

Abstract
Globoid cell leukodystrophy is a lysosomal storage disease characterized by the loss of galactocerebrosidase. Galactocerebrosidase loss leads to the accumulation of psychosine and subsequent oligodendrocyte cell death, demyelination, macrophage recruitment, and astroglial activation and proliferation. To date, no studies have elucidated the mechanism of glial cell activation and cytokine and chemokine up-regulation and release. We explored a novel explanation for the development of the pathological changes in the early stages of globoid cell leukodystrophy associated with toll-like receptor (TLR) 2 up-regulation in the hindbrain and cerebellum as a response to dying oligodendrocytes. TLR2 up-regulation on microglia/macrophages coincided with morphological changes consistent with activation at 2 and 3 weeks of age. TLR2 up-regulation on activated microglia/macrophages resulted in astrocyte activation and marked up-regulation of cytokines/chemokines. Because oligodendrocyte cell death is an important feature of globoid cell leukodystrophy, we tested the ability of TLR2 reporter cells to respond to oligodendrocyte cell death. These reporter cells responded in vitro to medium conditioned by psychosine-treated oligodendrocytes, indicating the likelihood that oligodendrocytes release a TLR2 ligand during apoptosis. TLRs are a member of the innate immune system and initiate immune and inflammatory events; therefore, the identification of TLR2 as a potential driver in the activation of central nervous system glial activity in globoid cell leukodystrophy may provide important insight into its pathogenesis.
Globoid cell leukodystrophy (GLD; Krabbe’s disease) is an autosomal recessive lysosomal storage disorder. It affects approximately 1 in 100,000 children born each year and often leads to mortality by 2 years of age. GLD results from the lack of metabolic enzyme galactocerebrosidase (GALC).1 In the absence of GALC, galactocerebroside (GalCer) undergoes alternative catabolism into galactosylsphingosine (alias psychosine) and a fatty acid instead of normal catabolism into its component parts, galactose and ceramide.2 Galactosylsphingosine has been shown to cause the terminal pathological changes in the central nervous system (CNS) of individuals affected by GLD: globoid cell formation,2,3 astroglial cytokine and chemokine secretion,4–8 and oligodendrocyte death and demyelination.9–14 In addition to these pathological changes, it is established that monocytes/macrophages are recruited to the CNS early in the disease process and continue to mobilize to the brain until the late stages of disease.15,16
Little is understood, however, about the mechanisms underlying these events, or which cells are involved in eliciting the initial pathological changes.7,17,18 This study investigated the mechanisms involved in monocyte/macrophage recruitment and cytokine/chemokine up-regulation and secretion, as well as which cell types are the earliest to undergo inflammatory cascades leading to terminal disease.
Inflammatory cytokines are up-regulated as a consequence of activation of the innate immune response.19–22 Given the connection, it is logical to link the innate immune response to inflammation in globoid cell leukodystrophy. Psychosine is a derivative of β-galactosylceramide and a ganglioside; therefore, it could potentially serve as a toll-like receptor (TLR) ligand and induce the up-regulation of cytokine/chemokine secretion and monocyte/macrophage recruitment through the initial activation of a TLR.
TLRs are most commonly associated with recognition of specific, exogenously derived recognition of microbial patterns. Examples of these patterns are lipopolysaccharide, peptidoglycan, lipoarabinomannan, and double-stranded RNA.23 However, TLRs also have known roles in exacerbating inflammation and inflammatory profiles in the brains of other neurodegenerative and demyelinating diseases, such as experimental autoimmune encephalitis (murine model for multiple sclerosis),24,25 amyotrophic lateral sclerosis,26 and Alzheimer disease.27,28 Investigating whether TLRs play a role in the initiation of inflammatory signaling pathways in the brains of twitcher mice (the murine model for globoid cell leukodystrophy) is important to understanding how inflammation in GLD is initiated. Elucidating how inflammation is induced in the course of disease will lead to more complete knowledge of the pathogenesis of GLD and the identification of novel therapeutic targets.
To date, few studies have been performed that investigate the early events in GLD that lead to the terminal changes associated with the disease. It is known that globoid cell appearance and demyelination occur caudally to rostrally in the twitcher brain,29 but the mechanism has not been studied. Our hypothesis was that TLR up-regulation on perivascular macrophages and/or microglia early in the disease initiates cytokine production and monocyte/macrophage recruitment to brains of affected individuals. After activation, microglia and macrophages activate neighboring astrocytes, which markedly exacerbate cytokine/chemokine secretion and inflammation. This study identifies early morphological changes in resident CNS glial cells and the innate immune system that explain, at least in part, how cells become activated, up-regulate cytokines/chemokines, and recruit monocytes/macrophages.
Materials and Methods
Animals
A total of 98 mice (twitcher and age-matched controls) were euthanized at the ages of 2, 3, 4, and 5 weeks. Each experiment used at least three animals per group and time point. At each time point, the brain was dissected into forebrain and cerebellum/brainstem (hindbrain). Twitcher and age-matched wild-type controls were genotyped using an established molecular beacon PCR assay.30 All procedures were performed according to an animal protocol approved by the Tulane Institutional Animal Care and Use Committee.
Tissue Dissection and Handling
After euthanasia via CO2 asphyxiation, the heads were removed with a scalpel blade and the skull was removed from the meninges with bone rongeurs. The brain was removed, and placed into an acrylic brain matrix with 1-mm divisions. The forebrain, immediately caudal to the olfactory bulbs, was trimmed coronally at 3 mm, and the entire cerebellum and underlying brainstem were taken together and trimmed along the sagittal midline. Tissues were: i) placed into 1 mL of RNAlater (Life Technologies, Grand Island, NY), ii) snap frozen on dry ice and maintained at −80°C, or iii) fixed in 2% paraformaldehyde for 4 hours. After fixation, the sections were washed in PBS and cryoprotected with 30% sucrose for 3 to 5 days. All cryoprotected sections were embedded in optimal cutting temperature compound (Tissue Tek, Torrance, CA) and divided into sections (16 μm thick) for confocal microscopy.
For paraffin-embedded tissues, mice were euthanized via CO2 asphyxiation and perfused with sterile PBS through the left ventricle of the heart. Tissues were removed and fixed for 24 hours to 1 week in 10% neutral-buffered formalin and subsequently embedded in paraffin. Paraffin-embedded tissues were divided into sections (5 μm thick) and mounted onto glass slides for immunohistochemical (IHC) or histochemical stains.
Confocal Microscopy
Optimal cutting temperature compound–embedded forebrain and hindbrain specimens were divided into sections (16 μm thick) for confocal microscopy. Immunodetection of TLR2 in combination with the cell-specific antibodies indicated in Table 1 and Topro-3 (model T3605; Life Technologies) to label nuclei was performed as described previously.33 Briefly, tissues were blocked with PBS containing 10% normal goat serum and 0.2% fish skin gelatin (Sigma-Aldrich, St. Louis, MO), incubated with the primary antibody for 1 hour, washed with PBS–fish skin gelatin–Triton X-100 (Sigma-Aldrich), and finally incubated for 1 hour with secondary antibodies against the appropriate primary antibody for 1 hour. TLR2 was always labeled with a secondary antibody tagged with Alexa 488 (Molecular Probes/Life Technologies, Grand Island, NY), whereas all cell-specific antibodies [glial fibrillary acidic protein (GFAP), ionized calcium-binding adapter protein (IBA)-1, S100, and neuronal nuclear antigen] were labeled with a secondary antibody tagged with either Cy3 (Sigma-Aldrich) or Alexa 568 (Molecular Probes/Life Technologies). Sections were fixed in 4% paraformaldehyde for 5 minutes after staining with the secondary antibody. When two primary rabbit polyclonal antibodies were used together (ie, TLR2 and IBA-1), 2.5 μg of the cell-specific antibody was prelabeled with Zenon labeling reagents (Molecular Probes/Life Technologies), according to the manufacturer’s instructions. Slides were mounted with an anti-quenching medium containing MOWIOL 4-88 (Calbiochem/EMD Millipore, Billerica, MA), glycerol (Sigma-Aldrich), and 2.5% DABCO Mountant (Sigma-Aldrich).
Table 1.
List of Antibodies Used in Immunofluorescence
| Antigen | Cell type | Source | Antibody type | Working dilution |
|---|---|---|---|---|
| TLR231,32 | NA | Abcam (Cambridge, MA) | Rb polyclonal | 1:50 |
| GFAP | Astrocyte | Sigma (St. Louis, MO) | Ms IgG1, labeled with Cy3 | 1:200 |
| S100 | Glial, ependymal | Sigma | Rb polyclonal | 2.5 μg used with zenon |
| IBA-1 | Microglia | Wako (Richmond, VA) | Rb polyclonal | 2.5 μg used with zenon |
| NeuN | Neuron | Chemicon (Billerica, MA) | Ms IgG1 | 1:10 |
| F4/80 | Microglia/macrophage | Santa Cruz (Dallas, TX) | Rat IgG1 | 1:10 |
| Glut-1 | Endothelial cells | Abcam | Ms IgG1 | 1:40 |
NA, not applicable.
All images were captured with a Leica SP2 true confocal system (Leica Microsystems, Buffalo Grove, IL) at ×40 magnification with an HCL planar apochromatic confocal system 2 oil lens objective. The numerical aperture of the lens objective was 1.3. All confocal microscopy was performed at 68°C to 70°C. Leica acquisition software was used to capture the confocal images. Volocity version 5 (Perkin Elmer, Boston, MA) and Adobe Photoshop CS5 were used for more in-depth image analysis and counting cells manually.
Image Analysis
To establish IBA-1 and GFAP up-regulation in the progression of disease, mean fluorescent intensity (MFI) was measured from 10 random, nonoverlapping fields from three twitcher and three normal mice at 2, 3, and 5 weeks of age. MFI was calculated with ImageJ software version 10.2 (NIH, Bethesda, MD) after thresholding was applied equally to each image to correct for apparent background and allow for determination of patterns in staining characteristics. Averages and SDs of MFI were calculated and reported. The same process was performed for glial nodule analysis within a diameter of >110 μm, which was determined to encompass the entire nodule in all cases examined.
To analyze TLR2 expression, 10 random, nonoverlapping fields from the forebrain and hindbrain were analyzed for three twitcher and three normal animals at 2, 3, and 5 weeks of age. Images were converted to three-dimensional (3D) images using Volocity software version 5, which allowed visualization of staining characteristics to determine whether colocalization was present for each antibody combination. Images were thresholded and analyzed. Cells were counted as positive if the signal was present on or near a stained nucleus, or if the staining had distinct cellular morphological characteristics.
Light Microscopy
3,3′-Diaminobenzidine/hematoxylin, luxol fast blue (for myelin), and PAS (for polysaccharides) stained slides were analyzed by standard light microscopy. Images were captured with a spectral camera (CRI-Nuance, Lincolnshire, UK), and the original red-green-blue colorspace image was spectrally unmixed to yield images with high contrast.34
Quantification of Astrocyte Morphological Features
All samples were coded and analyzed randomly by a researcher (H.A.S.) blinded to animal number and condition. Images of nonoverlapping fields in hindbrain and cerebellar sections were captured by fluorescence microscopy using a Leica confocal microscope at ×40 magnification and analyzed using Neurolucida software version 10.02 (MBF Bioscience, Williston, VT). A total of 28 astrocytes around or in a nodule within the twitcher hindbrain, 67 astrocytes from areas away from nodules within the hindbrain, and 26 astrocytes from control mice with well-defined cell bodies and processes were chosen for further analysis. The cells selected were fully intact and did not have processes that touched the edges of the field. The resulting files were analyzed with Neurolucida Explorer (MBF Bioscience), generating morphological data, including cell area, branching points (nodes), arbor length, and volume.
qPCR Analysis
Four twitcher and four normal mice per time point were euthanized at 2, 3, 4, and 5 weeks of age. Each section of brain was placed into RNAlater for preservation. For RNA extraction, the forebrain and hindbrain were halved and homogenized using standard TRIzol extraction (catalog no. 15596026; Life Technologies). RNA concentrations were measured with a spectrophotometer, and 2 mg of RNA was used for cDNA synthesis with random hexamers. TaqMan primers and probes from Life Technologies were used to quantify mRNA expression at each time point in the forebrain or hindbrain. The following primers and probes were used for real-time PCR: TLR1 (Mm00446095_m1), TLR2 (Mm00442346_m1), TLR3 (Mm0068112_m1), TLR4 (Mm00445273_m1), TLR5 (Mm00546288_s1), TLR6 (Mm02529782_s1), TLR7 (Mm00446590_m1), TLR8 (Mm01157262_m1), and TLR9 (Mm00446193_m1). Hypoxanthine guanine phosphoribosyl transferase (HPRT) 1 was used as the endogenous control (Mm00446968_m1).35 Real-time PCR was performed in an Applied Biosystems 7900 HT thermocycler (Life Technologies), and the accompanying software automatically selected the appropriate CT values. ΔΔCT values for use in fold change calculation were calculated using the following formula:
| (1) |
Fold change was determined with the formula 2-ΔΔCT, and the SD was calculated based on the final fold change values.36
Cell Culture
Human embryonic kidney (HEK)-blue (catalog no. hkb-mtlr2; Invivogen, San Diego, CA) murine (m)TLR2 (HEK-TLR2) overexpressing cells and HEK-blue null2 (catolog no. hkb-null2; Invivogen) (HEK-null) cells were propagated in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum, 10 U penicillin, 10 mg streptomycin, and normocin, as per the manufacturer’s instructions. HEK-TLR2 cells were placed in medium containing selective antibiotics—hygromycin, blasticidin, and zeocin—that induce expression of murine TLR2, CD14, and secreted alkaline phosphatase fused to five NF-κB and activator protein (AP)-1 binding sites. The HEK-null cells were placed in medium containing zeocin, which induces expression of secreted alkaline phosphatase fused to NF-κB and AP-1 binding sites. One packet of Quanti-blue (Invivogen) was mixed with 100 mL of ultrapure water and vacuum filtered through a 0.22-μm filter.
M03.13 cells (immortalized oligodendrocyte cell line) were cultured in high glucose (4.5 mg/dL) DMEM and 10% fetal bovine serum with no sodium pyruvate at 37°C and 5% CO2. Oligodendrocyte differentiation was induced by plating the cells onto 6-well dishes at approximately 1 × 106 cells/mL and cultured for 3 days in serum-free DMEM with 100 nmol/L phorbol 12-myristate 13-acetate. After differentiation, the cells were treated with psychosine at 100, 50, 20, 10, 5, and 0 μg/mL. Medium (200 μL) was removed at 24, 48, and 72 hours, and frozen at −80°C for use in the TLR2 reporter assay.
TLR2 Reporter Assay
The TLR2 reporter assay demonstrates completion of the TLR2 signaling pathway based on alkaline phosphatase production when stimulated with defined agents. Cell suspensions (180 μL) containing approximately 280,000 HEK-TLR2 or HEK-null cells per milliliter were incubated in 96-well plates for 20 hours with 20 μL of 1 μg/mL of tumor necrosis factor (TNF)-α, 100 ng/mL of PAM3CSK4, or 10 ng to 10 μg/mL psychosine. Supernatant (20 μL) was added to 180 μL of Quanti-blue reagent in 96-well plates and incubated at 37°C for 3 hours. Color change was measured with a Biotek Epoch (Winooski, VT) spectrophotometer at 655 nm. Alternatively, 20 μL of supernatant from psychosine-treated oligodendrocytes was added to 180 μL of HEK-mTLR2 or HEK-null2 cells and incubated for 20 hours. Analysis was performed as described.
Multiplex Bead-Based Cytokine Assay
Flash-frozen brain sections of four twitcher and three normal mice at 2, 3, 4, and 5 weeks of age were crushed with mortar and pestle on dry ice. A portion of ground tissue was weighed and added to 20 volumes of PBS with 0.2% NP-40 and protease/phosphatase inhibitors and ultrasonicated on ice briefly until dissolved. Samples were centrifuged twice at 10,000 × g for 10 minutes to remove debris. The supernatant was aliquoted and stored at −80°C. Protein was quantified with a bicinchoninic acid protein measurement kit. Samples were thawed and prepared for a cytokine kit according to the manufacturer’s instructions. Using this kit, 32 individual cytokines and chemokines were analyzed for up-regulation, including eotaxin [chemokine ligand (CCL) 11], granulocyte colony-stimulating factor (CSF), granulocyte-macrophage CSF, interferon (IFN)-γ, IL-1α, IL-β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-17, interferon γ-induced protein (IP)-10 (CXCL10), keratinocyte-chemoattractant (CXCL1), leukemia inhibitory factor, lipopolysaccharide-induced CXC chemokine, macrophage CSF, monocyte chemotactic protein-1 (CCL2), monokine induced by γ interferon (CXCL9), macrophage inflammatory protein (MIP)-1α (CCL3), MIP-1β (CCL4), MIP-2, regulated on activation normal T cell expressed and secreted (CCL5), TNF-α, and vascular endothelial growth factor. After analysis on a Bio-Rad bioplex analyzer, samples were normalized according to protein concentrations. Normalized values were used to quantify levels of cytokine expression.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism, version 5 (GraphPad Software, La Jolla, CA). For the quantitative PCR (qPCR) assay, data that conformed to normal distribution were analyzed with a Student’s t-test. Data that were not normally distributed were analyzed with a Wilcoxon signed rank test. In all other analyses, data were analyzed by one-way analysis of variance and Tukey’s post hoc test if normally distributed. If data were not normally distributed, they were analyzed with a Kruskal-Wallis test with Dunn’s post hoc multiple comparison test to determine significance between groups. Results are expressed as means ± SD. Significance was set at P < 0.05.
Results
IBA-1 and GFAP Expression Increases at 3 Weeks of Age
Microglia and, to a lesser extent, astrocytes are considered able to respond to innate immune system stimuli. To assess whether microglia or astrocytes are activated first, IBA-1 (microglia) and GFAP (astrocytes) immunoreactivities were examined at 2, 3, and 5 weeks of age. GFAP is a marker of astroglial activation, whereas IBA-1 identifies resting and activated microglia37; however, microglia demonstrate specific morphological changes when activated.38 At 2 weeks of age, IBA-1+ cells showed a moderate increase in the thickness and number of processes extending from the cell body compared with age-matched controls (Figure 1). By 3 weeks of age, microglial activation was common, characterized by aggregates of ameboid microglia forming discrete nodules within the neuropil. By using MFI as an arbitrary unit of measure, there was no significant change in IBA-1 or GFAP immunoreactivity in the twitcher mouse hindbrain at 2 weeks of age versus the normal hindbrain (Figure 2A). Beginning at 3 weeks of age, IBA-1 MFI in normal mice began to diminish significantly compared with age-matched twitcher mice (Figure 2B). At 5 weeks of age, IBA-1 MFI remained at similar expression levels as at 3 weeks of age; however, IBA-1 in normal mice was markedly diminished, resulting in a significant difference in both the hindbrain and forebrain (Figure 2C). GFAP immunoreactivity did not increase significantly over age-matched control mice until 3 weeks of age in the hindbrain of twitcher mice, and continued to increase in the twitcher brain through 5 weeks of age (Figure 2C). Similar to IBA-1, GFAP MFI diminished in normal mice over time in the brains of affected mice (Figure 2C). These results demonstrated morphological evidence of microglial activation before the activation of neighboring astrocytes as early as 2 weeks of age in the hindbrain. In normal mice, IBA-1 immunoreactivity appears to decrease over time as microglia begin to assume a quiescent state. In twitcher mice, microglia remained activated.
Figure 1.
Microglia exhibit morphological evidence of activation in the 2-week-old twitcher hindbrain. Confocal images were captured. Original magnification, ×40. Images are representative of three normal and three twitcher mouse hindbrains immunostained for microglia (IBA-1, green), astrocytes (GFAP, red), and cell nuclei (Topro-3, blue). A: Arrowhead identifies resting microglia in a normal mouse. The resting microglia has a small cell body and few, thin processes extending from the cell body. B: Activated microglial cells (arrowhead), characterized by enlarged cell bodies with numerous hypertrophied processes. Immunoreactivity for GFAP is minimal at this time point in normal and twitcher mice. Scale bar = 50 μm.
Figure 2.

Whole field analysis of IBA-1 (black bars) and GFAP (white bars) MFI demonstrated significant increases at 3 weeks of age in the twitcher (twi) mouse hindbrain. Ten random, ×40 fields were captured and analyzed with ImageJ software version 10.2 from three twitcher and three normal (N) mice at each time point. MFI was calculated and averaged among all 30 images in each group. A: No significant difference between MFI for IBA-1 and GFAP was found between twitcher mice and age-matched control mice at 2 weeks of age. B: At 3 weeks of age, significant differences between the MFI for IBA-1 and GFAP were observed in the hindbrain but not the forebrain. C: At 5 weeks of age, significant differences between the MFI for IBA-1 and GFAP were observed between twitcher mice and normal mice in both the hindbrain and forebrain. FB, forebrain; HB, hindbrain. Error bars = SD. ∗∗P < 0.01, ∗∗∗P < 0.001.
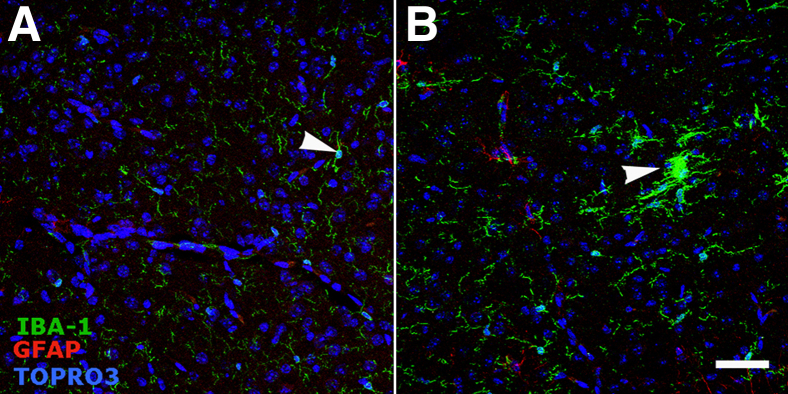
Formation of Glial Nodules and Activation of Surrounding Astrocytes
A novel pathological alteration was identified while examining images for IBA-1 and GFAP immunoreactivity. Microglia formed prominent, discrete nodules that were often surrounded by hypertrophied astrocytes (Figure 3A). At 2 weeks, these nodules occurred at a frequency of 0.5 per 0.14 mm2 (×40 field); the age-matched control mice demonstrated no evidence of glial nodules (Figure 3A). At 3 weeks, the nodules became more frequent—approximately 1.5 per 0.14 mm2—with numerous GFAP+ astrocytes surrounding them and interdigitating with the IBA-1+ microglia. No evidence of microglial or astroglial activation or nodule formation of any type was evident in age-matched control mice. By 5 weeks of age, nearly all microglia in twitcher mice appeared amoeboid; however, the activated astrocytes no longer appeared to be centered on microglial nodules. Instead, the GFAP+ cells appear to be diffusely activated with no clear, consistent interaction with nearby microglial cells. Age-matched controls for each group demonstrated morphological characteristics consistent with resting microglia and rare activated astrocytes. Based on high-resolution 3D confocal images, the microglial nodules were individual cells aggregated together rather than a single syncytial microglia surrounded by astrocytes (Figure 3B).
Figure 3.

Development of glial nodules and astrogliosis in the hindbrain of twitcher mice. A: Confocal images. Original magnification, ×40. Development of glial nodules and astrogliosis from weeks 2 to 5 compared with age-matched control mice. Arrowheads indicate glial nodules. Glial nodules (IBA-1, green) are seen as early as 2 weeks of age and then increase in frequency. Associated with the glial nodules are numerous astrocytes (GFAP, red) with thickened processes (astrogliosis). Over time, astrogliosis becomes diffuse. Scale bar = 50 μm. B: A representative 3D image from the hindbrain of a 3-week-old twitcher mouse captured with Volocity software version 5. Original magnification, ×63 with ×2 digital zoom. Images depict a central microglial aggregate (green) surrounded by interdigitating, hypertrophied astrocytes (red). Each square is 37.5 × 37.5 μm.
By using three twitcher and three normal mice per age group, MFI was measured in 110-μm-diameter areas centered on microglial nodules for both IBA-1 and GFAP at 2 and 3 weeks of age. At 2 weeks, there was a significant increase in MFI within the nodule and surrounding space compared with randomly chosen 110-μm-diameter locations within twitcher hindbrains and hindbrains of age-matched controls. GFAP did not demonstrate a meaningful change in MFI within any 110-μm-diameter area in any mice, whether around a nodule or randomly chosen in the hindbrain (Figure 4A). In 3-week-old twitcher mice, a significant elevation in the MFI of IBA-1 was noted in twitcher glial nodules when compared with randomly selected areas in twitcher hindbrains and with random areas in the age-matched controls (Figure 4B). However, the 3-week-old twitcher mice began to show a significant elevation of GFAP MFI around the glial nodules when compared with randomly located areas in twitcher hindbrains and age-matched controls. In addition, GFAP MFI was significantly higher in twitcher hindbrains when compared with randomly located areas within normal mice. These data demonstrated a significant, quantifiable elevation in IBA-1 immunoreactivity in local regions within hindbrains before activation of surrounding astrocytes, indicating that microglia are likely the first cells to demonstrate morphological and immunophenotypic markers of activation in the twitcher brain. Astrocytes were subsequently activated, first in association with glial nodules and then diffusely throughout the brain.
Figure 4.

Targeted MFI analysis of glial nodules shows that microglial activation precedes astroglial activation. MFI for IBA-1 (black bars) and GFAP (white bars) was measured within a 110-μm-diameter circular area centered on the microglial aggregate. A: At 2 weeks of age, 110-μm-diameter circles around glial nodules demonstrated significant increases in IBA-1 MFI compared with randomly chosen areas in twitcher (twi) brains and randomly chosen areas within normal (N) brains. B: At 3 weeks of age, 110-μm-diameter circles around glial nodules demonstrated significant increases in IBA-1 compared with randomly chosen areas in twitcher brains and normal brains. GFAP also demonstrated significant increases within 110-μm circles compared with randomly chosen areas in twitcher brains and normal brains. In addition, randomly chosen areas in twitcher brains also demonstrated significant elevations in MFI of GFAP when compared with randomly located areas in normal brains. Results are means of MFI across all nodules for area selected. C–H: Astrocyte cell bodies and dendrites were traced with Neurolucida to determine characteristics associated with hypertrophy. In this case, all characteristics demonstrated significant changes between the normal mouse hindbrain and twitcher mouse hindbrain. Cell body area was significantly larger between astrocytes around glial nodules and away from glial nodules within twitcher mouse hindbrains. Error bars = SD. A.U., arbitrary unit. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
Astrocytes Demonstrate Morphological Changes Associated with Activation in Twitcher Mice
Based on the MFI data, 3-week-old hindbrains were used to further quantify astroglial changes in twitcher and control brains. Fluorescent images of astrocytes were imported into Neurolucida and analyzed for process lengths (cell arbor), cell body area, the number of processes extending from the bodies, the number of tips associated with cell bodies, and the number of bifurcations of each process. Tracings of astrocytes from control and affected mice demonstrated numerous changes.
The sum of astrocyte process lengths (cell arbor) was increased greater than twofold in astrocytes near glial nodules and nearly twofold in astrocytes away from glial nodules compared with age-matched controls (Figure 4C). Similarly, by using the diameter of the astrocyte processes, frustra were generated in Neurolucida to determine arbor volume of astrocyte processes. These data also demonstrated significant increases between astrocytes near glial nodules and away from glial nodules, between astrocytes near glial nodules and astrocytes in normal control mice, and between astrocytes away from glial nodules and astrocytes in normal control mice (Figure 4D).
The area of the astrocyte cell bodies was measured in Neurolucida version 10.02 (MBF Bioscience). There was a significant increase in cell body area in astrocytes of twitcher mice hindbrains compared with age-matched controls (Figure 4E). Astrocytes near glial nodules had significantly enlarged bodies compared with astrocytes away from nodules and normal astrocytes. In addition, astrocytes away from glial nodules had significantly enlarged bodies compared with normal astrocytes (Figure 4E).
The number of processes leaving the cell body, bifurcations of processes, and tips of processes were all increased in twitcher mice compared with controls. Astrocytes in twitcher mice (both near and away from glial nodules) demonstrated increased mean numbers of processes (dendrites) extending from their cell bodies when compared with astrocytes in control mice (Figure 4F). Likewise, astrocytes in twitcher mice (near and away from glial nodules) demonstrated significant increases in bifurcations and tips compared with astrocytes in control mice (Figure 4, G and H).
Collectively, the data show astrocytes in twitcher mice have more processes, longer processes, and increased cell sizes compared with age-matched controls. All of the astrocyte morphological measurements support the hypothesis that glial nodules likely activate neighboring astrocytes. Although many of the parameters measured are not significantly elevated between astrocytes near glial nodules and those away from glial nodules, there is still an increasing trend in all parameters when astrocytes are near glial nodules. This could indicate that astrocytes near glial nodules are activated first, and subsequently activate neighboring astrocytes until astrocytes are diffusely activated near the terminal time point of 5 weeks old.
TLR1 and TLR2 Expression Levels Correlate with Microglial Activation
Based on data from previous studies indicating that gangliosides can induce TLR signaling,39–41 we examined the expression of TLRs 1 to 9 in the brain using qPCR. We found a statistically significant increase in TLR1 and TLR2 in the hindbrain at 2 weeks of age. In the forebrain, there was no significant change in TLRs 1 to 9 at 2 weeks of age compared with age-matched control mice. At 3 weeks of age, TLR2 demonstrated a ninefold change compared with age-matched control normal mice and remained markedly elevated through the life of the twitcher mice. In the forebrain, TLRs 1 and 2 began to increase at 3 weeks of age, mirroring the pattern in the hindbrain. By 5 weeks of age, TLR2 was markedly increased over normal mice in the forebrain. TLRs 7 and 8 showed increased expression, but only late in the disease process in the hindbrain (Figure 5A).
Figure 5.

TLR2 mRNA exhibited significant up-regulation at 2 weeks of age, primarily by microglia and macrophages. Results represent fold changes in mRNA levels of TLRs 1 to 9 in twitcher versus normal mice. A: Four twitcher and four normal brains were homogenized and qPCR was performed for each of the TLRs 1 to 9 at weekly intervals between 2 and 5 weeks of age. B: Confocal microscopy images analyzed with Volocity 3D imaging software version 5. Three-week-old twitcher hindbrain. Arrowhead indicates TLR2+/IBA-1+ cells. C: Five-week-old twitcher forebrain. Image depicts large bifurcating vessel with TLR2+/IBA-1+ cell. Arrowheads indicate TLR2+ cells. D: Five-week-old twitcher hindbrain. Arrowhead indicates TLR2+/F4/80+ cells. E: Three-week-old twitcher hindbrain. Arrowheads indicate TLR2+/S100+ cells. F: Five-week-old twitcher forebrain. Arrowhead indicates TLR2+ cell adjacent to Glut-1+ cells. G: Five-week-old normal mouse hindbrain. TLR2 immunoreactivity is not present. IBA-1+ cells are small, with few, narrow processes. H–J: Five-week-old twitcher hindbrain. Negative antibody control demonstrates no non-specific binding of the secondary antibody to tissues. Error bars = SD. ∗∗P < 0.01, ∗∗∗P < 0.001. Squares are 37.5 × 37.5 μm. Scale bar = 75 μm.
Microglia and Macrophages Express TLR2
To determine which cells expressed TLR2, multilabel confocal microscopy was performed. At 3 weeks of age, most TLR2-expressing cells were located around vessels and colocalized with IBA-1 (microglia and macrophages) (Figure 5, B and C) and F4/80 (macrophages) (Figure 5D). By 5 weeks of age, TLR2+ cells were frequently perivascular, had rounded morphological characteristics, and were intensely labeled (Figure 5C). Occasionally, colocalization with S100 was observed, indicating TLR2 expression by oligodendrocytes (Figure 5E). When stained simultaneously with Glut-1 (endothelial cells), TLR2 was often juxtaposed to Glut-1+ cells but failed to colocalize, confirming the perivascular nature of TLR2 expression (Figure 5F). In the age-matched control animals, TLR2 expression was characterized by minimal staining in a lightly stippled pattern on flattened cells in a perivascular location (consistent with perivascular macrophages) (Figure 5G). GFAP+ cells (astrocytes) and neuronal nuclear antigen positive cells (neurons) in both twitcher mice and normal mice never showed any TLR2 colocalization with images analyzed (data not shown). Negative antibody controls also demonstrated no identifiable immunoreactivity to any target in twitcher mice (Figure 5, H–J).
Quantitative analysis of immunofluorescent images (Figure 6) revealed TLR2+ cells in the normal mouse hindbrain and forebrain. In the twitcher mouse brain at 3 weeks of age, there was a twofold increase in total TLR2+ cells in the hindbrain, primarily in cells that did not colocalize with IBA-1 or S100 (Figure 6A). At 5 weeks of age, the total number of TLR2+ cells was nearly four times that of age-matched controls. The forebrain also showed increases in TLR2+ cells at both time points, including cells that did not label with typical glial markers. On average, there was approximately one TLR2+/S100+ cell per 0.14 mm2 in the twitcher mouse, with the exception of the 3-week-old forebrains (Figure 6B). No TLR2+/S100+ cells were detected in normal age-matched controls.
Figure 6.

TLR2 expression was markedly increased in macrophages and microglia. A: Ten random, nonoverlapping fields examined for three twitcher (twi) and three normal (N) mice in the hindbrain at both 3 and 5 weeks of age. There is marked up-regulation in TLR2 in the hindbrains at both 3 and 5 weeks of age, whereas TLR2 expression is relatively stable in age-matched control hindbrains. TLR2 expression is most up-regulated in the microglial/macrophage population, but occasional oligodendrocytes also express TLR2. B: Ten random, nonoverlapping fields examined for three twitcher and three normal mice in the forebrain at both 3 and 5 weeks of age. TLR2 expression lags behind expression in the hindbrain. The largest compartment expressing TLR2 is cells with morphological characteristics most consistent with macrophages, but IBA-1 negative. Oligodendrocytes only express TLR2 at 5 weeks of age in the forebrain. Avg, average.
TLR2 was up-regulated at 2 weeks of age, coincident with morphological evidence of microglial activation. At 2 weeks of age, there was minimal astroglial activation, based on GFAP immunoreactivity. In addition, microglia began to demonstrate evidence of aggregation in the form of nodules at 2 weeks of age. At 3 weeks of age, the microglial nodules were surrounded by numerous activated astrocytes. Taken together, these demonstrate the likelihood of TLR2 leading to macrophage and microglial activation as early as 2 weeks of age in twitcher hindbrains, before the activation of adjacent astrocytes.
TLR2+ Cells Reside in Areas of Demyelination among Aggregates of PAS+ Cells
IHC with 3,3′-diaminobenzidine was performed to demonstrate that TLR2 expression coincided with demyelination and aggregates of PAS+ cells (macrophages containing storage material from incomplete GALC catabolism) in the cerebrum and cerebellum that are characteristic of GLD. IHC analysis demonstrated that TLR2+ cells were found most often within white matter tracts of the hindbrain, cerebellum, and cerebrum (Figure 7, A and C). TLR2+ cells also were often located in areas that exhibited demyelination and PAS+ cells (Figure 7, B and D), consistent with being located in areas where lesions are present in the twitcher mouse. In this case, demyelination was demonstrated by decreased staining with luxol fast blue.29
Figure 7.

TLR2 was expressed in demyelinated areas and coincided with PAS+ cells. A: TLR2 is immunoreactive in the white matter tracts of the cerebellum. The highly nucleated areas near the top and bottom of the image represent the molecular layer of the cerebellum. The less cellular area in the center represents a white matter tract. B: In the same anatomical region as identified in A, arrowheads identify PAS+ cells scattered throughout. This area also demonstrates moderate to marked demyelination based on decreased luxol fast blue (LFB) staining in the area. C: White matter tracts in the cerebrum express TLR2+ cells most frequently. D: White matter tracts in the cerebrum demonstrate demyelination and PAS+ cell accumulation. E and F: Normal white matter tracts in the cerebellum and cerebrum, respectively. Both areas show normal levels of myelination and no PAS+ cells. Scale bar = 50 μm.
TLR2 Signaling Can Be Induced by Psychosine-Treated Oligodendrocytes
To determine whether TLR2 signaling could be activated by psychosine or dying oligodendrocytes, a TLR2 reporter assay was used that secretes alkaline phosphatase when NF-κB and AP-1 are activated. When the reporter cells were exposed to psychosine at concentrations ranging from 100 μg/mL to 1 ng/mL, there was no measurable increase in alkaline phosphatase production compared with HEK-null cells (negative control) (Figure 8A). The same cells responded appropriately to PAM3CSK4 (a TLR2 ligand), demonstrating the ability to respond when stimulated through TLR2. Psychosine also caused a morphological change in the treated cells: they appeared shrunken, with no evidence of replication or cell growth on the culture plate. Treating HEK-TLR2 cells with GalCer also did not induce TLR2 signaling (Figure 8B). However, when HEK-TLR2 cells were exposed to conditioned medium from oligodendrocytes cultured with 100 μg/mL psychosine, there was a significant fold change in TLR2 signaling over HEK-null cells (Figure 8C). PAM3CSK4 and TNF-α were used as positive controls to demonstrate the ability of HEK-TLR2 and HEK-null cells to respond to stimuli activating the TLR2 and extra-TLR pathways, respectively. These results show that as oligodendrocytes undergo cell death from psychosine treatment, they release a TLR2 ligand that activates the TLR2 signaling pathway and leads to NF-κB and AP-1 activation.
Figure 8.

TLR2 signaling was induced by psychosine-treated oligodendrocytes. A: Fold changes in HEK-TLR2 and HEK-null2 treated with psychosine at concentrations between 0 and 100 μg/mL. B: HEK-TLR2 and HEK-null2 cells treated with galactocerebroside in concentrations between 0 and 100 μg/mL. C: HEK-TLR2 and HEK-null2 reporter cells treated with conditioned medium from psychosine-treated oligodendrocytes in concentrations between 0 and 100 μg/mL. All experiments were repeated three times with two replicates per treatment. Fold values were calculated by dividing the spectrophotometry reading of HEK-TLR2 cells by the reading for HEK-null2 cells. Error bars = SD. ∗P < 0.05.
Cytokine/Chemokine Expression Lags Behind TLR2 Up-Regulation
Determining when cytokines and chemokines are up-regulated during the course of disease is important to establish the chronology of disease progression and whether they are the cause or effect of inflammatory activation. IL-6, CCL2 (monocyte chemotactic protein-1), CXCL10 (IP-10), and CXCL1 (keratinocyte chemoattractant; gro-α) all exhibited moderate to marked progressive increases in expression during the course of disease (Figure 9). None of these cytokines and chemokines demonstrated a significant increase in expression before 3 weeks of age in either the forebrain or the hindbrain/cerebellum; this was 1 week after elevations in TLR2 expression were observed (Figure 5A).
Figure 9.

Cytokine and chemokine up-regulation lags behind TLR2 up-regulation. Four twitcher (twi) mice and three normal (N) mice were evaluated at each age group for both the hindbrain and forebrain. Samples were prepared in duplicate for the cytokine/chemokine assay. Expression levels of IL-6, CXCL10, CXCL1, and CCl2 in the hindbrain and forebrain. Expression levels of IL-6, CXCL10, CXCL1, and CCL2 all increased after previous inflammation in the brains of twitcher mice, first in the hindbrain and then in the forebrain approximately a week later. This slight lag correlates well with the previous expression curve demonstrated for TLR2. In addition, TNF-α, IFN-γ, IL-α, and IL-β did not demonstrate any increase before the up-regulation of TLR2 in the hindbrain of twitcher mice. TNF-α only showed minimal up-regulation during the course of a twitcher mouse’s lifespan, whereas the other cytokines never demonstrated any evidence of up-regulation compared with age-matched controls. Error bars = SD. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
CXCL1 and CXCL10 had the earliest increase in expression levels at 3 weeks of age in the hindbrain. They did not increase significantly until 4 and 5 weeks, respectively, in forebrains of twitcher mice. IL-6 and CCL2 also demonstrated significant increases in expression levels, but not until 4 and 5 weeks of age in the hindbrain, respectively.
Additional chemokines that became elevated in the twitcher mouse during the course of disease were CCL3, CXCL9 (monokine induced by IFN-γ), CCL5 (regulated on activation normal T cell expressed and secreted), CCL11, and leukemia inhibitory factor (Supplemental Figure S1). Each of these increased significantly between 3 and 4 weeks of age in the hindbrain and cerebellum and lagged behind by approximately a week in the forebrain. Vascular endothelial growth factor decreased during the course of GLD in twitcher mice (data not shown).
In addition to the cytokines and chemokines previously mentioned, cytokines associated with the early immune response were tested: IL-1α, IL-1β, TNF-α, and IFN-γ. All demonstrated changes in expression between 2 and 5 weeks of age in the twitcher mouse. However, only TNF-α demonstrated any degree of significant expression compared with age-matched controls, and only increased to a significant level in the hindbrain at 3 weeks of age, but never increased significantly in the forebrain (Figure 9).
These findings support the hypothesis that TLRs and, specifically, TLR2 may serve as an initiator in the inflammatory profile of twitcher mouse brains. Based on the qPCR data, TLR2 was up-regulated as early as 2 weeks of age in the twitcher hindbrain, coinciding with morphological evidence of microglial/macrophage activation. However, the earliest cytokine/chemokine elevations observed in this study were at 3 weeks of age in the hindbrain. Cytokine/chemokine expression is often closely associated with the transcription factors NF-κB and AP-1. TLRs use those same transcription factors as the end point for their signaling pathway. The lag in cytokine/chemokine elevation behind TLR2 up-regulation is consistent with the possibility of TLR2 signaling contributing to the inflammatory milieu in GLD.
Discussion
The results of this study describe novel pathological findings related to the early events involved in the pathogenesis of GLD. The earliest change we identified was up-regulation of TLR2 on macrophages and microglia as early as 2 weeks of age in the hindbrain and cerebellum of twitcher mice. This occurred concurrent with the activation of microglia and resident macrophages. Astrocytes only became activated subsequent to formation of discrete microglial nodules. Subsequently, astrocytes were diffusely activated by 5 weeks of age when they no longer appear to be associated with microglial nodules.
Glial nodules or scars are most often presumed to be residual to resolution of disease or a quiescent state of the disease. In the case of GLD, these glial nodules appeared to be initiating foci that lead to activation of neighboring glial cells and inflammation in the brain. The concept of a glial nodule as a driver of cellular activation and inflammatory induction is novel and not completely understood. Further studies examining glial cell activation are warranted to clarify the cross talk that enables such interactions to occur. In the context of GLD, further studies should examine microglial TLR expression in spatial relationship to nearby oligodendrocytes undergoing apoptosis or necrosis.
The ability to elicit a TLR2 signaling response with psychosine-treated oligodendrocytes may represent a mechanism for cellular activation in vivo. Because oligodendrocytes accumulate psychosine in vivo from the inability to properly catabolize GALC, they undergo apoptosis.3,10–14 As they die, they may release an endogenous ligand for TLR2 that ultimately leads to macrophage and microglial activation, astrocyte activation, and cytokine and chemokine up-regulation. A recent study demonstrated such a possibility in that products of apoptosis can be identified through cross-reactivity with lipopolysaccharide and, ultimately, activation through TLR4.42 A similar mechanism may occur in this instance. Future studies are warranted to determine what causes TLR2 up-regulation in GLD. Possible causes of TLR2 up-regulation would be S10043 or some other damage-associated molecular pattern that is released on oligodendrocyte death.42 Proteomic analysis of oligodendrocytes undergoing apoptosis may yield valuable information about potential causes of innate immune up-regulation.
Previous studies have raised the question of what leads to the activation of glial cells and their subsequent secretion of cytokines and chemokines in GLD.5,8 Our study elucidated a potential series of steps leading to the ultimate up-regulation of cytokines/chemokines and the recruitment of monocytes/macrophages to affected areas. Based on existing data and the results of this study, the proposed sequence of events leading to the final pathological outcomes of this disease are as follows: i) oligodendrocytes cannot effectively catabolize GalCer due to absence of GALC, ii) psychosine accumulates in oligodendrocytes, iii) oligodendrocytes undergo apoptosis, leading to axonal demyelination, iv) a TLR ligand is released from oligodendrocytes, which leads to TLR2 signaling and macrophage/microglial activation, v) microglia form an early glial nodule, vi) astrocytes surrounding microglial nodules become activated, forming a mature glial nodule, and vii) cytokines/chemokines are released and inflammation continues to spread and increase.
Understanding the role of the innate immune system in up-regulation and activation of inflammatory cells is important to help identify potential targets for treatment. TLRs are important in inducing transcription of cytokines and chemokines39,44–48 but are also important in embryonic development and regeneration of the CNS.25,49–51 Understanding how TLR2 activation affects the pathological characteristics of the brains of individuals with GLD is important to understanding how to modify expression of the molecule and generate an effective treatment.
Because TLR up-regulation leads to cytokine and chemokine up-regulation in vivo, it is important to understand the chronological relationship of TLR up-regulation to cytokine and chemokine expression. Of the 32 cytokines and chemokines tested in this study, four of them (IL-6, CXCL1, CXCL10, and CCL2) demonstrated marked, significant up-regulation as early as 3 weeks of age. IL-6 is commonly associated with induction of the innate immune system.46,52,53 It has a wide range of properties, including induction of inflammation,46,54–56 and can affect neurogenesis.56,57 In GLD, previous studies have shown IL-6 to be produced by both microglia and astrocytes,17 and crossing a twitcher mouse with an IL-6 knockout mouse led to more severe pathological characteristics than the IL-6 competent twitcher mouse.58 It would seem that IL-6 plays an important protective role in the pathogenesis of the disease, but it is not clear how IL-6 is up-regulated or exactly how IL-6 is protective.
When considering CXCL1 in the context of GLD, it is helpful to understand that this molecule is most commonly associated with chemotaxis of neutrophils.55 In the case of GLD, there are no neutrophils or granulocytes present in the brain at any point during disease progression. According to other studies, CXCL1 and its receptor, CXCR2, can also have effects on oligodendrocyte progenitor maturation and migration and are produced at high levels at specific time points within the developing fetal brain.59–61 Another study demonstrated a protective ability to remyelinate neurons in the brains of mice engineered to overexpress CXCL1.62 Given the high levels of CXCL1 in GLD, it would seem that CXCL1 may be produced in an effort to replace the dying oligodendrocytes with new oligodendrocytes derived from resident oligodendrocyte progenitor cells.
Similarly, CXCL10 is most often associated with lymphocyte chemotaxis, yet there are no recognizable lymphocytes in the twitcher brain. Both CXCL9 and CXCL10 are ligands for the CXCR3 receptor, which is present on many of the resident glial cells in resting and pathological conditions.63–65 It may be that CXCR3 and CXCL10 have functions in the brain that are distinct from their roles in lymphocyte chemotaxis.
CCL2 (monocyte chemotactic protein-1) is most often associated with monocyte recruitment.66 In the case of GLD, there are numerous monocytes recruited to the brain during the disease process, presumably recruited by CCL2. In a previous study of GLD in macaques, CCL2 was produced primarily by astrocytes.7
Finally, it is important to mention the lack of TNF-α and IFN-γ elevation early in the course of GLD progression in the twitcher mouse. Both of these cytokines are associated with activation of the innate immune system and activation of signaling pathways associated with further up-regulation of other cytokines and chemokines. Neither of these cytokines was elevated before the increased expression of TLR2. This is in accordance with previous studies that demonstrated low TNF-α levels.18 In addition, when twitcher mice are crossed to TNF receptor 1 knockout mice, the progeny demonstrate no significant change in phenotype,67 leading to the conclusion that TNF-α does not appear to play a primary role in progression of disease.
The data support a mechanism of cellular activation and a sequence of events not previously described in the pathogenesis of globoid cell leukodystrophy. Earlier studies focused only on macrophages and microglia as they arrive in the CNS or begin to express markers of activation. This study indicates that microglia may be the primary cells of activation early in the disease process. Although it is possible that TLR2 could be a marker of inflammatory activation on microglia, its up-regulation occurs before the detection of all other markers of activation examined in this study, and occurs concurrently with morphological evidence of microglial and macrophage activation in the murine CNS. In addition, TLR2 signaling could be induced through a product released by oligodendrocytes when they are treated with psychosine. Phenotypic studies crossing TLR2 knockout mice to twitcher mice could be useful in further delineating the role of TLR2 in the progression of disease. Discovering that TLR2 may be the primary driver of inflammatory changes in the twitcher mouse could result in more effective, targeted treatments for GLD.
Acknowledgments
We thank Cyndi Trygg and Jessica Eiermann for their technical support throughout this study and Mary Barnes and Dr. Elizabeth Didier (Pathogen Detection and Quantification Core) at Tulane National Primate Research Center (Covington, LA) for being invaluable in setting up and running the multiplex cytokine assay.
Footnotes
Supported in part by the National Center for Research Resources, NIH, grants P51 OD011104 (Tulane National Primate Research Center) and T32 RR021309 (E.S.) and Tulane University grants.
Supplemental Data
Additional chemokines up-regulated during the course of globoid cell leukodystrophy. Shown chemokines are up-regulated significantly during the course of disease; however, they do not increase until late in the disease process. Their increased expression may play a role in the recruitment of monocytes to the brain, maturation/differentiation of progenitor cells in the brain, or a role in the activation of resident cells, such as astrocytes, in the course of disease. N, normal; twi, twitcher. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
References
- 1.Suzuki K., Suzuki Y. Globoid cell leucodystrophy (Krabbe’s disease): deficiency of galactocerebroside β-galactosidase. Proc Natl Acad Sci U S A. 1970;66:302–309. doi: 10.1073/pnas.66.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanazawa T., Nakamura S., Momoi M., Yamaji T., Takematsu H., Yano H., Sabe H., Yamamoto A., Kawasaki T., Kozutsumi Y. Inhibition of cytokinesis by a lipid metabolite, psychosine. J Cell Biol. 2000;149:943–950. doi: 10.1083/jcb.149.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Im D.-S., Heise C.E., Nguyen T., O’Dowd B.F., Lynch K.R. Identification of a molecular target of psychosine and its role in globoid cell formation. J Cell Biol. 2001;153:429–434. doi: 10.1083/jcb.153.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddy A.S., Kim J.H., Hawkins-Salsbury J.A., Macauley S.L., Tracy E.T., Vogler C.A., Han X., Song S.-K., Wozniak D.F., Fowler S.C., Klein R.S., Sands M.S. Bone marrow transplantation augments the effect of brain- and spinal cord-directed adeno-associated virus 2/5 gene therapy by altering inflammation in the murine model of globoid-cell leukodystrophy. J Neurosci. 2011;31:9945–9957. doi: 10.1523/JNEUROSCI.1802-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giri S., Jatana M., Rattan R., Won J.S., Singh I., Singh A.K. Galactosylsphingosine (psychosine)-induced expression of cytokine-mediated inducible nitric oxide synthases via AP-1 and C/EBP: implications for Krabbe disease. FASEB J. 2002;16:661–672. doi: 10.1096/fj.01-0798com. [DOI] [PubMed] [Google Scholar]
- 6.Giri S., Khan M., Nath N., Singh I., Singh A. The role of AMPK in psychosine mediated effects on oligodendrocytes and astrocytes: implication for Krabbe disease. J Neurochem. 2008;105:1820–1833. doi: 10.1111/j.1471-4159.2008.05279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borda J.T., Alvarez X., Mohan M., Ratterree M.S., Phillippi-Falkenstein K., Lackner A.A., Bunnell B.A. Clinical and immunopathologic alterations in rhesus macaques affected with globoid cell leukodystrophy. Am J Pathol. 2008;172:98–111. doi: 10.2353/ajpath.2008.070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bashir A., Haq E. Effect of psychosine on inducible nitric-oxide synthase expression under different culture conditions: implications for Krabbe disease. Eur Rev Med Pharmacol Sci. 2011;15:1282–1287. [PubMed] [Google Scholar]
- 9.Suzuki K. Twenty five years of the “psychosine hypothesis”: a personal perspective of its history and present status. Neurochem Res. 1998;23:251–259. doi: 10.1023/a:1022436928925. [DOI] [PubMed] [Google Scholar]
- 10.Taniike M., Mohri I., Eguchi N., Irikura D., Urade Y., Okada S., Suzuki K. An apoptotic depletion of oligodendrocytes in the twitcher, a murine model of globoid cell leukodystrophy. J Neuropathol Exp Neurol. 1999;58:644–653. doi: 10.1097/00005072-199906000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Zaka M., Wenger D.A. Psychosine-induced apoptosis in a mouse oligodendrocyte progenitor cell line is mediated by caspase activation. Neurosci Lett. 2004;358:205–209. doi: 10.1016/j.neulet.2003.12.126. [DOI] [PubMed] [Google Scholar]
- 12.Formichi P., Radi E., Battisti C., Pasqui A., Pompella G., Lazzerini P.E., Laghi-Pasini F., Leonini A., Stefano A.D., Federico A. Psychosine-induced apoptosis and cytokine activation in immune peripheral cells of Krabbe patients. J Cell Physiol. 2007;212:737–743. doi: 10.1002/jcp.21070. [DOI] [PubMed] [Google Scholar]
- 13.Haq E., Giri S., Singh I., Singh A.K. Molecular mechanism of psychosine-induced cell death in human oligodendrocyte cell line. J Neurochem. 2003;86:1428–1440. doi: 10.1046/j.1471-4159.2003.01941.x. [DOI] [PubMed] [Google Scholar]
- 14.Jatana M., Giri S., Singh A.K. Apoptotic positive cells in Krabbe brain and induction of apoptosis in rat C6 glial cells by psychosine. Neurosci Lett. 2002;330:183–187. doi: 10.1016/s0304-3940(02)00655-9. [DOI] [PubMed] [Google Scholar]
- 15.Yagi T., McMahon E.J., Takikita S., Mohri I., Matsushima G.K., Suzuki K. Fate of donor hematopoietic cells in demyelinating mutant mouse, twitcher, following transplantation of GFP+ bone marrow cells. Neurobiol Dis. 2004;16:98–109. doi: 10.1016/j.nbd.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Wu Y.P., Matsuda J., Kubota A., Suzuki K. Infiltration of hematogenous lineage cells into the demyelinating central nervous system of twitcher mice. J Neuropathol Exp Neurol. 2000;59:628–639. doi: 10.1093/jnen/59.7.628. [DOI] [PubMed] [Google Scholar]
- 17.LeVine S.M., Brown D.C. IL-6 and TNF-α expression in brains of twitcher, quaking and normal mice. J Neuroimmunol. 1997;73:47–56. doi: 10.1016/s0165-5728(96)00166-x. [DOI] [PubMed] [Google Scholar]
- 18.Ripoll C.B., Flaat M., Klopf-Eiermann J., Fisher-Perkins J.M., Trygg C.B., Scruggs B.A., McCants M.L., Leonard H.P., Lin A.F., Zhang S., Eagle M.E., Alvarez X., Li Y.T., Li S.C., Gimble J.M., Bunnell B.A. Mesenchymal lineage stem cells have pronounced anti-inflammatory effects in the twitcher mouse model of Krabbe’s disease. Stem Cells. 2011;29:67–77. doi: 10.1002/stem.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alvarez Y., Valera I., Municio C., Hugo E., Padrón F., Blanco L., Rodriguez M., Fernández N., Crespo M.S. Eicosanoids in the innate immune response: TLR and non-TLR routes. Mediators Inflamm. 2010;2010 doi: 10.1155/2010/201929. doi: pii:201929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bajramovic J.J. Regulation of innate immune responses in the central nervous system. CNS Neurol Disord Drug Targets. 2011;10:4–24. doi: 10.2174/187152711794488610. [DOI] [PubMed] [Google Scholar]
- 21.Bhat R., Steinman L. Innate and adaptive autoimmunity directed to the central nervous system. Neuron. 2009;64:123–132. doi: 10.1016/j.neuron.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 22.Finsen B., Owens T. Innate immune responses in central nervous system inflammation. FEBS Lett. 2011;585:3806–3812. doi: 10.1016/j.febslet.2011.05.030. [DOI] [PubMed] [Google Scholar]
- 23.Akira S., Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 24.Farez M.F., Quintana F.J., Gandhi R., Izquierdo G., Lucas M., Weiner H.L. Toll-like receptor 2 and poly(ADP-ribose) polymerase 1 promote central nervous system neuroinflammation in progressive EAE. Nat Immunol. 2009;10:958–964. doi: 10.1038/ni.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sloane J.A., Batt C., Ma Y., Harris Z.M., Trapp B., Vartanian T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci U S A. 2010;107:11555–11560. doi: 10.1073/pnas.1006496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Casula M., Iyer A.M., Spliet W.G.M., Anink J.J., Steentjes K., Sta M., Troost D., Aronica E. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience. 2011;179:233–243. doi: 10.1016/j.neuroscience.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 27.Chen K., Iribarren P., Hu J., Chen J., Gong W., Cho E.H., Lockett S., Dunlop N.M., Wang J.M. Activation of toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid beta peptide. J Biol Chem. 2006;281:3651–3659. doi: 10.1074/jbc.M508125200. [DOI] [PubMed] [Google Scholar]
- 28.Jana M., Palencia C.A., Pahan K. Fibrillar amyloid-beta peptides activate microglia via TLR2: implications for Alzheimer’s disease. J Immunol. 2008;181:7254–7262. doi: 10.4049/jimmunol.181.10.7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LeVine S.M., Wetzel D.L., Eilert A.J. Neuropathology of twitcher mice: examination by histochemistry, immunohistochemistry, lectin histochemistry and Fourier transform infrared microspectroscopy. Int J Dev Neurosci. 1994;12:275–288. doi: 10.1016/0736-5748(94)90075-2. [DOI] [PubMed] [Google Scholar]
- 30.Terrell K.A., Rasmussen T.A., Trygg C., Bunnell B.A., Buck W.R. Molecular beacon genotyping for globoid cell leukodystrophy from hair roots in the twitcher mouse and rhesus macaque. J Neurosci Methods. 2007;163:60–66. doi: 10.1016/j.jneumeth.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang B., Tian C., Zhang Z.G., Han F.C., Azem R., Yu H., Zheng Y., Jin G., Arnold J.E., Zheng Q.Y. Sh3pxd2b mice are a model for craniofacial dysmorphology and otitis media. PLoS One. 2011;6:e22622. doi: 10.1371/journal.pone.0022622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davidson L.B., Nessar R., Kempaiah P., Perkins D.J., Byrd T.F. Mycobacterium abscessus glycopeptidolipid prevents respiratory epithelial TLR2 signaling as measured by HβD2 gene expression and IL-8 release. PLoS One. 2011;6:e29148. doi: 10.1371/journal.pone.0029148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borda J.T., Alvarez X., Mohan M., Hasegawa A., Bernardino A., Jean S., Aye P., Lackner A.A. CD163, a marker of perivascular macrophages, is up-regulated by microglia in simian immunodeficiency virus encephalitis after haptoglobin-hemoglobin complex stimulation and is suggestive of breakdown of the blood-brain barrier. Am J Pathol. 2008;172:725–737. doi: 10.2353/ajpath.2008.070848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farkas D.L., Du C., Fisher G.W., Lau C., Niu W., Wachman E.S., Levenson R.M. Non-invasive image acquisition and advanced processing in optical bioimaging. Comput Med Imaging Graph. 1998;22:89–102. doi: 10.1016/s0895-6111(98)00011-1. [DOI] [PubMed] [Google Scholar]
- 35.Meldgaard M., Fenger C., Lambertsen K.L., Pedersen M.D., Ladeby R., Finsen B. Validation of two reference genes for mRNA level studies of murine disease models in neurobiology. J Neurosci Methods. 2006;156:101–110. doi: 10.1016/j.jneumeth.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 36.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Lewis S.D., Butchi N.B., Khaleduzzaman M., Morgan T.W., Du M., Pourciau S., Baker D.G., Akira S., Peterson K.E. Toll-like receptor 7 is not necessary for retroviral neuropathogenesis but does contribute to virus-induced neuroinflammation. J Neurovirol. 2008;14:492–502. doi: 10.1080/13550280802345723. [DOI] [PubMed] [Google Scholar]
- 38.Sun J., Zheng J.H., Zhao M., Lee S., Goldstein H. Increased in vivo activation of microglia and astrocytes in the brains of mice transgenic for an infectious R5 human immunodeficiency virus type 1 provirus and for CD4-specific expression of human cyclin T1 in response to stimulation by lipopolysaccharides. J Virol. 2008;82:5562–5572. doi: 10.1128/JVI.02618-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jou I., Lee J.H., Park S.Y., Yoon H.J., Joe E.-H., Park E.J. Gangliosides trigger inflammatory responses via TLR4 in brain glia. Am J Pathol. 2006;168:1619–1630. doi: 10.2353/ajpath.2006.050924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoon H.J., Jeon S.B., Suk K., Choi D.K., Hong Y.J., Park E.J. Contribution of TLR2 to the initiation of ganglioside-triggered inflammatory signaling. Mol Cells. 2008;25:99–104. [PubMed] [Google Scholar]
- 41.Hung L.-C., Lin C.-C., Hung S.-K., Wu B.-C., Jan M.-D., Liou S.-H., Fu S.-L. A synthetic analog of α-galactosylceramide induces macrophage activation via the TLR4-signaling pathways. Biochem Pharmacol. 2007;73:1957–1970. doi: 10.1016/j.bcp.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 42.Tennant I., Pound J., Marr L., Willems J., Petrova S., Ford C., Paterson M., Devitt A., Gregory C. Innate recognition of apoptotic cells: novel apoptotic cell-associated molecular patterns revealed by crossreactivity of anti-LPS antibodies. Cell Death Differ. 2013;20:698–708. doi: 10.1038/cdd.2012.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foell D., Wittkowski H., Vogl T., Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. 2007;81:28–37. doi: 10.1189/jlb.0306170. [DOI] [PubMed] [Google Scholar]
- 44.Barton G.M., Medzhitov R. Toll-like receptors and their ligands. Curr Top Microbiol Immunol. 2002;270:81–92. doi: 10.1007/978-3-642-59430-4_5. [DOI] [PubMed] [Google Scholar]
- 45.Carty M., Bowie A.G. Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem Pharmacol. 2011;81:825–837. doi: 10.1016/j.bcp.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 46.Jack C.S., Arbour N., Manusow J., Montgrain V., Blain M., McCrea E., Shapiro A., Antel J.P. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- 47.Janssens S., Beyaert R. Role of toll-like receptors in pathogen recognition. Clin Microbiol Rev. 2003;16:637–646. doi: 10.1128/CMR.16.4.637-646.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaisho T., Akira S. Toll-like receptors and their signaling mechanism in innate immunity. Acta Odontol Scand. 2001;59:124–130. doi: 10.1080/000163501750266701. [DOI] [PubMed] [Google Scholar]
- 49.Du X., Fleiss B., Li H., D’Angelo B., Sun Y., Zhu C., Hagberg H., Levy O., Mallard C., Wang X. Systemic stimulation of TLR2 impairs neonatal mouse brain development. PLoS One. 2011;6:e19583. doi: 10.1371/journal.pone.0019583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Okun E., Griffioen K.J., Son T.G., Lee J.H., Roberts N.J., Mughal M.R., Hutchison E., Cheng A., Arumugam T.V., Lathia J.D., van Praag H., Mattson M.P. TLR2 activation inhibits embryonic neural progenitor cell proliferation. J Neurochem. 2010;114:462–474. doi: 10.1111/j.1471-4159.2010.06778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bsibsi M., Nomden A., van Noort J.M., Baron W. Toll-like receptors 2 and 3 agonists differentially affect oligodendrocyte survival, differentiation, and myelin membrane formation. J Neurosci Res. 2012;90:388–398. doi: 10.1002/jnr.22767. [DOI] [PubMed] [Google Scholar]
- 52.Aravalli R.N., Hu S., Rowen T.N., Palmquist J.M., Lokensgard J.R. Cutting edge: TLR2-mediated proinflammatory cytokine and chemokine production by microglial cells in response to herpes simplex virus. J Immunol. 2005;175:4189–4193. doi: 10.4049/jimmunol.175.7.4189. [DOI] [PubMed] [Google Scholar]
- 53.Chiu Y.-C., Lin C.-Y., Chen C.-P., Huang K.-C., Tong K.-M., Tzeng C.-Y., Lee T.-S., Hsu H.-C., Tang C.-H. Peptidoglycan enhances IL-6 production in human synovial fibroblasts via TLR2 receptor, focal adhesion kinase, Akt, and AP-1-dependent pathway. J Immunol. 2009;183:2785–2792. doi: 10.4049/jimmunol.0802826. [DOI] [PubMed] [Google Scholar]
- 54.Miranda-Hernandez S., Gerlach N., Fletcher J.M., Biros E., Mack M., Korner H., Baxter A.G. Role for MyD88, TLR2 and TLR9 but not TLR1, TLR4 or TLR6 in experimental autoimmune encephalomyelitis. J Immunol. 2011;187:791–804. doi: 10.4049/jimmunol.1001992. [DOI] [PubMed] [Google Scholar]
- 55.Roy M., Richard J.F., Dumas A., Vallières L. CXCL1 can be regulated by IL-6 and promotes granulocyte adhesion to brain capillaries during bacterial toxin exposure and encephalomyelitis. J Neuroinflammation. 2012;9:18. doi: 10.1186/1742-2094-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spooren A., Kolmus K., Laureys G., Clinckers R., De Keyser J., Haegeman G., Gerlo S. Interleukin-6, a mental cytokine. Brain Res Rev. 2011;67:157–183. doi: 10.1016/j.brainresrev.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 57.Bowen K.K., Dempsey R.J., Vemuganti R. Adult interleukin-6 knockout mice show compromised neurogenesis. Neuroreport. 2011;22:126–130. doi: 10.1097/WNR.0b013e3283430a44. [DOI] [PubMed] [Google Scholar]
- 58.Pedchenko T.V., LeVine S.M. IL-6 deficiency causes enhanced pathology in twitcher (globoid cell leukodystrophy) mice. Exp Neurol. 1999;158:459–468. doi: 10.1006/exnr.1999.7125. [DOI] [PubMed] [Google Scholar]
- 59.Jakovcevski I., Filipovic R., Mo Z., Rakic S., Zecevic N. Oligodendrocyte development and the onset of myelination in the human fetal brain. Front Neuroanat. 2009;3:5. doi: 10.3389/neuro.05.005.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Omari K.M., John G., Lango R., Raine C.S. Role for CXCR2 and CXCL1 on glia in multiple sclerosis. Glia. 2006;53:24–31. doi: 10.1002/glia.20246. [DOI] [PubMed] [Google Scholar]
- 61.Tsai H.H., Frost E., To V., Robinson S., Ffrench-Constant C., Geertman R., Ransohoff R.M., Miller R.H. The chemokine receptor CXCR2 controls positioning of oligodendrocyte precursors in developing spinal cord by arresting their migration. Cell. 2002;110:373–383. doi: 10.1016/s0092-8674(02)00838-3. [DOI] [PubMed] [Google Scholar]
- 62.Omari K.M., Lutz S.E., Santambrogio L., Lira S.A., Raine C.S. Neuroprotection and remyelination after autoimmune demyelination in mice that inducibly overexpress CXCL1. Am J Pathol. 2009;174:164–176. doi: 10.2353/ajpath.2009.080350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu L., Callahan M.K., Huang D., Ransohoff R.M. Chemokine receptor CXCR3: an unexpected enigma. In: Gerald P.S., editor. Current Topics in Developmental Biology. Academic Press; Salt Lake City, UT: 2005. pp. 149–181. [DOI] [PubMed] [Google Scholar]
- 64.Omari K.M., John G.R., Sealfon S.C., Raine C.S. CXC chemokine receptors on human oligodendrocytes: implications for multiple sclerosis. Brain. 2005;128:1003–1015. doi: 10.1093/brain/awh479. [DOI] [PubMed] [Google Scholar]
- 65.Sørensen T.L., Trebst C., Kivisäkk P., Klaege K.L., Majmudar A., Ravid R., Lassmann H., Olsen D.B., Strieter R.M., Ransohoff R.M., Sellebjerg F. Multiple sclerosis: a study of CXCL10 and CXCR3 co-localization in the inflamed central nervous system. J Neuroimmunol. 2002;127:59–68. doi: 10.1016/s0165-5728(02)00097-8. [DOI] [PubMed] [Google Scholar]
- 66.Conductier G., Blondeau N., Guyon A., Nahon J.-L., RovËre C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J Neuroimmunol. 2010;224:93–100. doi: 10.1016/j.jneuroim.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 67.Pedchenko T.V., Bronshteyn I.G., LeVine S.M. TNF-receptor 1 deficiency fails to alter the clinical and pathological course in mice with globoid cell leukodystrophy (twitcher mice) but affords protection following LPS challenge. J Neuroimmunol. 2000;110:186–194. doi: 10.1016/s0165-5728(00)00345-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional chemokines up-regulated during the course of globoid cell leukodystrophy. Shown chemokines are up-regulated significantly during the course of disease; however, they do not increase until late in the disease process. Their increased expression may play a role in the recruitment of monocytes to the brain, maturation/differentiation of progenitor cells in the brain, or a role in the activation of resident cells, such as astrocytes, in the course of disease. N, normal; twi, twitcher. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.