (d, J = 8.4 Hz, 2H), 5.40 (t, J = 1.6 Hz, 1H), 5.16 (d, J = 1.6 Hz, 1H, Man H-1), 4.89-4.51 (m, 12H), 4.32 (t, J = 2.0 Hz, 1H), 4.02-3.94 (m, 4H), 3.84-3.79 (m, 4H), 3.69 (dd, J = 11.6, 2.4 Hz, 1H), 3.46 (d, J = 10.8 Hz, 1H), 3.36-3.29 (m, 2H), 3.17 (dd, J = 9.6, 2.0 Hz, 1H), 2.07 (s, 3H), 0.89 (s, 9H, tBu), 0.04 (s, 3H, SiMe), 0.01 (s, 3H, SiMe). 13C NMR (100 MHz, CDCl3) δ 170.0, 159.4, 139.0, 138.7, 138.6, 138.1, 138.0, 129.7, 129.4, 128.5, 128.4, 128.3, 128.1, 128.0, 127.9, 127.7, 127.6, 127.5, 127.2, 113.9, 98.5 (Man C-1), 83.2, 80.9, 79.7, 79.1, 77.6, 75.7, 75.6, 75.1, 73.9, 72.9, 72.48, 72.44, 72.0, 71.9, 71.3, 68.9, 61.8, 55.3, 25.9, 21.0, 18.3, −5.0, −5.3. HR MS (ESI-TOF) m/z: calcd for C63H76O13SiNa [M + Na]+ 1091.4953; found, 1091.4956. MS (MALDI-TOF): found, 1092.1.
(2-O-Acetyl-3,4-di-O-benzyl-6-O-allyl-α-D-mannopyranosyl)-(1→6)-3,4,5-tri-O-benzyl-2-O-(2-O-acetyl-3,4-di-O-benzyl-6-O-tert-butydimethylsilyl-α-D-mannopyranosyl)-1-O-(para-methoxybenzyl)-D-myo-inositol (2)
General Procedure A was used to parepare 2 (173 mg, 83 %) from 9 (84 mg, 0.154 mmol) and 17 (150 mg, 0.140 mmol). 1H NMR (500 MHz, CDCl3) δ 7.42-7.17 (m, 35H), 7.11 (t, J = 7.5 Hz, 2H), 6.88 (d, J = 8.5 Hz, 2H), 5.80 (m, 1H, H-All), 5.49 (m, 2H), 5.46 (s, 1H, ManA H-1), 5.16 (m, 2H, ManB H-1, H-All), 5.05 (d, J = 10.0 Hz, 1H, H-All), 4.95-4.48 (m, 16H), 4.32 (s, 1H), 4.08 (t, J = 9.5 Hz, 1H), 4.02-3.94 (m, 7H), 3.88 (t, J = 9.5 Hz, 1H), 3.82 (s, 3H), 3.75-3.70 (m 2H), 3.52 (d, J = 11.5 Hz, 1H), 3.36-3.24 (m, 5H), 2.10 (s, 6H), 0.92 (s, 9H), 0.06 (s, 3H), 0.02 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 170.3, 169.7, 159.4, 139.2, 139.0, 138.4, 138.2, 138.1, 137.9, 134.9, 129.9, 129.2, 128.6, 128.3, 128.2, 128.1, 128.0, 127.9, 127.6, 127.5, 127.3, 127.2, 116.6, 113.8, 98.6 (ManB C-1), 98.3 (ManA C-1), 81.4, 80.9, 78.9, 78.2, 77.4, 76.0, 75.7, 75.5, 75.06, 75.00, 74.1, 73.8, 72.6, 72.4, 72.3, 71.7, 71.6, 71.3, 70.6, 68.6, 68.5, 68.1, 61.9, 55.2, 25.9, 21.2, 21.0, 18.3, −5.1, −5.3. HR MS (ESI-TOF) m/z: calcd for C88H104O19SiNa [M + Na]+ 1515.6839; found, 1515.6865. MS (MALDI-TOF): found, 1516.6.
(2-O-Acetyl-3,4-di-O-benzyl-α-D-mannopyranosyl)-(1→6)-3,4,5-tri-O-benzyl-2-O-(2-O-acet yl-3,4-di-O-benzyl-6-O-tert-butydimethylsilyl-α-D-mannopyranosyl)-1-O-(para-methoxyben zyl)-D-myo-inositol (18)
General Procedure B was used to prepare 18 (124 mg, 91 %) from 2 (140 mg, 0.094 mmol). 1H NMR (500 MHz, CDCl3) δ 7.40-7.19 (m, 35H), 7.10 (t, J = 7.5 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 5.49 (dd, J = 2.5, 1.0 Hz, 1H), 5.46-5.43 (m, 2H, ManA H-1, ManB H-2), 5.19 (d, J = 1.0 Hz, 1H, ManB H-1), 4.92-4.47 (m, 16H), 4.35 (s, 1H), 4.10 (t, J = 9.5 Hz, 1H), 4.02-3.91 (m, 5H), 3.87 (t, J = 9.5 Hz, 1H), 3.83-3.78 (m, 4H), 3.76 (dd, J = 11.0, 2.0 Hz, 1H), 3.55 (d, J = 11.5 Hz, 1H), 3.49 (dd, J = 12.0, 2.0 Hz, 1H), 3.40 (dd, J = 12.0, 3.0 Hz, 1H), 3.35-3.27 (m, 3H), 2.11 (s, 3H), 2.09 (s, 3H), 0.92 (s, 9H), 0.08 (s, 3H), 0.04 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 170.2, 170.1, 159.4, 138.9, 138.8, 138.4, 138.1, 138.0, 137.8, 130.0, 129.1, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.6, 127.5, 127.4, 127.3, 113.8, 98.2 (ManB C-1), 98.0 (ManA C-1), 81.5, 81.4, 80.9, 78.8, 77.9, 76.2, 75.7, 75.1, 74.0, 73.9, 72.6, 72.5, 71.6, 71.5, 70.0, 68.7, 68.6, 61.9, 61.4, 55.2, 25.9, 21.1, 21.0, 18.3, −5.1, −5.3. HR MS (ESI-TOF) m/z: calcd for C85H100O19SiNa [M + Na]+ 1475.6526; found, 1475.6366. MS (MALDI-TOF): found, 1476.4.
p-Tolyl (2-O-acetyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2-O-acetyl-3,4 -di-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2-O-acetyl-3,4-di-O-benzyl-α-D-mannopyranosyl)-(1→6)-3,4-di-O-benzyl-2-O-acetyl-α-D-mannopyranoside (3)
A mixture of 8 (370 mg, 0.619 mmol) and activated MS 4Å in anhydrous CH2Cl2 (8 mL) was stirred at room temperature for 40 min, and then cooled to −78 °C. A solution of AgOTf (477 mg, 1.856 mmol) in acetonitrile (1.5 mL) was added. After 10 min of stirring, p-TolSCl (89 μL, 0.619 mmol) was added dropwise. Fifteen minutes later, a solution of 6 (286 mg, 0.563mmol) and TTBP (140 mg, 0.563 mmol) in anhydrous CH2Cl2 (1.5 mL) was added. The reaction mixture was warmed to room temperature slowly in 1 h, stirred for another 20 min, and then cooled to −78 °C, which was followed by the same sequence of addition of AgOTf (434 mg, 1.689 mmol) in acetonitrile (1 mL), p-TolSCl (81 μL, 0.563 mmol), a solution of 6 (260 mg, 0.512 mmol) and TTBP (127 mg, 0.512 mmol) in anhydrous CH2Cl2 (1.5 mL). The reaction mixture was warmed to room temperature slowly in 1 h, stirred for another 20 min, and then cooled to −78 °C, and again was followed by the same sequential addition of AgOTf (395 mg, 1.536 mmol) in acetonitrile (1 mL), p-TolSCl (74 μL, 0.512 mmol), a solution of 6 (236 mg, 0.465 mmol) and TTBP (115 mg, 0.465 mmol) in anhydrous CH2Cl2 (1.5 mL). The reaction mixture was warmed to room temperature slowly in 1 h, stirred for another 20 min, and then quenched with Et3N, diluted with CH2Cl2, and finally filtered. The filtrate was concentrated in vacuum, and the residue purified by silica gel column chromatography with EtOAc and toluene (1:12) as the eluent to give 3 (317 mg, 39% for three glycosylation steps) as a foamy solid. 1H NMR (600 MHz, CDCl3) δ 7.35-7.08 (m, 49H), 5.61 (dd, J = 2.5, 1.2 Hz, 1H), 5.47 (m, 2H), 5.46 (dd, J = 2.5, 1.2 Hz, 1H), 5.38 (s, 1H, ManD C-1), 4.98 (s, 1H, ManC H-1), 4.93-4.82 (m, 6H, ManA H-1, ManB H-1, 4 × Bn-CH2-), 4.74-4.39 (m, 14H), 4.31 (dd, J = 10.2, 4.8 Hz, 1H), 3.98 (dd, J = 9.6, 3.0 Hz, 1H), 3.96-3.80 (m, 8H), 3.77 (dd, J = 11.4, 3.0 Hz, 1H), 3.74-3.68 (m, 2H), 3.68-3.58 (m, 4H), 3.57-3.50 (m, 3H), 2.19 (s, 3H), 2.15 (s, 3H), 2.14 (s, 6H), 2.13 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 170.3, 170.2, 170.19, 170.18, 138.5, 138.4, 138.2, 137.9, 137.7, 137.6, 137.5, 132.0, 129.9, 128.5, 128.4, 128.3, 128.2, 127.9, 127.8, 127.7, 127.6, 127.5, 127.4, 127.3, 98.0 (ManC C-1), 97.98 (ManA C-1), 97.97 (ManB C-1), 86.6 (ManD C-1), 78.5, 77.9, 77.7, 77.6, 75.2, 75.1, 75.0, 74.9, 74.3, 74.1, 73.8, 73.7, 73.4, 72.1, 71.8, 71.6, 71.4, 71.3, 71.15, 71.11, 70.2, 68.6, 68.2, 68.1, 68.0, 66.3, 65.5, 65.3, 29.7, 21.1, 21.04, 21.03, 21.00. HR MS (ESI-TOF) m/z: calcd for C102H110O24SNa [M + Na]+ 1773.7005; found, 1773.7000. MS (MALDI-TOF): found, 1774.6.
6-O-[(2-O-acetyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2-O-acetyl-3,4-di-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2-O-acetyl-3,4-di-O-benzyl-α-D-mannopyranosyl)]-3,4-di-O-benzyl-2-O-acetyl-α-D-mannopyranosyl trichloroacetimidate (19)
To a solution of 3 (310 mg, 0.177 mmol) and TTBP (132 mg, 0.532 mmol) in wet CH2Cl2 was added N-iodosuccinimide (80 mg, 0.354 mmol) and silver triflate (91 mg, 0.354 mmol) at 0 °C. The reaction mixture was stirred at room temperature for 2 h, quenched with saturated aq. Na2S2O3 solution, and diluted with CH2Cl2 (150 mL). The organic phase, after being washed with saturated aq. NaCl solution and H2O, was dried over Na2SO4 and concentrated in vacuo to afford a residue that was purified with silica gel column chromatography with EtOAc and toluene (1:5) as the eluent to give a white solid. It was dissolved in 10 mL of anhydrous CH2Cl2, and then DBU (14 μL, 0.089 mmol) and trichloroacetonitrile (91μL, 0.89 mmol) were added at 0 °C. After 1.5 h of stirring, the mixture was concentrated in vacuum and the residue was purified on a Et3N-neutralized silica gel column with EtOAc and toluene (1:8) as the eluent to give 19 (234 mg, 74% for two steps) as a solid. 1H NMR (600 MHz, CDCl3) δ 8.67 (s, 1H), 7.32-7.07 (m, 45H), 6.19 (d, J = 1.8 Hz, 1H), 5.48 (dd, J = 3.0, 1.8 Hz, 1H), 5.46 (dd, J = 3.0, 1.8 Hz, 1H), 5.45 (dd, J = 3.0, 1.8 Hz, 1H), 5.41 (dd, J = 3.0, 1.8 Hz, 1H), 4.98 (d, J = 1.2 Hz, 1H, Man H-1), 4.91-4.38 (m, 20H, 3 × Man H-1, 17 × Bn-CH2-), 4.02 (dd, J = 9.0, 3.0 Hz, 1H), 3.94-3.87 (m, 4H), 3.86-3.86 (m, 9H), 3.67 – 3.59 (m, 3H), 3.56 (d, J = 11.4 Hz, 2H), 3.51 (d, J = 10.8, 1.2 Hz, 1H), 2.19 (s, 3H), 2.15 (s, 3H), 2.13 (s, 3H), 2.12 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 170.3, 170.2, 170.17, 170.15, 159.6, 138.4, 138.2, 137.9, 137.7, 137.6, 137.4, 128.4, 128.3, 128.2, 127.9, 127.8, 127.5, 127.4, 98.0 (Man C-1), 97.9 (Man C-1), 97.6 (Man C-1), 94.9 (Man C-1), 90.7, 77.8, 77.7, 77.6, 77.5, 75.3, 75.1, 74.97, 74.95, 74.1, 73.9, 73.76, 73.72, 73.4, 73.3, 72.0, 71.5, 71.45, 71.41, 71.13, 71.11, 68.6, 68.2, 68.1, 68.0, 67.2, 65.42, 65.40, 65.3, 21.1, 21.04, 21.03, 20.9.
6-O-[(2-O-acetyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2-O-acetyl-3,4-di-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2-O-acetyl-3,4-di-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2-O-acetyl-3,4-di-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2-O-acetyl-3,4-di-O-benzyl-α-D-mannopyranosyl)]-3,4,5-tri-O-benzyl-2-O-(2-O-acetyl-3,4-di-O-benzyl-6-O-tert-butyldimethylsilyl-α-D-mannopyranosyl)-1-O-(para-methoxybenzyl)-D-myo-inositol (20)
To a stirred mixture of 18 (60 mg, 0.041 mmol), 19 (96 mg, 0.054 mmol), and MS 4Å in anhydrous CH2Cl2 (4 mL) was added TMSOTf (1 μL, 5.4 μmol) under N2 protection at 0 °C. After the reaction mixture was stirred for another 30 min, it was neutralized with Et3N, filtered and concentrated. The residue was subjected to silica column chromatography with EtOAc and toluene (1:10) as the eluent to afford 20 (97 mg, 77%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 7.38 (d, J = 7.8 Hz, 2H), 7.35-7.01 (m, 78H), 6.99 (d, J = 7.2 Hz, 2H), 6.84 (d, J = 7.2 Hz, 2H), 5.52 (s, 1H), 5.49 (m, 4H), 5.41 (s, 1H), 5.38 (s, 1H, ManA H-1), 5.14 (s, 1H, ManB H-1), 5.01 (d, J = 10.8 Hz, 1H), 4.97 (s, 1H, ManC H-1), 4.93-4.20 (m, 36H, 3 × Man H-1, 33 × Bn-CH2-), 4.01-3.69 (m, 21H), 3.64-3.58 (m, 3H), 3.51-3.37 (m, 7H), 3.31-3.22 (m, 6H), 3.15 (d, J = 12.0 Hz, 1H), 3.12 (d, J = 12.0 Hz, 1H), 2.14 (s, 3H), 2.11 (s, 3H), 2.10 (s, 9H), 2.05 (s, 3H), 0.88 (s, 9H), 0.03 (s, 3H), −0.003 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 170.18, 170.13, 170.0, 169.9, 169.7, 159.4, 139.0, 138.9, 138.6, 138.5, 138.3, 138.2, 138.1, 138.0, 137.9, 137.8, 137.7, 137.5, 130.0, 129.0, 128.7, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.6, 127.5, 127.3, 127.2, 127.1, 126.9, 126.6, 113.8, 98.6 (ManA C-1), 98.3 (ManB C-1), 98.2 (Man C-1), 98.1 (ManC C-1), 98.0 (Man C-1), 97.9 (Man C-1), 81.4, 81.3, 80.6, 78.8, 77.8, 77.7, 77.67, 77.61, 76.4, 75.7, 75.1, 74.9, 74.8, 74.6, 74.4, 74.0, 73.8, 73.6, 73.5, 73.4, 73.3, 72.6, 71.68, 71.65, 71.5, 71.44, 71.40, 71.3, 71.2, 72.1, 71.0, 70.8, 70.5, 69.9, 68.6, 68.5, 68.2, 67.9, 67.8, 67.7, 65.3, 65.2, 61.9, 55.2, 25.9, 21.1, 21.09, 21.03, 21.00, 18.3, −5.1, −5.4. HR MS (ESI-TOF) m/z: calcd for C180H202O43SiNa2 [M + 2Na]2+ 1562.6593; found, 1562.6454. MS (MALDI-TOF) m/z: calcd for C180H202O43SiNa [M + Na]+ 3102.3; found, 3103.2.
6-O-[(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)]-3,4,5-tri-O-benzyl-2-O-(2,3,4-tri-O-benzyl-6-O-tert-butyldimethylsilyl-α-D-mannopyranosyl)-1-O-(para-methoxybenzyl)-D-myo-inositol (22)
To a solution of 20 (90 mg, 0.029 mmol) in CH2Cl2 and CH3OH (1:2, 9 mL) was added CH3ONa solution (0.5 M in CH3OH) until pH reached 11. The solution was stirred at 35 °C for 24 h before the solvent was removed in vacuo. The residue was dissolved in a mixture of CH2Cl2 and CH3OH (1:10) and filtered, and the filtrate was concentrated under vacuum to give crude 21. To a solution of crude 21 in dry DMF was added BnBr (42 μL, 0.35 mmol) and TBAI (4.0 mg, 10.8 μmol). After the mixture was stirred for 10 min, NaH was added (5.6 mg, 0.232 mmol) at 0 °C, which was stirred for 1.5 h. Then, MeOH was added to quench the reaction before water was added. The aqueous phase was extracted with CH2Cl2 (3×50 mL), and the organic layer was dried over Na2SO4, concentrated. The residue was purified by silica gel column chromatography with EtOAc and toluene (1:16) as the eluent to give 22 (66 mg, 67% for two steps) as syrup. 1H NMR (600 MHz, CDCl3) δ 7.40-6.98 (m, 112H), 6.58 (d, J = 8.4 Hz, 2H), 5.39 (s, 1H, ManA H-1), 5.29 (s, 1H, ManF H-1), 5.09 (s, 1H, ManE H-1), 5.02 (d, J = 11.4 Hz, 1H), 5.00 (s, 1H, ManC H-1), 4.94 (s, 1H, ManB H-1), 4.93-4.27 (m, 46H, ManD H-1, 45 × Bn-CH2-), 4.05 (t, J = 9.6 Hz, 1H), 4.03-3.78 (m, 21H), 3.73 (s, 1H), 3.68 (d, J = 9.6 Hz, 2H), 3.62-3.54 (m, 6H), 3.53-3.48 (m, 3H), 3.46-3.43 (m, 2H), 3.38 (d, J = 9.0 Hz, 1H), 3.35-3.25 (m, 6H), 3.17 (d, J = 11.4 Hz, 1H), 3.10 (d, J = 10.8 Hz, 1H), 0.85 (s, 9H), −0.002 (s, 3H), −0.01 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 159.4, 139.1, 138.9, 138.8, 138.7, 138.6, 138.5, 138.3, 138.2, 138.1, 137.9, 129.5, 129.0, 128.5, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.6, 127.5, 127.4, 127.3, 127.2, 127.1, 126.9, 126.8, 113.9, 99.1 (ManA C-1), 98.6 (ManB C-1), 98.4 (ManC C-1), 98.2 (ManD C-1), 98.15 (ManE C-1), 98.10 (ManF C-1), 82.0, 81.4, 80.6, 79.6, 79.4, 79.3, 79.2, 79.1, 78.9, 76.0, 75.9, 75.7, 75.6, 75.0, 74.92, 74.90, 74.8, 74.7, 74.67, 74.62, 74.52, 74.50, 74.4, 74.2, 73.9, 73.8, 73.6, 73.2, 72.8, 72.63, 72.61, 72.5, 72.4, 72.3, 72.2, 72.1, 71.9, 71.8, 71.5, 71.4, 71.26, 71.22, 71.20, 71.1, 71.0, 69.0, 65.8, 65.7, 65.6, 62.2, 55.0, 53.4, 25.9, 18.3, −5.1, −5.4.HR MS (ESI-TOF) m/z: calcd for C210H226O37SiNa2 [M + 2Na]2+ 1706.7684; found, 1706.7743. MS (MALDI-TOF) m/z: calcd for C210H226O37SiNa [M + Na]+ 3390.5; found, 3291.2.
6-O-[(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)]-3,4,5-tri-O-benz yl-2-O-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)-1-O-(para-methoxybenzyl)-D-myo-inositol (23)
A solution of 22 (60.0 mg, 0.018 mmol) in THF and CH3CN (2:13, mL) and triethylamine trihydrofluoride (1.0 mL) was stirred at room temperature overnight under Ar. The solution was quenched with dropwise addition of saturated aq. NaHCO3 solution. The aq. phase was extracted with CH2Cl2 (3×40 mL), and the organic layer was dried over Na2SO4, concentrated. The residue was purified by silica gel column chromatography with EtOAc and toluene (1:10) as the eluent to give 23 (50.4 mg, 87%) as syrup. 1H NMR (600 MHz, CDCl3) δ 7.40-7.04 (m, 112H), 6.65 (dd, J = 7.8 Hz, 2H), 5.44 (d, J = 3.6 Hz, 1H, ManA H-1), 5.21 (d, J = 4.2 Hz, 1H, ManB H-1), 5.12 (d, J = 4.2 Hz, 1H, ManE H-1), 5.07-5.04 (d, J = 10.8 Hz, 1H), 5.02 (d, J = 3.6 Hz, 1H, ManD H-1), 4.96-4.85 (m, 8H, ManC H-1, ManF H-1, 6 × Bn-CH2-), 4.77 (d, J = 10.8 Hz, 1H), 4.72-4.29 (m, 38H), 4.07-3.42 (m, 36H), 3.40 (d, J = 8.4 Hz, 1H), 3.36-3.25 (m, 6H), 3.18 (d, J = 10.8 Hz, 1H), 3.12 (d, J = 10.8 Hz, 1H), 2.11 (s, 1H). 13C NMR (150 MHz,CDCl3) δ 159.4, 138.9, 138.8, 138.7, 138.6, 138.5, 138.4, 138.3, 138.2, 138.1, 138.0, 137.7, 129.2, 128.9, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.6, 127.5, 127.4, 127.3, 127.2, 127.1, 127.0, 126.8, 113.9, 99.1 (ManA C-1), 99.0 (ManB C-1), 98.7 (ManC C-1), 98.4 (ManD C-1), 98.3 (ManE C-1), 98.2 (ManF C-1), 81.8, 81.3, 80.7, 79.6, 79.4, 79.3, 79.2, 79.0, 78.7, 75.9, 75.8, 75.7, 75.1, 74.9, 74.8, 74.7, 74.6, 74.5, 74.2, 74.0, 73.9, 73.7, 73.2, 72.8, 72.6, 72.5, 72.4, 72.26, 72.21, 72.1, 72.0, 71.8, 71.6, 71.5, 71.3, 71.2, 71.1, 70.9, 69.1, 65.8, 65.7, 65.6, 62.1, 55.1. HR MS (ESI-TOF) m/z: calcd for C204H212O37Na2 [M + 2Na]2+ 1649.7252; found, 1649.7317. MS (MALDI-TOF) m/z: calcd for C204H212O37Na [M + Na]+ 3276.4; found, 3277.1.
6-O-[(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)]-3,4,5-tri-O-benzyl-2-O-(2,3,4-tri-O-benzyl-6-O-stearoyl-α-D-mannopyranosyl)-D-myo-inositol (25)
To a solution of 23 (49.0 mg, 0.015 mmol) in anhydrous CH2Cl2 (2 mL) was added stearic acid (21.3 mg, 0.075 mmol), DCC (15.5 mg, 0.075 mmol), and DMAP (9.2 mg, 0.075 mmol) at room temperature. After having been stirred overnight, the mixture was filtered off through a Celite pad, and the filtrate was concentrated to get a residue that was purified by silica gel column chromatography with toluene and EtOAc (15:1) as the eluent to give 24 as a white solid. To a solution of 24 in CH2Cl2 (1.5 mL) was added 20% TFA/CH2Cl2, giving a final concentration of about 10% of TFA. The mixture was stirred for 3 h, at which time TLC indicated the completion of the reaction. The solution was co-evaporated with toluene 3 times to remove TFA completely. Purification of the residue by silica gel column chromatography with EtOAc and toluene (1:12) as the eluent gave 25 (35.3 mg, 69% from 23) as a white solid. 1H NMR (600 MHz, CDCl3) δ 7.31-7.01 (m, 110H), 5.36 (s, 1H, ManA H-1), 5.09 (s, 1H, ManB H-1), 5.04 (s, 1H, ManF H-1), 4.96 (s, 2H, ManC H-1, ManD H-1), 4.85-4.73 (m, 8H, ManE H-1, 7 × Bn-CH2-), 4.65 (d, J = 10.8 Hz, 1H), 4.60-4.27 (m, 36H), 4.16 (s, 1H), 4.08 (d, J = 9.6 Hz, 1H), 4.05 (dd, J = 12.0, 3.6 Hz, 1H), 3.96-3.64 (m, 24H), 3.60 (d, J = 12.0, 2.4 Hz, 1H), 3.58-3.53 (m, 2H), 3.50-3.35 (m, 9H), 3.30-3.27 (m, 1H), 3.22 (dd, J = 9.6, 1.8 Hz, 1H), 3.12 (t, J = 9.6 Hz, 1H), 2.10 (t, J = 7.8 Hz, 2H), 1.48-1.41 (m, 2H), 1.24-1.12 (m, 28H), 0.80 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 173.6, 138.8, 138.7, 138.6, 138.5, 138.4, 138.3, 138.2, 138.1, 138.0, 137.9, 137.6, 128.5, 128.4, 128.3, 128.2, 128.1, 127.9, 127.8, 127.7, 127.6, 127.5, 127.4, 127.3, 127.2, 98.6 (ManA C-1), 98.5 (ManB C-1, ManC C-1, ManD C-1), 98.4 (ManE C-1), 98.2 (ManF C-1), 81.2, 80.4, 80.3, 79.4, 79.3, 79.2, 78.8, 78.3, 75.5, 75.3, 75.2, 75.1, 75.0, 74.9, 74.87, 74.81, 74.7, 74.6, 74.3, 74.1, 74.0, 73.9, 73.2, 73.0, 72.67, 72.61, 72.5, 72.2, 72.0, 71.9, 71.8, 71.7, 71.67, 71,61, 71.3, 71.28, 71.26, 71.20, 70.1, 69.1, 66.5, 65.8, 65.7, 65.6, 63.1, 34.1, 31.9, 29.7, 29.65, 29.60, 29.5, 29.3, 29.25, 29.20, 24.8, 22.7, 14.1. HR MS (ESI-TOF) m/z: calcd for C214H238O37Na2 [M + 2Na]2+ 1722.8269; found, 1722.8318. MS (MALDI-TOF) m/z: calcd for C214H238O37Na [M + Na]+ 3422.6; found, 3423.0
6-O-[(2,3,4,6-tetra-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)-(1→6)-(2,3,4-tri-O-benzyl-α-D-mannopyranosyl)]-3,4,5-tri-O-benzyl-2-O-(2,3,4-tri-O-benzyl-6-O-stearoyl-α-D-mannopyranosyl)-1-O-(1,2-di-O-stearoyl-sn-glycero-3-benzylphosphoryl)-D-myo-inositol (26)
To a mixture of 25 (16.0 mg, 4.7 μmol), freshly prepared glycerylphosphoramidite 4 (20.2 mg, 0.024 mmol), and MS 4 Å in CH2Cl2 and CH3CN (2:1, 3 mL) was added 1H-tetrazole (0.45 M in CH3CN, 105 μL, 0.047 mmol). After stirring at room temperature under Ar for 40 min, the reaction mixture was cooled to −20 °C, and m-CPBA (4.1 mg, 0.024 mmol) was added. The reaction mixture was slowly warmed to room temperature in 1 h, and then quenched with saturated aq. NaS2O3 solution. The aq. layer was extracted with CH2Cl2 (3×30 mL), and the organic phase, after being dried over Na2SO4, was concentrated, with the residue purified by silica gel column chromatography with EtOAc and toluene (1:13) as the eluent to give 26 (14.1 mg, 72%, mixture of two isomers in about 1:3 ratio) as a white solid. 1H and 13C NMR spectroscopic data for the major stereoisomer: 1H NMR (600 MHz, CDCl3) δ 5.33 (s, 1H, ManA H-1), 5.19 (s, 1H, ManB H-1), 5.03 (s, 1H, ManC H-1), 4.93 (s, 1H, ManD H-1), 4.86 (s, 1H, ManE H-1), 4.76 (s, 1H, ManF H-1); 13C NMR (150 MHz, CDCl3) δ 99.2 (ManB C-1), 99.1 (ManA C-1), 99.0 (ManE C-1), 98.8 (ManD C-1), 98.5 (ManF C-1), 98.4 (ManC C-1). 31P NMR (162 MHz, CDCl3) δ −0.42 (minor isomer), −0.19 (major isomer). HR MS (ESI-TOF) m/z: calcd for C260H319O44PNa2 [M + 2Na]2+ 2111.1129; found, 2111.1270.
6-O-[(α-D-mannopyranosyl)-(1→6)-(α-D-mannopyranosyl)-(1→6)-(α-D-mannopyranosyl)-(1→6)-(α-D-mannopyranosyl)-(1→6)-(α-D-mannopyranosyl)]-2-O-[(6-O-stearoyl-α-D-mann opyranosyl)]-1-O-(1,2-di-O-stearoyl-sn-glycero-3-phosphoryl)-D-myo-inositol (1)
The mixture of 26 (10 mg, 2.4 μmol) and 10% Pd/C (15 mg) in CHCl3, MeOH and H2O (3:3:1, 3 mL) was stirred under a 50 psi H2 atmosphere for 3 days. The reaction solution was filtered off through a pad of Celite, and the filtrate was concentrated in vacuum to give the target compound 1 (3.5 mg, 69%) as a pale yellow solid. 1H NMR (600 MHz, CD3OD, CDCl3 and D2O 3:3:1) δ 5.33 (s, 1H, ManA H-1), 5.19 (s, 1H, ManB H-1), 5.12 (s, 1H, ManC H-1), 4.90 (s, 1H, ManD H-1), 4.84 (s, 1H, ManE H-1), 4.83 (s, 1H, ManF H-1), 4.60-2.98 (m, 47H), 2.38-2.16 (m, 6H), 1.65-1.55 (m, 6H), 1.40-1.15 (m, 84H), 0.90-0.80 (m, 9H). 31P NMR (162 MHz, CD3OD/CDCl3/D2O 3:3:1) δ 0.54. MS (MALDI-TOF) m/z: calcd for C99H180O44PK3 [M + 3K - H]2+ 1110.5; found, 1109.2.
Supplementary Material
Acknowledgments
This work was supported in part by National High Technology Research and Development (863) Program of China (No. 2012AA021504) and National Major Scientific and Technological Special Project for New Drugs Development (2012ZX09502001) of China and the National Institutes of Health (R01 GM090270) of the United States.
Footnotes
Supporting Information 1H, 13C, 31P, 1H-1H gCOSY, and 1H-13C gHMQC NMR spectra of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Neyrolles O, Guilhot C. Tuberculosis. 2011;91:187–195. doi: 10.1016/j.tube.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Daffe M, Draper P. Adv Microb Physiol. 1998;39:131–203. doi: 10.1016/s0065-2911(08)60016-8. [DOI] [PubMed] [Google Scholar]
- 3.Vignal C, Guerardel Y, Kremer L, Masson M, Legrand D, Mazurier J, Elass E. J Immunol. 2003;171:2014–2013. doi: 10.4049/jimmunol.171.4.2014. [DOI] [PubMed] [Google Scholar]
- 4.Guerardel Y, Maes E, Briken V, Chirat F, Leroy Y, Locht C, Strecker G, Kremer L. J Biol Chem. 2003;278:36637–36651. doi: 10.1074/jbc.M305427200. [DOI] [PubMed] [Google Scholar]
- 5.Quesniaux VJ, Nicolle DM, Torres D, Kremer L, Guerardel Y, Nigou J, Puzo G, Erard F, Ryffel B. J Immunol. 2004;172:4425–4434. doi: 10.4049/jimmunol.172.7.4425. [DOI] [PubMed] [Google Scholar]
- 6.Doz E, Rose S, Nigou J, Gilleron M, Puzo G, Erard F, Ryffel B, Quesniaux VF. J Biol Chem. 2007;282:26014–26025. doi: 10.1074/jbc.M702690200. [DOI] [PubMed] [Google Scholar]
- 7.Liu X, Stocker BL, Seeberger PH. J Am Chem Soc. 2006;128:3638–3648. doi: 10.1021/ja0565368. [DOI] [PubMed] [Google Scholar]
- 8.Boonyarattanakalin S, Liu X, Michieletti M, Lepenies B, Seeberger PH. J Am Chem Soc. 2008;130:16791–16799. doi: 10.1021/ja806283e. [DOI] [PubMed] [Google Scholar]
- 9.Jayaprakash KN, Lu J, Fraser-Reid B. Angew Chem Int Ed. 2005;44:5894–5898. doi: 10.1002/anie.200500505. [DOI] [PubMed] [Google Scholar]
- 10.Fraser-Reid B, Chaudhuri SR, Jayaprakash KN, Lu J, Ramarnurty CVS. J Org Chem. 2008;73:9732–9743. doi: 10.1021/jo802000p. [DOI] [PubMed] [Google Scholar]
- 11.Fraser-Reid B, Lu J, Jayaprakash KN, Lopez JC. Tetrahedron: Asymmetry. 2006;17:2449–2463. [Google Scholar]
- 12.Ainge GD, Compton BJ, Hayman CM, Martin WJ, Toms SM, Larsen DS, Harper JL, Painter GF. J Org Chem. 2011;76:4941–4951. doi: 10.1021/jo200588u. [DOI] [PubMed] [Google Scholar]
- 13.Ainge GD, Hudson J, Larsen DS, Painter GF, Gill GS, Harper JL. Bioorg Med Chem. 2006;14:5632–5642. doi: 10.1016/j.bmc.2006.04.037. [DOI] [PubMed] [Google Scholar]
- 14.Ainge GD, Parlane NA, Denis M, Hayman CM, Larsen DS, Painter GF. Bioorg Med Chem. 2006;14:7615–7624. doi: 10.1016/j.bmc.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Joe M, Bai Y, Nacario RC, Lowary TL. J Am Chem Soc. 2007;129:9885–9901. doi: 10.1021/ja072892+. [DOI] [PubMed] [Google Scholar]
- 16.Stadelmaier A, Biskup MB, Schmidt RR. Eur J Org Chem. 2004:3292–3303. [Google Scholar]
- 17.Ainge GD, Parlane NA, Denis M, Dyer BS, Harer A, Hayman CM, Larsen DS, Painter GF. J Org Chem. 2007;72:5291–5296. doi: 10.1021/jo070639m. [DOI] [PubMed] [Google Scholar]
- 18.Dyer BS, Jones JD, Ainge GD, Denis M, Larsen DS, Painter GF. J Org Chem. 2007;72:3282–3288. doi: 10.1021/jo0625599. [DOI] [PubMed] [Google Scholar]
- 19.Huang X, Huang L, Wang H, Ye XS. Angew Chem Int Ed. 2004;43:5221–5224. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z, Zhou LY, El-Boubbou K, Ye XS, Huang X. J Org Chem. 2007;72:6409–6420. doi: 10.1021/jo070585g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crich D, Banerjee A. J Am Chem Soc. 2006;128:8078–8086. doi: 10.1021/ja061594u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swarts BM, Guo ZW. J Am Chem Soc. 2010;132:6648–6650. doi: 10.1021/ja1009037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu F, Guo ZW. Bioorg Med Chem Lett. 2009;19:3852–3855. doi: 10.1016/j.bmcl.2009.03.151. [DOI] [PubMed] [Google Scholar]
- 24.Oltvoort JJ, Vanboeckel CAA, Dekoning JH, Vanboom JH. Synthesis. 1981:305–308. [Google Scholar]
- 25.Podlasek CA, Wu J, Stripe WA, Bondo PB, Serianni AS. J Am Chem Soc. 1995;117:8635–8644. [Google Scholar]
- 26.Lee JC, Greenberg WA, Wong CH. Nat Protoc. 2006;1:3143–3152. doi: 10.1038/nprot.2006.489. [DOI] [PubMed] [Google Scholar]
- 27.Burkhart F, Zhang Z, Wacowich-Sgarbi S, Wong CH. Angew Chem Int Ed. 2001;113:1314–1317. doi: 10.1002/1521-3773(20010401)40:7<1274::aid-anie1274>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 28.Zhang ZY, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. J Am Chem Soc. 1999;121:734–753. [Google Scholar]
- 29.Tam PH, Lowary TL. Carbohydr Res. 2007;342:1741–1772. doi: 10.1016/j.carres.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Chao CS, Lin CY, Mulani S, Hung WC, Mong KK. Chem Eur J. 2011;17:12193–12202. doi: 10.1002/chem.201100732. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.