

Abstract
Blood–retinal barrier (BRB) breakdown and related vascular changes are implicated in several ocular diseases. The molecules and mechanisms regulating BRB integrity and pathophysiology are not fully elucidated. Caveolin-1 (Cav-1) ablation results in loss of caveolae and microvascular pathologies, but the role of Cav-1 in the retina is largely unknown. We examined BRB integrity and vasculature in Cav-1 knockout mice and found a significant increase in BRB permeability, compared with wild-type controls, with branch veins being frequent sites of breakdown. Vascular hyperpermeability occurred without apparent alteration in junctional proteins. Such hyperpermeability was not rescued by inhibiting eNOS activity. Veins of Cav-1 knockout retinas exhibited additional pathological features, including i) eNOS-independent enlargement, ii) altered expression of mural cell markers (eg, down-regulation of NG2 and up-regulation of αSMA), and iii) dramatic alterations in mural cell phenotype near the optic nerve head. We observed a significant NO–dependent increase in retinal artery diameter in Cav-1 knockout mice, suggesting that Cav-1 plays a role in autoregulation of resistance vessels in the retina. These findings implicate Cav-1 in maintaining BRB integrity in retinal vasculature and suggest a previously undefined role in the retinal venous system and associated mural cells. Our results are relevant to clinically significant retinal disorders with vascular pathologies, including diabetic retinopathy, uveoretinitis, and primary open-angle glaucoma.
The retina is a photosensitive neural tissue lining the back of the eye that develops as an extension of the diencephalon.1 It is supported by two distinct blood supplies: the fenestrated choroidal vasculature (which supports the nutrient and waste exchange needs of the photoreceptors in the outer retina) and the inner retinal vasculature (which supports similar needs of the inner retinal neurons). The endothelial cells of the inner retinal vasculature provide a tight inner blood–retinal barrier (BRB), which is structurally and functionally analogous to the blood–brain barrier; the outer BRB is provided by a network of lateral junctional complexes that border adjacent cells of the monolayer retinal pigment epithelium.2 An intact BRB is essential for the maintenance of normal retinal structure and function, and loss of BRB structure and function is a pathological hallmark of several major vision-threatening diseases, including diabetic retinopathy, age-related macular degeneration, and retinopathy of prematurity.2,3 The discovery of vascular endothelial growth factor (VEGF) and the application of anti-VEGF therapies represent major breakthroughs in the clinical management of retinal vascular diseases.4,5 Not all patients respond to such therapy, however, and antagonizing VEGF activity could affect trophic support to the ciliary body and retina.6,7 Thus, a more complete understanding of the mechanisms and molecules that regulate BRB integrity and pathophysiology is crucial for the development of improved therapeutic interventions for such diseases.
Caveolin-1 (Cav-1) is the primary structural protein of the cholesterol- and sphingolipid-rich, flask-shaped membrane domains known as caveolae.8 Cav-1 intrinsically participates in multiple caveolar functions, including lipid trafficking, transcytosis, mechanosensing, and cell signaling.8,9 Mice in which Cav-1 is globally deleted are viable, but they exhibit several abnormalities, including insulin resistance, alterations in lipid metabolism, defective albumin uptake, pulmonary hypertension, and hypertrophic cardiomyopathy.10–13 Loss of Cav-1 also increases pulmonary hyperpermeability14,15 and induces abnormal angiogenic responses to VEGF.16–18
Several of these pathologies are mediated by endothelial nitric oxide synthase (eNOS), which is normally negatively regulated by its interaction with Cav-1.19 Thus, Cav-1 knockout (KO) mice expectedly exhibit hyperactive eNOS and impaired nitric oxide (NO) signaling.10,11 Furthermore, vascular permeability and other cardiovascular phenotypes in Cav-1 KO mice can be rescued either by pharmacological inhibition of NO production or by re-expression of Cav-1 in the vascular endothelium (which also inhibits NO production).14,15 Cav-1 also promotes atherosclerotic lesion formation, potentially by mediating lipoprotein trafficking across the vascular endothelium.20 These various studies highlight the important roles that Cav-1 plays both in normal vascular physiology and in pathophysiology. Less is known about the function or functions of Cav-1 in tight barrier–forming vascular beds, such as the inner BRB.
The inner BRB is composed of a monolayer of tightly sealed endothelial cells with well-developed interendothelial tight junctions (TJs) and adherens junctions (AJs).2 Furthermore, the retinal vascular endothelium actively interacts with several additional support cells, including mural cells (vascular smooth muscle cells and pericytes) and glia (Müller glia and astrocytes).2,21 In the neural retina, vascular cells (both endothelial and mural) and Müller glia predominantly express Cav-1.22 Cav-1 is also found in photoreceptors.23,24 Interestingly, in retinal (and brain) vascular endothelium and pericytes, caveolae are largely polarized abluminally,25,26 raising doubts about whether caveolae perform similar transcellular transport roles in the central nervous system as are reported in the lung.27 Up-regulation of Cav-1 transcripts in experimental diabetic retinopathy correlates with BRB breakdown,28 as does an increase in the number of caveolae on the abluminal surface of pericytes.29 Although this up-regulation correlates with enhanced pathological permeability, it is unclear whether Cav-1 plays a compensatory protective role or a pathological one. Of note, similar Cav-1 up-regulation occurs in cerebral ischemic injury; in that case, loss of Cav-1 enhances ischemic infarct volumes, indicating a protective role for Cav-1 expression.30 However, direct, rigorous analysis of the role of Cav-1 in BRB integrity has not been reported previously.
Our research group recently reported that Cav-1 KO mice exhibit reduced retinal neuronal function in vivo as a consequence of changes in the retinal microenvironment.31 Because the retinal microenvironment is largely dependent on an intact BRB, we hypothesized that Cav-1 might be important in the maintenance of the BRB. In the present study, we tested this idea using mice deficient in Cav-1 and found that loss of Cav-1 causes BRB hyperpermeability. This BRB disruption generally localized to large branch retinal veins in the superficial retina, which were also abnormally enlarged. Although neither hyperpermeability nor venous enlargement was dependent on eNOS expression and activity, we did find alterations in the association of mural cells with these enlarged veins, suggesting that Cav-1 plays an important role in endothelial–mural cell interactions in the retina.
Materials and Methods
Animals
All procedures were performed in accord with the Association for Research in Vision and Ophthalmology’s Statement for the Use of Animals in Ophthalmic and Visual Research and were approved by the Institutional Animal Care and Use Committees of the University of Oklahoma Health Sciences Center and the Dean A. McGee Eye Institute. Experiments were performed on Cav-1 KO mice,11 Cav-1 and eNOS double KO (DKO) mice,32 and eNOS KO mice (stock no. 002684; Jackson Laboratory, Bar Harbor, ME) on a C57BL/6J background. C57BL/6J mice (stock no. 000664; Jackson Laboratory) were used as wild-type (WT) controls.
Quantification of BRB Breakdown
Mice were anesthetized and injected with 100 μL of fluorescein isothiocyanate (FITC)–dextran (4 kDa, 50 mg/mL; Sigma-Aldrich, St. Louis, MO) into the circulation. After 15 minutes, approximately 100 μL of blood was collected, and mice were perfused with PBS. Retinas were collected and weighed, and FITC-dextran was extracted and filtered as described previously.33 Blood samples were centrifuged (7000 × g), and plasma was collected and diluted 1:1000 in water. Fluorescence was measured (excitation 485 nm; emission 538 nm) in a plate reader (BMG LABTECH, Ortenberg, Germany), and the amount of FITC-dextran in the retina and plasma was calculated based on a FITC-dextran standard curve. Retinal autofluorescence was corrected based on uninjected controls. BRB breakdown was calculated using the equation
| (1) |
Results are expressed in units of (μL plasma/g retina wet weight)/hour.
L-NAME Treatment and NO Measurement
Cav-1 KO and control mice were given an intraperitoneal injection of 30 mg/kg N-ω-nitro-l-arginine methyl ester (L-NAME; Cayman Chemical, Ann Arbor, MI) or vehicle control (PBS) at 2 hours before quantification of BRB breakdown. L-NAME dosage was determined to be effective by measuring plasma nitrite and nitrate concentration with a commercial colorimetric kit (Cayman Chemical) according to the manufacturer’s instructions.
Retinal Flatmount Preparation, IHC, and Morphometry
Eyes were enucleated and immersion-fixed in 4% paraformaldehyde in PBS. Retinas were carefully dissected under a stereomicroscope (Carl Zeiss Microscopy, Jena, Germany), permeabilized with 1% Triton X-100 in PBS, blocked with 10% normal horse serum, and incubated with the following primary antibodies at 4°C overnight: hamster anti-CD31 (1:200; EMD Millipore, Billerica, MA), rabbit anti–caveolin-1 (1:200; BD Pharmingen, San Diego, CA), FITC-labeled goat anti-albumin (1:200; Bethyl Laboratories, Montgomery, TX), rabbit and/or guinea pig anti-NG2 (1:200),34,35 rabbit anti-PDGFRβ (1:200),36 rat anti-PDGFRβ (1:500; eBioscience, San Diego, CA), Cy3-conjugated anti-alpha smooth muscle actin (αSMA) (1:400; Sigma-Aldrich), rat anti–ZO-1 (1:50; EMD Millipore), mouse anti-occludin (1:50; Life Technologies, Carlsbad, CA), rat anti–VE-cadherin (1:50; BD Pharmingen), and FITC-conjugated mouse anti–claudin-5 (1:200; Life Technologies). After washing with 0.1% Triton X-100 in PBS, the retinas were incubated with the appropriate fluorophore-conjugated secondary antibodies [1:500 (Life Technologies) and/or 1:200 (Jackson ImmunoResearch Laboratories, West Grove, PA)] at 4°C overnight. After another washing with 0.1% Triton X-100 in PBS, four to five radial cuts were made in the retinas, from the edge toward the optic nerve head (ONH), ganglion cell layer up, for flat-mounting in glycerol:PBS (1:1, v/v). For junction protein visualization, eyes were hemisected and fixed in 4% paraformaldehyde containing calcium and magnesium for 15 minutes.
For visualization of BRB breakdown, retinal frozen sections were prepared as described previously, with minor modification.37 In brief, eyecups (with cornea and lens removed) were fixed in 2% paraformaldehyde in PBS for 20 minutes, cryoprotected by sequential incubations in 10%, 15%, and 30% sucrose in PBS, and frozen in optimal cutting temperature compound (Tissue-Tek OCT; Sakura Finetek, Auburn, CA). Frozen sections of 14-μm thickness were cut using a cryostat (Leica Biosystems, Buffalo Grove, IL) and processed for staining with rabbit anti-collagen IV (1:800; EMD Millipore) and FITC-labeled goat anti-albumin (1:200; Bethyl Laboratories); nuclei were counterstained with DAPI (Sigma-Aldrich). Imaging was performed using confocal laser scanning microscopes FV500 and FV1200 (Olympus, Tokyo, Japan); images were processed with Photoshop CS5 (Adobe Systems, San Jose, CA).
For morphometric analysis of vein and artery diameters, images were processed using ImageJ version 10.2 software (NIH, Bethesda, MD). Defined and reproducible locations were chosen for measurements. For measurement of branch retinal vein diameter, the starting point was chosen at the bifurcation and extended toward the ONH for approximately 500 μm. Four branch retinal veins were measured in each mouse (two per retina), and values were averaged and counted as one sample. For artery diameter, the starting point was chosen at the ONH and extended toward the retinal periphery for approximately 500 μm. If the artery bifurcated before 500 μm of distance, only the common artery trunk was measured. In each mouse, 10 to 12 arteries were measured, averaged, and counted as one sample. (Usually one retina has five to six arteries entering from the ONH.)
RNA Extraction and RT-qPCR
Total RNA was isolated from previously snap-frozen retinas with TRIzol reagent (Life Technologies) and treated with DNase (DNA-free kit; Life Technologies). One microgram of RNA was used for cDNA synthesis (iScript cDNA synthesis kit; Bio-Rad Laboratories, Hercules, CA). The levels of NG2 and RPL19 (housekeeping gene) transcripts were measured by quantitative real-time PCR (RT-qPCR) using a SYBR Green PCR mix (SsoFast EvaGreen supermix; Bio-Rad Laboratories) and a CFX96 real-time PCR detection system (Bio-Rad Laboratories) according to the manufacturer’s instructions. Primers to NG2 and RPL19 were designed to span intron–exon boundaries, to avoid the chance of amplifying potentially residual genomic DNA contamination. Primers used were as follows: mouse NG2 forward primer (5′-CGTGATGGTGTCTTTCGATG-3′) and reverse primer (5′-GAGTACATCATGCCGACTGC-3′); mouse/rat/human RPL19 forward primer (5′-TCACAGCCTGTACCTGAAGG-3′) and reverse primer (5′-TCGTGCTTCCTTGGTCTTAG-3′). Relative quantities of NG2 expression were calculated by the comparative CT value method.37
Detergent-Resistant Membrane Fractionation
Detergent-resistant membranes were isolated from retinas as described previously38,39 with slight modifications. Four retinas each from WT and Cav-1 KO mice were pooled and homogenized in TNE buffer [10 mmol/L Tris-Cl (pH 7.4), 100 mmol/L NaCl, 1 mmol/L EDTA, supplemented with protease inhibitor cocktail] (Calbiochem; EMD Millipore). After centrifugation in a benchtop microcentrifuge (Eppendorf, Hamburg, Germany), the supernatant was discarded and the pellet was saved. The pellet was resuspended in 250 μL of ice-cold TNE buffer containing 0.5% Triton X-100, diluted with another 250 μL of TNE buffer, and homogenized. The homogenates were transferred to 4-mL centrifuge tubes (Beckman Coulter, Brea, CA), placed at the bottom of a discontinuous sucrose gradient [500 μL 80% (w/v), 3 mL 35% (w/v), and 500 μL 5% (w/v)]. The resulting 5% to 40% discontinuous sucrose gradient was centrifuged at 40,000 rpm for 20 hours in a swing-bucket rotor (model SW60Ti; Beckman Coulter). Five-hundred-microliter fractions collected from top to bottom of the gradient (fractions 1 to 8) were used for Western blotting.
Western Blotting and Immunoprecipitation
Retinas were lysed in radioimmunoprecipitation assay buffer [50 mmol/L Tris (pH 7.4), 150 mmol/L NaCl, 1% NP-40, 0.1% sodium deoxycholate, 2.5 mmol/L EDTA] supplemented with protease inhibitor cocktail (Calbiochem; EMD Millipore). Protein content was determined with a Pierce BCA assay (Thermo Fisher Scientific, Rockford, IL), using bovine serum albumin as a standard. Equal amounts of protein were resolved on 4% to 20% or 4% to 12% Tris-glycine gradient gels (Life Technologies). Proteins were transferred to nitrocellulose membranes and then probed with antibodies, using standard methods. Primary antibodies and dilutions were as follows: rat anti–ZO-1 (1:1000; EMD Millipore), rabbit anti-occludin (1:500; Life Technologies), rabbit anti–claudin-5 (1:500; Life Technologies), rabbit anti–VE cadherin (1:1000; Abcam, Cambridge, MA), rabbit anti–caveolin-1 (1:3000; BD Pharmingen), and mouse anti–α-tubulin (1:500; Sigma-Aldrich). Immunoreactivity was detected using horseradish peroxidase (HRP)–conjugated secondary antibodies (1:5000; GE Healthcare, Little Chalfont, UK) and imaged with a Kodak In Vivo F-Pro system (Carestream, Rochester, NY).
Occludin phosphorylation was assessed by Western blotting coupled with phosphatase treatment, as described previously.40 Phosphorylation of occludin results in its reduced migration on SDS-PAGE gels, and these more slowly migrating bands collapse to a single band after phosphatase treatment. We therefore treated retinal lysates from Cav-1 KO and WT mice with or without 20 units of alkaline phosphatase (New England Biolabs, Ipswich, MA), to determine differential phosphorylation between genotypes.
Tyrosine phosphorylation of tight junction protein ZO-1 was determined by phosphotyrosine immunoprecipitation followed by ZO-1 and phosphotyrosine immunoblotting. Retinas were lysed as described above, and 240 μg of protein was diluted to 1 μg/μL with radioimmunoprecipitation assay buffer. A portion of the protein (40 μg) was saved as input; the remaining 200 μg was precleared with protein A/G PLUS-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) before incubation with 2 μg anti-phosphotyrosine antibody (clone 4G10; EMD Millipore) at 4°C overnight. Protein A/G PLUS-agarose beads (10 μL) were added to the samples and incubated under rotation for 1 hour at 4°C. Protein-bound beads were collected by centrifugation, and the resulting supernatants were saved as flow-through. Beads were washed three times with 1× radioimmunoprecipitation assay buffer and resuspended in 2× Laemmli sample buffer for Western blotting analyses using both ZO-1 and phosphotyrosine antibodies.
HRP Tracing and Transmission Electron Microscopy
HRP tracing was performed as described previously.41,42 HRP (type II, 500 mg/kg; Sigma-Aldrich) was injected intravenously and allowed to circulate for 15 minutes. Eyes were enucleated and immersion-fixed in Karnovsky’s fixative [2% glutaraldehyde, 2% paraformaldehyde, 0.02% CaCl2 in 0.1 mol/L cacodylate buffer (pH 7.2)] for 3 hours. The eyecups were embedded in 2% agarose (Life Technologies) and cut into 100-μm slabs using a vibratome (Leica Biosystems). Slabs were washed in PBS (pH 7.4) at 4°C overnight, to remove any fixative, and then were incubated with 3,3′-diaminobenzidine (DAB) solution [5 mg DAB in 10 mL 50 mmol/L Tris-Cl buffer (pH 7.6)] at room temperature for 3 hours. The sections were then incubated with fresh DAB solution containing 1% H2O2 (Sigma-Aldrich) at room temperature for an additional 3 hours. Sections were then washed and refixed in Karnovsky’s fixative and 1% OsO4 (Electron Microscopy Sciences, Hatfield, PA), dehydrated in ethanol gradients, and embedded in Spurr's resin. Sections were cut with a diamond knife, placed on copper grids, and viewed with a JEOL 1200 EX transmission electron microscope (JEOL USA, Peabody, MA). Mice without HRP injection were used as controls for endogenous peroxidase activity.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism software version 5 (GraphPad Software, La Jolla, CA). For comparison of two means, Student’s t-test was used; for comparing more than two means, one-way or two-way analysis of variance with Newman–Keuls post hoc analysis was used. P values of < 0.05 were considered significant. Data are expressed as means ± SEM.
Results
Loss of Cav-1 Results in BRB Breakdown
We recently reported that loss of Cav-1 results in reduced retinal photoresponses measured in vivo.31 However, when Cav-1 KO retinas are placed in controlled medium in vitro, photoresponses are similar to those of normal WT retinas. This indicates that the reduced retinal function results from perturbation of the retinal milieu in situ. Because a tight, intact BRB is essential to the maintenance of the retinal environment and because Cav-1 has been shown to modulate permeability in other vascular beds,14,16,17 we hypothesized that loss of Cav-1 would result in BRB breakdown. To quantitatively measure BRB integrity, we adapted a previously published FITC-dextran assay33 and observed a significant increase in FITC-dextran accumulation in Cav-1 KO retinal tissues, compared with WT controls [WT, 14.05 ± 2.185 (μL/g)/hour; KO, 30.59 ± 2.850 (μL/g)/hour] (P = 0.0017) (Figure 1A), indicating a loss in BRB integrity.
Figure 1.

Loss of Cav-1 results in BRB hyperpermeability. A: Cav-1 KO and WT mice were intravenously injected with 4-kDa FITC-dextran, and BRB permeability was quantified. Cav-1 KO retinas exhibit a two-fold increase in leakage. B and C: Endogenous albumin staining (green) of retinal flatmount from WT (B) and Cav-1 KO (C) mice reveals leakage site (arrow) in the Cav-1 KO mouse. D and E: FITC-conjugated albumin was injected into WT (D) and Cav-1 KO (E) mice; retinas were flat-mounted and visualized for albumin extravasation. Compared with WT, the Cav-1 KO retina exhibits more leakage (arrow), and over a larger area, around a vein. F and G: IHC of albumin (green) on frozen sections of WT (F) and Cav-1 KO retinas (G). Vessels were immunolabeled with anti–collagen IV antibody (red). Extravasation of albumin is seen around the large vessels in the Cav-1 KO retina (arrows), but not in the WT or in nonleaky vessels in the Cav-1 KO retina. Representative images are shown. Data are expressed as means ± SEM. n = 5. ∗∗P < 0.01 versus WT. Scale bar = 50 μm. Alb, albumin; Col IV, collagen IV.
To localize the barrier defect, we directly stained for endogenous albumin in retinal flatmounts (Figure 1, B and C) or intravenously injected FITC-conjugated albumin (Figure 1, D and E). The majority of leakage sites in the superficial layer were associated with branch retinal veins in Cav-1 KO retinas. Each mouse retina typically has two large branch retinal veins that drain the circumferential vein. We observed eight leakage sites in 20 KO branch retinal veins, but only two sites in 20 WT branch retinal veins (n = 10 retinas per genotype). We observed only rare leakage sites associated with branch retinal arteries. There are typically five to six such arteries stemming from the ONH; we observed only three leakage sites in KO arteries and one leakage site in WT arteries (>50 arteries per genotype; n = 10 retinas). Thus, these large branch retinal veins were hotspots for extravasation.
We stained frozen sections from Cav-1 KO and WT retinas for albumin and observed that Cav-1 KO retinas exhibited diffuse immunopositive albumin staining around the large vessels in the superficial layer and deep capillary plexus (Figure 1G), whereas albumin was confined to the vessel lumen both in control retinal sections (Figure 1F) and in nonleaky areas of Cav-1 KO retinal sections. This leakage pattern is consistent with previous reports that early BRB breakdown localizes to veins and capillaries in diabetic retinopathy and uveoretinitis.43,44 The venous localization of BRB breakdown in Cav-1 KO retinas correlated with the vessels exhibiting the highest expression of Cav-1 in the WT retinal vasculature (Figure 2). Cav-1 was highly expressed in branch retinal veins and venules and was expressed less predominantly in branch retinal arteries and capillaries. These results indicate that loss of Cav-1 leads to BRB impairment, and suggests a potential specific role of Cav-1 in the venous system.
Figure 2.
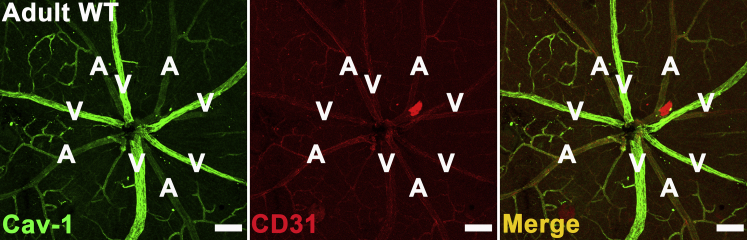
Cav-1 predominantly localizes to branch retinal veins in adult WT retinal vasculature. Retinas from adult WT mice were stained with antibodies to Cav-1 (green) and CD31 (red) to label the vasculature. Cav-1 strongly localizes to branch retinal veins and is weakly expressed by arteries and capillaries. Scale bar = 100 μm. A, artery; V, vein.
Loss of Cav-1 Results in Paracellular Permeability
Endothelial permeability can occur via paracellular or transcellular routes; the former is determined by the complexity of TJs and AJs and the latter involves caveolae trafficking.45 Loss of Cav-1 impairs transcytosis of albumin across the endothelium.46 However, if active albumin transport requires caveolae and Cav-1 in the retinal vasculature, why do we observe increased albumin extravasation in the BRB of Cav-1 KO mice? Previous studies have demonstrated crosstalk between paracellular and transcellular pathways,45,47 but the existence of caveolae-mediated transport in retinal vessels has been disputed.26 We therefore set out to determine, using ultrastructural localization of HRP extravasation, whether the loss of caveolae results in increased paracellular permeability.
HRP reaction product stained retinal vascular basement membranes and leaked into the retinal parenchyma in Cav-1 KO mice (Figure 3B), but was confined to the vessel lumen in the retinas of WT controls (Figure 3A). The presence of caveolae was evident in the WT retinas, with many of them facing the abluminal side of endothelium, as previously reported25,26 (Figure 3, A and C, and Supplemental Figure S1). Although HRP was apparent in vesicular compartments with caveolar characteristics in WT retinal endothelium (Figure 3C), we could not determine whether these were stable caveolae on the luminal surface or were trafficking vesicles. The first possibility is suggested by the lack of HRP reaction product on the abluminal side. Importantly, we did not observe HRP passing through endothelial junctions in WT vessels (Figure 3C). In Cav-1 KO vessels, morphologically identifiable caveolae were undetectable (Figure 3, B and D, and Supplemental Figure S1). HRP reaction product was frequently found abluminal to morphologically identifiable TJs (Figure 3D) and to prominently stain vascular basement membranes in Cav-1 KO retinal vessels (Figure 3, B and D). Staining abluminal to TJs indicated paracellular permeability, but prominent basement membrane staining was also observed at locations distant from TJs. We therefore cannot exclude additional increases in transcellular permeability in Cav-1 KO retinal vessels. Because the harsh reaction conditions necessary for the peroxidase reaction often resulted in poorly preserved ultrastructural morphology, we also examined vasculature in WT and Cav-1 KO retinas that were not processed for HRP; we found no overt changes in junctional appearance (Supplemental Figure S1).
Figure 3.

Hyperpermeability in Cav-1 KO retina occurs, at least in part, through the paracellular pathway, as can be seen in electron micrographs. A and B: HRP reaction products stain the basement membrane (arrows) in Cav-1 KO vessels (B), but are confined to the lumen in WT vessels (A). Caveolae (arrowheads) are evident in WT vasculature, but absent in KO vasculature. C and D: At higher magnification, it can be seen that HRP reaction product remains on the luminal side of the TJ (boxed region) in a WT vessel (C), but passes through the junction (boxed region) in a Cav-1 KO vessel (D). In WT vessels (C), abluminal caveolae (arrowheads) and luminal, HRP-laden caveolae (v) are apparent. Scale bar = 500 nm. BM, basement membrane; EC, endothelial cell; L, lumen; P, pericyte.
Although the ultrastructural localization of HRP extravasation provided unequivocal evidence of junction dysfunction, we did not detect significant changes in the protein levels of several TJ proteins (ZO-1, occludin, and claudin-5) and AJ proteins (VE-cadherin) by Western blot analysis (Figure 4, A–E). Because BRB breakdown was largely localized to retinal veins, we suspected that Western blot analysis of whole retinal protein extracts might not detect such local changes in junction proteins; we therefore examined TJ and AJ protein localization specifically in branch retinal veins by immunostaining of flatmounts. However, this analysis also failed to detect any changes in localization (Figure 4, F–I) of junctional proteins in Cav-1 KO retinal vasculature, compared with WT controls.
Figure 4.

Junctional proteins are not affected in Cav-1 KO retina. A: Representative blots of TJ proteins (claudin-5, ZO-1, and occludin) and an AJ protein, VE-cadherin, from retinal lysates from Cav-1 KO and WT retinas. B–E: Quantitative densitometric analyses of junctional proteins reveal no statistical differences in protein levels between Cav-1 KO and WT retinas. F–I: IHC of claudin-5 (Cldn5) (F), ZO-1 (G), occludin (H), and VE-cadherin (I) labeled in green, in Cav-1 KO and WT retinal vasculature. Vessels are labeled in red with anti-CD31 antibody. All assessed proteins localized to the vascular endothelium properly, without apparent breaks or mislocalization. Data are expressed as means ± SEM. n = 6–9. Scale bar = 20 μm. VE-Cad, VE-cadherin.
Phosphorylation of TJ proteins is proposed to affect junctional stability.40,48 Given that the loss of Cav-1 causes basal hyperphosphorylation of several proteins [notably, extracellular signal-regulated kinase (ERK), Akt, and eNOS15,49] we examined the effect of Cav-1 loss on the phosphorylation status of the TJ proteins ZO-1 and occludin. We did not recover any detectable ZO-1 in immunoprecipitates of tyrosine phosphorylated proteins from Cav-1 KO and WT retinal lysates (Supplemental Figure S2A), using an antibody previously demonstrated to react with phosphotyrosine sites in ZO-1.40 Interestingly, we did recover several phosphotyrosine-positive proteins of unknown identity in Cav-1 KO retinas, but not in controls, validating our immunoprecipitation strategy and suggesting that loss of Cav-1 results in hyperactive tyrosine phosphorylation (Supplemental Figure S2A). In separate experiments, we examined the phosphorylation of occludin by phosphatase-induced SDS-PAGE mobility shift.40 A proportion of occludin was phosphorylated in both Cav-1 KO and WT retinas, to a similar extent (Supplemental Figure S2B). Taken together, these results suggest that loss of Cav-1 does not alter the phosphorylation status of two major TJ proteins.
It has also been shown that Cav-1 mediates claudin-5 internalization during TJ remodeling in brain endothelial cells.50 We therefore prepared detergent-resistant membranes to determine whether claudin-5 distribution is altered in the absence of Cav-1. Claudin-5 fractionated similarly in WT and Cav-1 KO retinas (Supplemental Figure S3). Overall, our results suggest that, although some extravasation in the Cav-1 KO occurs through the paracellular pathway, there are no overt concomitant alterations in expression, localization, phosphorylation, or detergent-resistant membrane distribution of junctional proteins. Our results are consistent with recent work by Armstrong et al,47 who showed that inhibiting the transcellular pathway rapidly increases paracellular leakage without affecting junctional proteins.
BRB Breakdown Is Independent of eNOS Expression and Hyperactivity
Cav-1 is a well-established regulator of eNOS activity,19 and several vascular abnormalities observed when Cav-1 is lost, including lung hyperpermeability, are rescued when eNOS is inhibited.14 Furthermore, excess NO production is linked to BRB breakdown in experimental diabetic retinopathy.51 To assess whether excessive NO production is a mechanism for BRB breakdown in response to Cav-1 loss, we administered L-NAME (a well-characterized NOS inhibitor) to Cav-1 KO and WT control mice and assessed acute permeability with FITC-dextran permeability assay. Although L-NAME effectively inhibited nitrate and nitrite levels by 65% in Cav-1 KO serum (PBS, 11.89 ± 1.937 μmol/L; L-NAME, 4.253 ± 0.6186 μmol/L) (P = 0.0021), it did not rescue BRB hyperpermeability (P = 0.7305) (Figure 5A).
Figure 5.
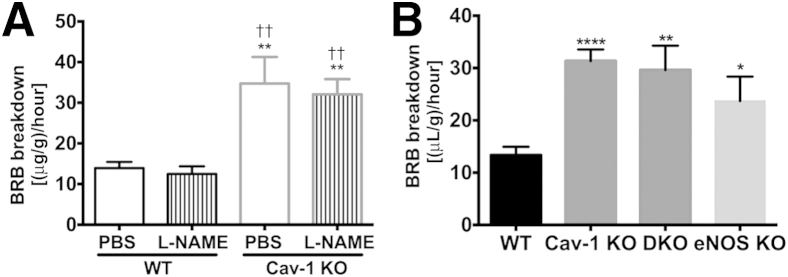
BRB hyperpermeability in Cav-1 KO retinas is independent of eNOS action. A: Cav-1 KO and WT mice were treated with L-NAME for 2 hours and injected intravenously with FITC-dextran. Quantitative analysis of BRB integrity was performed. L-NAME treatment did not rescue Cav-1–induced vascular hyperpermeability, nor did it increase permeability in WT controls. B: WT, Cav-1 KO, Cav-1 and eNOS DKO, and eNOS KO mice were injected intravenously with FITC-dextran and BRB breakdown was evaluated. Loss of eNOS in the Cav-1 KO background did not correct BRB hyperpermeability. Data are expressed as means ± SEM. n = 9 (WT PBS), n = 8 (WT L-NAME), and n = 5 (Cav-1 KO PBS and L-NAME) (A). n = 4 (DKO and eNOS KO), n = 7 (WT), and n = 10 (Cav-1 KO) (B). ∗∗P < 0.01 versus WT PBS; ††P < 0.01 versus WT L-NAME (A). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗∗P < 0.0001 versus WT (B). One-way analysis of variance followed by Newman–Keuls post hoc test.
Because the effect of chronic eNOS hyperactivity on the BRB of Cav-1 KO retinas might not be corrected by transient inhibition of NOS activity by L-NAME, we also examined BRB breakdown in DKO mice, in which both Cav-1 and eNOS were deleted. BRB hyperpermeability was not rescued in DKO retinas (P = 0.0028 WT versus DKO; P = 0.7146 Cav-1 KO versus DKO) (Figure 5B), reinforcing the idea that eNOS expression or hyperactivity is not responsible for the BRB breakdown observed in Cav-1 KO retinas. This is in contrast to what has been reported for other vascular beds14–16 and thus suggests a different mechanism at play in the retinal vasculature. Interestingly, we did observe increased BRB permeability in eNOS KO retinas (P = 0.0293 WT versus eNOS KO) (Figure 5B), which agrees with a previous report that eNOS KO mice have slightly higher basal BRB leakage and develop significantly higher permeability under diabetic conditions.52 Of note, transient inhibition of NOS activity in control mice did not induce BRB hyperpermeability (Figure 5A), supporting the idea that the increased permeability observed in eNOS KO mice results from vascular changes due to chronic loss of eNOS.
Loss of Cav-1 Increases Venous Diameter Independent of eNOS Action
Because retinal veins were identified as hotspots for BRB breakdown (Figure 1), and because Cav-1 is primarily localized to branch retinal veins in WT retinal vasculature (Figure 2), we focused our attention on other venous pathologies. Interestingly, we observed apparent increases in venous diameter in Cav-1 KO retinas, compared with WT controls (Figure 6B). To quantify these changes, we measured venous diameters at a defined and reproducible location just adjacent to the bifurcation where peripheral circumferential veins converge on branch retinal veins (Figure 6A). Cav-1 KO branch retinal veins were significantly enlarged, compared with WT controls (Figure 6, B and C). We hypothesized that this enlargement results from eNOS hyperactivity and overproduction of the potent vasodilator NO. To test this hypothesis, we measured venous diameters in DKO mice; contrary to our expectations, the venous enlargement was not corrected by eNOS deletion (Figure 6, B and C). Similarly, transient inhibition of NO production by L-NAME did not normalize venous diameters to WT levels, nor did L-NAME treatment alter venous diameter in WT retinas (Figure 6D). These results indicate that loss of Cav-1 induces eNOS-independent changes in venous diameter.
Figure 6.

Loss of Cav-1 results in venous enlargement independent of eNOS action. Retinal flatmounts were prepared from WT, Cav-1 KO, Cav-1 and eNOS DKO, and eNOS KO mice and were immunostained with anti-CD31 antibody to label the vasculature. A: Schematic representation of locations used for obtaining diameter measurements. B: Representative images of branch retinal veins showing enlarged vessels in mice in which Cav-1 is deleted (ie, Cav-1 KO and DKO mice). Scale bar = 50 μm. C: Quantification of venous enlargement shows the lack of eNOS effect on enlargement. D: Quantification of venous diameter in the L-NAME- and vehicle-treated WT and Cav-1 KO retinas shows that transient NOS inhibition does not correct venous enlargement. Data are expressed as means ± SEM. n = 13 (WT), n = 15 (Cav-1), n = 11 (DKO), and n = 7 (eNOS KO) (A). n = 4–7 per treatment group (B). ∗∗P < 0.01, ∗∗∗∗P < 0.0001 versus WT; †P < 0.05, †††P < 0.001 versus eNOS KO (C). ∗∗P < 0.01, ∗∗∗P < 0.001 versus WT PBS; †††P < 0.001, ††††P < 0.0001 versus WT L-NAME (D). Scale bar = 50 μm. One-way analysis of variance followed by Newman–Keuls post hoc test.
Because the influence of eNOS might be more pronounced on the arterial circulation, we also examined artery diameters in Cav-1 KO and WT mice. Similar to retinal veins, branch retinal arteries in Cav-1 KO mice were markedly dilated, compared with WT controls (Supplemental Figures S4 and S5). Unlike veins, however, this dilation was corrected by L-NAME treatment in both WT and Cav-1 KO retinas (Supplemental Figure S4), suggesting that dilation of retinal arteries is attributable to constitutive release of NO, as has been shown for isolated bovine retinal arteries.53 This is also consistent with an earlier report of NO-mediated flow-induced arteriole dilation in isolated porcine retinal vessels.54 Similar observations have been made in Cav-1 KO cerebral arteries, which were significantly dilated, compared with those of WT mice; NOS inhibition constricted arteries in both genotypes.55 In the present study, however, artery diameter in DKO retinas was not normalized to WT level and was also surprisingly increased in eNOS KO mice (Supplemental Figure S5). Such effects may be due to multiple compensatory mechanisms resulting from chronic loss of eNOS by genetic ablation. Several local factors that induce arterial dilation have been identified in the retina, including glial-derived NO, prostaglandins, and an as yet unidentified retinal-derived relaxing factor (reviewed by Delaey and Van De Voorde56).
Taken together, these results indicate that venous enlargement is not due to the excessive production of NO from hyperactive eNOS action and suggest a novel mechanism for venous changes in the retina resulting from Cav-1 loss. Our results are also consistent with the idea that Cav-1 may regulate flow-dependent autoregulation in retinal arterioles, but this remains to be determined empirically.
Loss of Cav-1 Causes Mural Cell Changes on Retinal Veins
Given these results, we sought to determine what factors might contribute to venous enlargement. Mural cells (ie, pericytes and vascular smooth muscle cells) share the same basement membrane with endothelial cells and perform key functions to maintain vascular stability and integrity.57–59 Mural cells also possess contractile ability,57–59 which suggests the possibility that, if Cav-1 deficiency causes a loss or dysfunction of venous smooth muscle cells, the veins may be enlarged. To test this possibility, we immunolabeled Cav-1 KO and WT retinas with NG2, a chondroitin sulfate proteoglycan expressed by mural cells in the retinal vasculature.36 Surprisingly, the enlarged veins in the Cav-1 KO retinas exhibited a dramatic reduction in NG2 immunoreactivity (Figure 7, E, F, K, L, and O), compared with WT controls (Figure 7, A, B, I, J, and M). We initially interpreted this result to indicate a loss of mural cell coverage; however, subsequent analysis of veins labeled for another mural cell marker, platelet-derived growth factor receptor β (PDGFRβ), revealed no significant difference in coverage of vessels (Figure 7, C, G, I, K, and Q). When signal intensities for NG2 were normalized to those of PDGFRβ, we observed a significant reduction in NG2 levels in Cav-1 KO branch retinal veins (Figure 7Q). The NG2 reduction was most pronounced in these large veins; postcapillary venules did not exhibit significant decreases in NG2 immunoreactivity in either WT (Figure 7, J and M) or KO (Figure 7, L and O) retinas.
Figure 7.

Loss of Cav-1 reduces NG2 on enlarged branch retinal veins without affecting mural cell coverage and increases concomitant venous αSMA immunoreactivity. A–H: Retinas from WT and Cav-1 KO mice were immunostained with antibodies against mural cell markers, NG2 (green) (A and F), PDGFRβ (blue) (C and G), and αSMA (red) (D and H). Cav-1 deficiency in branch retinal veins significantly reduced NG2 expression and increased αSMA expression (F and H), compared with WT (B and D), but loss of Cav-1 did not noticeably decrease PDGFRβ in either genotype (C and G). I–L: Higher magnification images correspond to boxed regions in panels A and E. Cav-1 KO veins are not devoid of NG2+ cells (K and L), with weakly NG2+ cells visible on branch retinal veins (arrowheads), but the intensity is significantly weaker than in WT (I and J). M–P: Retinas from WT (M and N) and Cav-1 KO (O and P) mice were stained against CD31 (red) and NG2 (green). CD31 immunoreactivity was similar in both genotypes. Q: Fluorescence intensity was quantified from CD31, NG2, PDGFRβ, and αSMA staining in WT and Cav-1 KO retinal vasculature. Results suggest an intrinsic mural cell phenotype alteration due to the loss of Cav-1, rather than to altered mural cell coverage. Data are expressed as means ± SEM. n = 5–8 per group. ∗P < 0.05, ∗∗∗∗P < 0.0001 versus WT. Scale bars: 50 μm (A–H); 10 μm (I–L); and 20 μm (M–P).
The NG2 reduction remained evident in enlarged branch retinal veins from DKO retinas, indicating that eNOS is unlikely to contribute to this decrease (Supplemental Figure S6). We also used RT-qPCR to examine the relative expression of NG2 transcripts from Cav-1 KO and WT retinal total RNA. Although we observed a decrease in NG2 mRNA levels, this reduction did not reach our statistical threshold (P = 0.1142) (Supplemental Figure S7). In addition to the reduction in NG2, we observed a concomitant significant increase in immunoreactivity for the venous smooth muscle contractile protein αSMA in both Cav-1 KO (Figure 7) and DKO mice (Supplemental Figure S8). Compared with eNOS KO retinas, DKO retinas had an increase in αSMA (Supplemental Figure S8, A, C, E, and G) and a concomitant reduction in NG2 immunoreactivity (Supplemental Figure S8, A, B, E, and F), suggesting that loss of Cav-1, rather than eNOS deficiency, mediates the observed phenotypes. We did not observe any appreciable differences in CD31 immunoreactivity in either WT (Figure 7, M and N) or KO (Figure 7, O and P) retinas. The similarity of the PDGFRβ/CD31 ratios in Cav-1 KO and WT retinal vasculature strongly argues against the loss of mural cells per se around the enlarged veins; however, the dramatic reduction in the NG2/PDGFRβ ratio in the absence of Cav-1 suggests possible intrinsic alterations in mural cell phenotype (Figure 7Q).
Strikingly, we identified additional mural cell morphological alterations on the branch retinal veins proximal to their entering the ONH. In this location, NG2 staining in Cav-1 KO veins was not dramatically reduced, compared with WT controls, but the Cav-1 KO mural cells had a spindly morphology with reduced coverage of attached veins and numerous disorganized processes (Figure 8, C, D, I, and J) not seen in WT controls (Figure 8, A, B, E, and F). Higher magnification revealed that NG2+ mural cells in Cav-1 KO retinas had thinner processes, compared with WT retinas (Figure 8, B and D). NG2 staining on the arterial smooth muscle cells, on the other hand, appeared normal in both Cav-1 KO (Figure 8, I and J) and WT (Figure 8, E and F) retinas. However, αSMA staining was more intense on the Cav-1 KO retinal veins (Figure 8, I and L), compared with WT (Figure 8, E and H), suggesting increased stress fiber formation.
Figure 8.

Loss of Cav-1 alters the mural cell phenotype in enlarged branch retinal vein proximal to the ONH. WT (A and B) and Cav-1 KO (C and D) retinas were immunostained with antibodies against CD31 (red) and NG2 (green). Boxed areas in A and C are enlarged as panels B and D, respectively. NG2+ cells on Cav-1 KO retinal veins have a spindly appearance (arrowhead) (D). These cells, compared with counterparts on WT retinal veins, had thinner processes (arrows) (B and D). NG2 staining on arteries seemed normal. Immunostaining of WT (E–H) and Cav-1 KO (I–L) retinas with antibodies against mural cell markers NG2 (green) (F and J), PDGFRβ (blue) (G and K), and αSMA (red) (H and L) confirmed the spindly appearance of venous smooth muscle cells on branch retinal veins immediately proximal to entering the ONH in Cav-1 KO retina (I and J), compared with WT retina (E and F). αSMA staining further supported the observed phenotypic changes in mural cell morphology in WT (H) and KO (L) retinas. Mural cell staining on arteries was similar in WT and Cav-1 KO retinas. E and I represent merged images. Scale bars: 10 μm (A, C, and E–L); 5 μm (B and D). a, artery; v, vein.
As expected, another mural cell marker (PDGFRβ) exhibited similarly disorganized mural cell morphology in Cav-1 KO retinas (Figure 8K). We also examined mural cell morphology on the veins entering the ONH in eNOS KO and DKO retinas. In the eNOS KO retinas, αSMA+ smooth muscle cells possessed an organized ensheathment of the veins (Supplemental Figure S8, I and K), whereas in DKO retinas, these cells appeared disorganized and αSMA staining was more intense (Supplemental Figure S8, M and O), indicating that eNOS hyperactivity resulting from Cav-1 ablation does not play a role in the mural cell morphological alterations.
Taken together, these results indicate that loss of Cav-1 results in spatially defined mural cell abnormalities in branch retinal veins. Distal to the ONH, Cav-1–deficient veins exhibited reduced NG2 and increased αSMA immunoreactivity; proximal to the ONH, Cav-1 deficiency resulted in mural cells with a pronounced myofibroblast-like phenotype with prominent αSMA stress fibers (Figure 8E).
Discussion
Caveolin and caveolar alterations are associated with several ocular pathologies, including diabetic retinopathy, posterior uveitis, and primary open-angle glaucoma, but the pathophysiology linking caveolins to these diseases is not understood.28,60,61 Notably, however, all of these diseases exhibit pathological changes in retinal vasculature that are associated with disease severity. We recently reported that ablation of Cav-1 impairs retinal function via changes in the retinal microenvironment.31 Because a tight BRB helps to maintain the retinal extracellular milieu, we asked whether loss of Cav-1 function might affect BRB integrity. In the course of this first detailed study of retinal vasculature in Cav-1 KO mice, we have demonstrated that i) vascular BRB integrity is impaired, and this impairment is not dependent on eNOS expression or hyperactivity; ii) hyperpermeability occurs, at least in part, paracellularly, but junctional protein components appear unaffected; iii) Cav-1 expression in the adult retina is predominantly localized to the branch retinal veins; iv) branch retinal veins in the superficial retina, which are hotspots of leakage, are abnormally enlarged, and this enlargement is not dependent on eNOS expression or hyperactivity; and v) these enlarged veins have region-specific mural cell phenotypic changes consistent with a transition to a more contractile, myofibroblast-like phenotype.
Caveolae are known to be critical for transendothelial albumin transport.46 However, loss of caveolae leads to paradoxical microvascular hyperpermeability in several organs, and this hyperpermeability occurs through the opening of paracellular junctions in response to loss of the transcellular pathway mediated by caveolae.14,45 This crosstalk mechanism involves eNOS, which, in the inactive state, is anchored to the cell membrane and inhibited by Cav-1 in caveolae.8 Thus, hyperactivated eNOS is implicated in several reported cardiovascular and pulmonary pathologies in Cav-1 KO mice, and inhibiting eNOS rescues these pathologies.14,15 Unlike the vascular beds in these other organs, those in the retina and the brain both have a tight barrier with a relatively polarized abluminal distribution of caveolae in both endothelial and mural cells. This brings up a long-debated question in the field: that is, whether caveolae are static structures or whether they participate in transcellular transport in the retina.25,26 If they are static structures that do not participate in albumin transport in the retina, would their loss activate the eNOS-mediated crosstalk pathway? Consistent with previous reports of microvascular hyperpermeability,14–16 in the present study, Cav-1 KO retinas exhibited paracellular hyperpermeability; however, this hyperpermeability could not be rescued by pharmacologically inhibiting eNOS activity, nor by genetically deleting the eNOS gene in the Cav-1 KO background. We did observe an increase in BRB permeability in eNOS KO mice, suggesting that the effects of Cav-1 and eNOS on the BRB may be through different mechanisms.
Studies of brain microvascular endothelium have shown that transient knockdown of Cav-1 causes reduction in the levels of both TJ and AJ proteins,62 and that caveolae mediate the internalization of claudin-5 and occludin during CCL2-induced TJ remodeling in brain endothelial cells.50 In the present study, however, we could not detect differences in junctional protein expression or localization in Cav-1 KO retinal vessels, not even in the context of paracellular hyperpermeability. We also found no differences in the distribution of claudin-5 in detergent-resistant membrane fractions from Cav-1 KO and WT retinas. Although phosphorylation of junctional proteins affects junctional stability,40 we found no evidence that the phosphorylation status of occludin and ZO-1 is altered as a consequence of Cav-1 loss. Although at the ultrastructural level we observed clear examples of endothelial junctions permeable to HRP, we could not detect ultrastructural differences in TJ morphology. Thus, the present findings support the conclusion that the hyperpermeability in Cav-1 KO retinas occurs (at least in part) paracellularly, but does not affect the expression, distribution, or phosphorylation of junctional proteins. This conclusion is consistent with a recent report by Armstrong et al,47 who found that blockade of Cav-1–dependent transcytosis rapidly increased paracellular leakage without affecting junctional proteins or eNOS activity; instead, they found that actin cytoskeletal remodeling was involved. Caveolae dynamics are intricately related to the cytoskeleton,9 but whether the cytoskeleton of the Cav-1 KO retinal endothelium is remodeled to account for the hyperpermeability needs further investigation.
It is tempting to speculate that the increased permeability associated with Cav-1 KO branch retinal veins results from poor venous return and blood pooling, as is seen in venous stasis retinopathy. Consistent with this idea, loss of Cav-1 resulted in venous enlargement. The mechanisms that regulate retinal venous diameter under physiological or pathophysiological conditions are poorly understood (reviewed in63), but our present findings imply a role for Cav-1 that is independent of NO synthesis and eNOS activity. It is possible that loss of Cav-1 results in up-regulation of other endothelium-derived vasodilators, such as prostacyclin. We consider this unlikely, however, given that prostacyclin levels in arterial segments of Cav-1 KO mice have been found to be lower than in WT mice, not higher,64 and that vasodilation via cyclooxygenase activity has greater dilatory effects on retinal arteries than on retinal veins.65 In fact, endothelium-derived autoregulatory signals such as NO, prostacyclin, and endothelium-derived hyperpolarizing factor are more likely to affect arterial, not venous, caliber.
Similar to our findings for veins, we observed significant enlargement of arteries in Cav-1 KO retinas, but (unlike the case with veins) this enlargement was corrected by transient NOS inhibition by L-NAME. Similar results have been observed in isolated and perfused cerebral arteries,55 in which NOS inhibition constricted control and Cav-1 KO vessels (albeit without affecting myogenic tone). In the lung, inhibition of NOS activity exacerbates pulmonary hypertension in Cav-1 KO mice, suggesting that eNOS hyperactivity compensates for the gross pathological changes to pulmonary vasculature.66 Although considerably less is known about retinal vessel autoregulation, there is evidence that NO regulates both basal53 and flow-dependent54 arterial tone. Our present findings provide compelling complementary evidence that retinal arteries are subject to intrinsic NO-dependent control and, more importantly, provide the novel finding that Cav-1 plays an endogenous regulatory role. The implications of these findings for retinal vascular disorders that result in altered autoregulation or abnormal vessel diameters remain to be determined, but it is intriguing to note that Cav-1 expression and/or polymorphisms are linked to diabetic retinopathy and primary open-angle glaucoma, two diseases with significant pathological alterations in vascular diameter and/or autoregulation.
Mural cells possess contractile properties and thus contribute to the mechanical stability of vessel walls.57–59 We therefore chose to localize a panel of retinal mural cell markers (ie, NG2, PDGFRβ, and αSMA67) in Cav-1 KO and WT retinas. Surprisingly, the enlarged branch retinal veins were significantly deficient in NG2, but had similar PDGFRβ immunoreactivity and PDGFRβ/CD31 ratios, indicating that reduced NG2 staining was not due to a loss in mural cell coverage. In our analysis of NG2 transcript levels from total retinal mRNA, differences approached, but did not reach, our statistical threshold. However, given that our cDNA templates for RT-qPCR were amplified from total retinal RNA, the transcriptional reduction could nonetheless be significant in the subset of retinal vessels (ie, branch retinal veins) in which NG2 protein levels were reduced.
NG2 is a chondroitin sulfate proteoglycan that requires proper targeting to the membrane to function68 and that is known to associate with basement membrane components.69,70 Caveolae are polarized to the abluminal side of central nervous system pericytes26 and have been shown to colocalize with NG2 at the ultrastructural level in both pericytes and endoneurial cells.71 Interestingly, Hughes and Chan-Ling67 found that “pericyte maturation was characterized by the restriction of NG2 expression to abluminal cell bodies.” Thus, it is conceivable (although this remains to be rigorously tested) that Cav-1 may play a role in NG2 trafficking or NG2 function at the cell surface, and possibly also in pericyte maturation.
In addition to reductions in NG2 in branch retinal vein smooth muscle cells, we also observed significant increases in αSMA immunolabeling in Cav-1 KO retinal vessels (Figure 7). The extent of αSMA expression by smooth muscle cells in the retina normally increases with maturation.67 However, we observed markedly increased αSMA immunoreactivity along the entire branch retinal vein, as well as morphological changes in smooth muscle cells near the ONH, which suggested a transition to a myofibroblast-like phenotype. Intriguingly, increased αSMA staining has been observed in lung parenchyma, although (in contrast to our findings), pulmonary postcapillary vessels did not exhibit such increase.66 Whether the increase in αSMA levels associated with retinal veins originates from existing mural cells that have changed phenotype or from endothelial cells that have undergone endothelial–mesenchymal transition remains to be determined. However, spontaneous endothelial–mesenchymal transition has recently been shown in pulmonary endothelial cells.72
We can only speculate on the spatial differences in mural cell morphology. At present, we favor the hypothesis that differences in mechanical forces along the retinal veins are not sensed appropriately in the absence of caveolae. There is abundant evidence of rhythmic, spontaneous venous pulsations in the central retinal vein (the major vein that drains the branch retinal veins) of healthy human subjects. These regular cycles of venous collapse and opening are associated in a complex way with pressure gradients resulting from the cardiac cycle and rhythmic fluctuations of intraocular and/or intracranial pressures.73,74 These fluctuations are more commonly observed in healthy subjects than in glaucoma patients.75 Caveolae are now recognized as mechanosensors in vascular cell membranes,9,76,77 and it is conceivable that endothelial and mural cells in retinal veins of Cav-1 KO mice are unable to cope with mechanical stresses resulting from continuous venous pulsations. This speculation remains to be tested, but could have profound clinical relevance, because vascular changes near the ONH are strongly associated with glaucoma progression, and because CAV1 polymorphisms have recently been associated with risk of developing primary open-angle glaucoma.61
Acknowledgments
We thank Dr. Jan Ryerse and Barbara Nagel (Saint Louis University) for technical support and helpful discussions concerning the ultrastructural studies, Drs. David Antonetti and Arivu Muthusamy for suggestions regarding TJ analyses, Mark Dittmar in the vivarium (Dean McGee Eye Institute) for support, and Louisa Williams and Linda Boone in the DMEI/NEI Cellular Imaging Core Facility (OUHSC) for outstanding technical assistance.
Footnotes
Supported by NIH grants R01-EY019494 (M.H.E.), P01-HL077806 (Y.Z.), R01-HL085462 (Y.Z.), R01-CA95287 (W.B.S.), and R01-EY007631 (S.J.F.); and P30-EY021725 (Center Core Grant for Vision Research), a Sigma Xi Grant-in-Aid of Research (X.G.), unrestricted grants from Research to Prevent Blindness (Dean McGee Eye Institute and SUNY Eye Institute), and facilities and resources provided by the Veterans Affairs Western New York Healthcare System (S.J.F.). M.H.E. is the recipient of the Thomas R. Lee Award for Glaucoma Research from the BrightFocus Foundation and an Alcon Research Institute Young Investigator Award.
The views expressed herein do not necessarily reflect those of the Department of Veterans Affairs.
Supplemental Data
WT and Cav-1 KO vessels have TJ morphology. Electron micrographs reveal abundant caveolae in WT vascular endothelial cells (arrows) and absent caveolae in Cav-1 KO vessels. Junctions (boxed regions) have similar gross appearance in both Cav-1 KO and WT retinas. BM, basement membrane; EC, endothelial cells; P, pericytes. Scale bar = 500 nm.
Phosphorylation status of ZO-1 and occludin was not altered in Cav-1 KO retinas. A: Lysates from WT and Cav-1 KO retinas were immunoprecipitated using phosphotyrosine 4G10 antibody and blotted for ZO-1 and phosphotyrosine (4G10). Multiple proteins exhibited enhanced tyrosine phosphorylation in Cav-1 KO retinas (arrows) but ZO-1 was not recovered in phosphotyrosine immunoprecipitates from either KO or WT retinas. B: Occludin phosphorylation identified by the reduced migration on SDS-PAGE gels. Upon treatment with alkaline phosphatase, the slower migrating bands collapsed and occludin ran as a single 60-kDa band. No changes in occludin phosphorylation were detected between Cav-1 KO and WT retinas. AP, alkaline phosphatase; B, beads; FT, flow-through; IN, input; pY, phosphotyrosine.
Sucrose gradient analysis revealed similar fractionation pattern of claudin-5 in the Cav-1 KO and WT retinas. A: Western blotting shows that claudin-5 fractionation pattern in the Cav-1 KO retinas does not differ from that in the WT retinas. B: Densitometry analysis of the Western blotting results.
eNOS hyperactivity in Cav-1 KO retinas contributes to arterial dilation. WT and Cav-1 KO mice were treated with 30 mg/kg body weight L-NAME or PBS for 2 hours and retinas were stained using anti-CD31 antibody (red). Artery diameters were measured to 500 μm from the ONH, or to the first bifurcation before 500 μm, measuring only the common artery trunk. A–D: Representative images show that PBS-injected Cav-1 KO arteries (C) are more dilated than WT arteries (A). In response to L-NAME treatment, arteries in WT (B) and Cav-1 KO (D) retinas constricted to a similar extent. E: Quantification of artery diameters. Five to six arteries from each retina were measured, averaged, and collected as one sample. Data are expressed as means ± SEM. n = 4 per group. ∗∗P < 0.01 versus WT PBS. ††††P < 0.0001 versus WT L-NAME. ‡‡‡P < 0.001 versus Cav-1 KO PBS. Two-way analysis of variance with Newman–Keuls post hoc analysis. Scale bar = 50 μm.
Cav-1–dependent arterial dilation is not corrected by eNOS deletion. Arteries from WT (A), Cav-1 KO (C), eNOS KO (D) and Cav-1 and eNOS DKO (B) mice were measured according to the criteria described for Supplemental Figure S4 and quantified (E). In contrast to the transient pharmacological inhibition of NOS activity shown in Supplemental Figure S4, chronic genetic deletion of eNOS did not correct artery diameter. In fact, eNOS KO retinas also had enlarged diameters, suggesting a compensatory mechanism to dilate arteries when eNOS is constitutively deleted. Data are expressed as means ± SEM. n = 7 or 8 per genotype. ∗P < 0.05 versus WT. One-way analysis of variance with Newman–Keuls post hoc analysis was used. Scale bar = 50 μm.
NG2 deficiency is not dependent on eNOS activity. Cav-1 and eNOS DKO retinas were stained with mural cell markers, NG2 (green) and PDGFRβ (blue) and endothelial marker, CD31 (red). Compared with WT, DKO veins have obvious NG2 deficiency, as also observed in Cav-1 KO retinas (Figure 7). As in the Cav-1 KO mice, another mural cell marker (PDGFRβ) and an endothelial marker (CD31) did not differ between DKO and WT retinas, suggesting that such NG2 reduction is not due to eNOS activity but rather depends on Cav-1, reinforcing the possibility that Cav-1 might play an important role in NG2 biology (eg, trafficking, membrane targeting, or polarization). Scale bar = 50 μm.
Total NG2 transcript levels in Cav-1 KO retinas. Gene expression was measured by RT-qPCR, normalized to housekeeping gene RPL19 and presented as fold change over control (WT). The results did not reach our statistical threshold (P = 0.11 Student’s t-test); however, given that NG2 is reduced on a subset of retinal vessels (the venous system, and notably the branch veins), NG2 transcripts from total retinal mRNA may not reveal vessel-specific reductions. Data are expressed as means ± SEM. n = 10 (WT) or 14 (Cav-1 KO).
Mural cell phenotypic changes observed in Cav-1 KO branch retinal veins are also apparent in Cav-1 and eNOS DKO mice but not in eNOS KO mice. Retinas were stained with the mural cell markers NG2 (blue) and αSMA (red) and the endothelial marker CD31 (green). DKO branch retinal veins exhibited reduced NG2 and enhanced αSMA immunoreactivity distal to the ONH, as well as spindly mural cell morphology proximal to the ONH, as was observed for Cav-1 KO mice. Scale bar = 50 μm (A–H); 10 μm (I–P).
References
- 1.Patton N., Aslam T., Macgillivray T., Pattie A., Deary I.J., Dhillon B. Retinal vascular image analysis as a potential screening tool for cerebrovascular disease: a rationale based on homology between cerebral and retinal microvasculatures. J Anat. 2005;206:319–348. doi: 10.1111/j.1469-7580.2005.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Runkle E.A., Antonetti D.A. The blood-retinal barrier: structure and functional significance. Methods Mol Biol. 2011;686:133–148. doi: 10.1007/978-1-60761-938-3_5. [DOI] [PubMed] [Google Scholar]
- 3.Kaur C., Foulds W.S., Ling E.A. Blood-retinal barrier in hypoxic ischaemic conditions: basic concepts, clinical features and management. Prog Retin Eye Res. 2008;27:622–647. doi: 10.1016/j.preteyeres.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 4.Chung A.S., Ferrara N. Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol. 2011;27:563–584. doi: 10.1146/annurev-cellbio-092910-154002. [DOI] [PubMed] [Google Scholar]
- 5.Kim L.A., D’Amore P.A. A brief history of anti-VEGF for the treatment of ocular angiogenesis. Am J Pathol. 2012;181:376–379. doi: 10.1016/j.ajpath.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurihara T., Westenskow P.D., Bravo S., Aguilar E., Friedlander M. Targeted deletion of Vegfa in adult mice induces vision loss. J Clin Invest. 2012;122:4213–4217. doi: 10.1172/JCI65157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ford K.M., Saint-Geniez M., Walshe T.E., D’Amore P.A. Expression and role of VEGF-a in the ciliary body. Invest Ophthalmol Vis Sci. 2012;53:7520–7527. doi: 10.1167/iovs.12-10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Razani B., Woodman S.E., Lisanti M.P. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54:431–467. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 9.Parton R.G., del Pozo M.A. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. 2013;14:98–112. doi: 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- 10.Drab M., Verkade P., Elger M., Kasper M., Lohn M., Lauterbach B., Menne J., Lindschau C., Mende F., Luft F.C., Schedl A., Haller H., Kurzchalia T.V. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- 11.Razani B., Engelman J.A., Wang X.B., Schubert W., Zhang X.L., Marks C.B., Macaluso F., Russell R.G., Li M., Pestell R.G., Di Vizio D., Hou H., Jr., Kneitz B., Lagaud G., Christ G.J., Edelmann W., Lisanti M.P. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- 12.Cohen A.W., Hnasko R., Schubert W., Lisanti M.P. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–1379. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Y.Y., Liu Y., Stan R.V., Fan L., Gu Y., Dalton N., Chu P.H., Peterson K., Ross J., Chien K.R. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci USA. 2002;99:11375–11380. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schubert W., Frank P.G., Woodman S.E., Hyogo H., Cohen D.E., Chow C.W., Lisanti M.P. Microvascular hyperpermeability in caveolin-1 (−/−) knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, L-NAME, restores normal microvascular permeability in Cav-1 null mice. J Biol Chem. 2002;277:40091–40098. doi: 10.1074/jbc.M205948200. [DOI] [PubMed] [Google Scholar]
- 15.Murata T., Lin M.I., Huang Y., Yu J., Bauer P.M., Giordano F.J., Sessa W.C. Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J Exp Med. 2007;204:2373–2382. doi: 10.1084/jem.20062340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin M.I., Yu J., Murata T., Sessa W.C. Caveolin-1-deficient mice have increased tumor microvascular permeability, angiogenesis, and growth. Cancer Res. 2007;67:2849–2856. doi: 10.1158/0008-5472.CAN-06-4082. [DOI] [PubMed] [Google Scholar]
- 17.Chang S.H., Feng D., Nagy J.A., Sciuto T.E., Dvorak A.M., Dvorak H.F. Vascular permeability and pathological angiogenesis in caveolin-1-null mice. Am J Pathol. 2009;175:1768–1776. doi: 10.2353/ajpath.2009.090171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woodman S.E., Ashton A.W., Schubert W., Lee H., Williams T.M., Medina F.A., Wyckoff J.B., Combs T.P., Lisanti M.P. Caveolin-1 knockout mice show an impaired angiogenic response to exogenous stimuli. Am J Pathol. 2003;162:2059–2068. doi: 10.1016/S0002-9440(10)64337-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.García-Cardeña G., Martasek P., Masters B.S., Skidd P.M., Couet J., Li S., Lisanti M.P., Sessa W.C. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem. 1997;272:25437–25440. doi: 10.1074/jbc.272.41.25437. [DOI] [PubMed] [Google Scholar]
- 20.Frank P.G., Lee H., Park D.S., Tandon N.N., Scherer P.E., Lisanti M.P. Genetic ablation of caveolin-1 confers protection against atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:98–105. doi: 10.1161/01.ATV.0000101182.89118.E5. [DOI] [PubMed] [Google Scholar]
- 21.Antonetti D.A., Klein R., Gardner T.W. Diabetic retinopathy. N Engl J Med. 2012;366:1227–1239. doi: 10.1056/NEJMra1005073. [DOI] [PubMed] [Google Scholar]
- 22.Gu X., Reagan A., Yen A., Bhatti F., Cohen A.W., Elliott M.H. Spatial and temporal localization of caveolin-1 protein in the developing retina. Adv Exp Med Biol. 2014 doi: 10.1007/978-1-4614-3209-8_3. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elliott M.H., Fliesler S.J., Ghalayini A.J. Cholesterol-dependent association of caveolin-1 with the transducin alpha subunit in bovine photoreceptor rod outer segments: disruption by cyclodextrin and guanosine 5′-O-(3-thiotriphosphate) Biochemistry. 2003;42:7892–7903. doi: 10.1021/bi027162n. [DOI] [PubMed] [Google Scholar]
- 24.Kachi S., Yamazaki A., Usukura J. Localization of caveolin-1 in photoreceptor synaptic ribbons. Invest Ophthalmol Vis Sci. 2001;42:850–852. [PubMed] [Google Scholar]
- 25.Raviola G., Butler J.M. Unidirectional vesicular transport mechanism in retinal vessels. Invest Ophthalmol Vis Sci. 1983;24:1465–1474. [PubMed] [Google Scholar]
- 26.Gardiner T.A., Archer D.B. Does unidirectional vesicular transport occur in retinal vessels? Br J Ophthalmol. 1986;70:249–254. doi: 10.1136/bjo.70.4.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oh P., Borgström P., Witkiewicz H., Li Y., Borgström B.J., Chrastina A., Iwata K., Zinn K.R., Baldwin R., Testa J.E., Schnitzer J.E. Live dynamic imaging of caveolae pumping targeted antibody rapidly and specifically across endothelium in the lung [Erratum appeared in Nat Biotechnol 2007, 25:478] Nature Biotechnol. 2007;25:327–337. doi: 10.1038/nbt1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klaassen I., Hughes J.M., Vogels I.M., Schalkwijk C.G., Van Noorden C.J., Schlingemann R.O. Altered expression of genes related to blood-retina barrier disruption in streptozotocin-induced diabetes. Exp Eye Res. 2009;89:4–15. doi: 10.1016/j.exer.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Caldwell R.B., Slapnick S.M. Freeze-fracture and lanthanum studies of the retinal microvasculature in diabetic rats. Invest Ophthalmol Vis Sci. 1992;33:1610–1619. [PubMed] [Google Scholar]
- 30.Jasmin J.F., Malhotra S., Singh Dhallu M., Mercier I., Rosenbaum D.M., Lisanti M.P. Caveolin-1 deficiency increases cerebral ischemic injury. Circ Res. 2007;100:721–729. doi: 10.1161/01.RES.0000260180.42709.29. [DOI] [PubMed] [Google Scholar]
- 31.Li X., McClellan M.E., Tanito M., Garteiser P., Towner R., Bissig D., Berkowitz B.A., Fliesler S.J., Woodruff M.L., Fain G.L., Birch D.G., Khan M.S., Ash J.D., Elliott M.H. Loss of caveolin-1 impairs retinal function due to disturbance of subretinal microenvironment. J Biol Chem. 2012;287:16424–16434. doi: 10.1074/jbc.M112.353763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Y.Y., Zhao Y.D., Mirza M.K., Huang J.H., Potula H.H., Vogel S.M., Brovkovych V., Yuan J.X., Wharton J., Malik A.B. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest. 2009;119:2009–2018. doi: 10.1172/JCI33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishida S., Usui T., Yamashiro K., Kaji Y., Ahmed E., Carrasquillo K.G., Amano S., Hida T., Oguchi Y., Adamis A.P. VEGF164 is proinflammatory in the diabetic retina. Invest Ophthalmol Vis Sci. 2003;44:2155–2162. doi: 10.1167/iovs.02-0807. [DOI] [PubMed] [Google Scholar]
- 34.Burg M.A., Pasqualini R., Arap W., Ruoslahti E., Stallcup W.B. NG2 proteoglycan-binding peptides target tumor neovasculature. Cancer Res. 1999;59:2869–2874. [PubMed] [Google Scholar]
- 35.Grako K.A., Ochiya T., Barritt D., Nishiyama A., Stallcup W.B. PDGF (alpha)-receptor is unresponsive to PDGF-AA in aortic smooth muscle cells from the NG2 knockout mouse. J Cell Sci. 1999;112:905–915. doi: 10.1242/jcs.112.6.905. [DOI] [PubMed] [Google Scholar]
- 36.Ozerdem U., Grako K.A., Dahlin-Huppe K., Monosov E., Stallcup W.B. NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev Dyn. 2001;222:218–227. doi: 10.1002/dvdy.1200. [DOI] [PubMed] [Google Scholar]
- 37.Chucair-Elliott A.J., Elliott M.H., Wang J., Moiseyev G.P., Ma J.X., Politi L.E., Rotstein N.P., Akira S., Uematsu S., Ash J.D. Leukemia inhibitory factor coordinates the down-regulation of the visual cycle in the retina and retinal-pigmented epithelium. J Biol Chem. 2012;287:24092–24102. doi: 10.1074/jbc.M112.378240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abedinpour P., Jergil B. Isolation of a caveolae-enriched fraction from rat lung by affinity partitioning and sucrose gradient centrifugation. Anal Biochem. 2003;313:1–8. doi: 10.1016/s0003-2697(02)00561-4. [DOI] [PubMed] [Google Scholar]
- 39.Martin R.E., Elliott M.H., Brush R.S., Anderson R.E. Detailed characterization of the lipid composition of detergent-resistant membranes from photoreceptor rod outer segment membranes. Invest Ophthalmol Vis Sci. 2005;46:1147–1154. doi: 10.1167/iovs.04-1207. [DOI] [PubMed] [Google Scholar]
- 40.Antonetti D.A., Barber A.J., Hollinger L.A., Wolpert E.B., Gardner T.W. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occludens 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem. 1999;274:23463–23467. doi: 10.1074/jbc.274.33.23463. [DOI] [PubMed] [Google Scholar]
- 41.Reese T.S., Karnovsky M.J. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol. 1967;34:207–217. doi: 10.1083/jcb.34.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gardiner T.A., Archer D.B. Endocytosis in the retinal and choroidal microcirculation. Br J Ophthalmol. 1986;70:361–372. doi: 10.1136/bjo.70.5.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qaum T., Xu Q., Joussen A.M., Clemens M.W., Qin W., Miyamoto K., Hassessian H., Wiegand S.J., Rudge J., Yancopoulos G.D., Adamis A.P. VEGF-initiated blood-retinal barrier breakdown in early diabetes. Invest Ophthalmol Vis Sci. 2001;42:2408–2413. [PubMed] [Google Scholar]
- 44.Lin W.L., Essner E., Shichi H. Breakdown of the blood-retinal barrier in S-antigen-induced uveoretinitis in rats. Graefes Arch Clin Exp Ophthalmol. 1991;229:457–463. doi: 10.1007/BF00166310. [DOI] [PubMed] [Google Scholar]
- 45.Komarova Y., Malik A.B. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 2010;72:463–493. doi: 10.1146/annurev-physiol-021909-135833. [DOI] [PubMed] [Google Scholar]
- 46.Schubert W., Frank P.G., Razani B., Park D.S., Chow C.W., Lisanti M.P. Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J Biol Chem. 2001;276:48619–48622. doi: 10.1074/jbc.C100613200. [DOI] [PubMed] [Google Scholar]
- 47.Armstrong S.M., Khajoee V., Wang C., Wang T., Tigdi J., Yin J., Kuebler W.M., Gillrie M., Davis S.P., Ho M., Lee W.L. Co-regulation of transcellular and paracellular leak across microvascular endothelium by dynamin and Rac. Am J Pathol. 2012;180:1308–1323. doi: 10.1016/j.ajpath.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 48.Murakami T., Frey T., Lin C., Antonetti D.A. Protein kinase Cβ phosphorylates occludin regulating tight junction trafficking in vascular endothelial growth factor-induced permeability in vivo. Diabetes. 2012;61:1573–1583. doi: 10.2337/db11-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cohen A.W., Park D.S., Woodman S.E., Williams T.M., Chandra M., Shirani J., Pereira de Souza A., Kitsis R.N., Russell R.G., Weiss L.M., Tang B., Jelicks L.A., Factor S.M., Shtutin V., Tanowitz H.B., Lisanti M.P. Caveolin-1 null mice develop cardiac hypertrophy with hyperactivation of p42/44 MAP kinase in cardiac fibroblasts. Am J Physiol Cell Physiol. 2003;284:C457–C474. doi: 10.1152/ajpcell.00380.2002. [DOI] [PubMed] [Google Scholar]
- 50.Stamatovic S.M., Keep R.F., Wang M.M., Jankovic I., Andjelkovic A.V. Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J Biol Chem. 2009;284:19053–19066. doi: 10.1074/jbc.M109.000521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.El-Remessy A.B., Behzadian M.A., Abou-Mohamed G., Franklin T., Caldwell R.W., Caldwell R.B. Experimental diabetes causes breakdown of the blood-retina barrier by a mechanism involving tyrosine nitration and increases in expression of vascular endothelial growth factor and urokinase plasminogen activator receptor. Am J Pathol. 2003;162:1995–2004. doi: 10.1016/S0002-9440(10)64332-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Q., Verma A., Han P.Y., Nakagawa T., Johnson R.J., Grant M.B., Campbell-Thompson M., Jarajapu Y.P., Lei B., Hauswirth W.W. Diabetic eNOS-knockout mice develop accelerated retinopathy. Invest Ophthalmol Vis Sci. 2010;51:5240–5246. doi: 10.1167/iovs.09-5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benedito S., Prieto D., Nielsen P.J., Nyborg N.C. Role of the endothelium in acetylcholine-induced relaxation and spontaneous tone of bovine isolated retinal small arteries. Exp Eye Res. 1991;52:575–579. doi: 10.1016/0014-4835(91)90059-n. [DOI] [PubMed] [Google Scholar]
- 54.Hein T.W., Rosa R.H., Jr., Yuan Z., Roberts E., Kuo L. Divergent roles of nitric oxide and rho kinase in vasomotor regulation of human retinal arterioles. Invest Ophthalmol Vis Sci. 2010;51:1583–1590. doi: 10.1167/iovs.09-4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adebiyi A., Zhao G., Cheranov S.Y., Ahmed A., Jaggar J.H. Caveolin-1 abolishment attenuates the myogenic response in murine cerebral arteries [Erratum appeared in Am J Physiol Heart Circ Physiol 2007, 292:H2556–H2557] Am J Physiol Heart Circ Physiol. 2007;292:H1584–H1592. doi: 10.1152/ajpheart.00584.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Delaey C., Van De Voorde J. Regulatory mechanisms in the retinal and choroidal circulation. Ophthalmic Res. 2000;32:249–256. doi: 10.1159/000055622. [DOI] [PubMed] [Google Scholar]
- 57.Armulik A., Genové G., Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 58.Dore-Duffy P., Cleary K. Morphology and properties of pericytes. Methods Mol Biol. 2011;686:49–68. doi: 10.1007/978-1-60761-938-3_2. [DOI] [PubMed] [Google Scholar]
- 59.Hirschi K.K., D’Amore P.A. Pericytes in the microvasculature. Cardiovasc Res. 1996;32:687–698. [PubMed] [Google Scholar]
- 60.Hauck S.M., Dietter J., Kramer R.L., Hofmaier F., Zipplies J.K., Amann B., Feuchtinger A., Deeg C.A., Ueffing M. Deciphering membrane-associated molecular processes in target tissue of autoimmune uveitis by label-free quantitative mass spectrometry. Mol Cell Proteomics. 2010;9:2292–2305. doi: 10.1074/mcp.M110.001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thorleifsson G., Walters G.B., Hewitt A.W., Masson G., Helgason A., DeWan A. Common variants near CAV1 and CAV2 are associated with primary open-angle glaucoma. Nat Genet. 2010;42:906–909. doi: 10.1038/ng.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song L., Ge S., Pachter J.S. Caveolin-1 regulates expression of junction-associated proteins in brain microvascular endothelial cells. Blood. 2007;109:1515–1523. doi: 10.1182/blood-2006-07-034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun C., Wang J.J., Mackey D.A., Wong T.Y. Retinal vascular caliber: systemic, environmental, and genetic associations. Surv Ophthalmol. 2009;54:74–95. doi: 10.1016/j.survophthal.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 64.Murata T., Lin M.I., Stan R.V., Bauer P.M., Yu J., Sessa W.C. Genetic evidence supporting caveolae microdomain regulation of calcium entry in endothelial cells. J Biol Chem. 2007;282:16631–16643. doi: 10.1074/jbc.M607948200. [DOI] [PubMed] [Google Scholar]
- 65.Ogawa N., Mori A., Hasebe M., Hoshino M., Saito M., Sakamoto K., Nakahara T., Ishii K. Nitric oxide dilates rat retinal blood vessels by cyclooxygenase-dependent mechanisms. Am J Physiol Regul Integr Comp Physiol. 2009;297:R968–R977. doi: 10.1152/ajpregu.91005.2008. [DOI] [PubMed] [Google Scholar]
- 66.Maniatis N.A., Shinin V., Schraufnagel D.E., Okada S., Vogel S.M., Malik A.B., Minshall R.D. Increased pulmonary vascular resistance and defective pulmonary artery filling in caveolin-1−/− mice. Am J Physiol Lung Cell Mol Physiol. 2008;294:L865–L873. doi: 10.1152/ajplung.00079.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hughes S., Chan-Ling T. Characterization of smooth muscle cell and pericyte differentiation in the rat retina in vivo. Invest Ophthalmol Vis Sci. 2004;45:2795–2806. doi: 10.1167/iovs.03-1312. [DOI] [PubMed] [Google Scholar]
- 68.Stallcup W.B., Dahlin-Huppe K. Chondroitin sulfate and cytoplasmic domain-dependent membrane targeting of the NG2 proteoglycan promotes retraction fiber formation and cell polarization. J Cell Sci. 2001;114:2315–2325. doi: 10.1242/jcs.114.12.2315. [DOI] [PubMed] [Google Scholar]
- 69.Burg M.A., Nishiyama A., Stallcup W.B. A central segment of the NG2 proteoglycan is critical for the ability of glioma cells to bind and migrate toward type VI collagen. Exp Cell Res. 1997;235:254–264. doi: 10.1006/excr.1997.3674. [DOI] [PubMed] [Google Scholar]
- 70.Fukushi J., Makagiansar I.T., Stallcup W.B. NG2 proteoglycan promotes endothelial cell motility and angiogenesis via engagement of galectin-3 and alpha3beta1 integrin. Mol Biol Cell. 2004;15:3580–3590. doi: 10.1091/mbc.E04-03-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rezajooi K., Pavlides M., Winterbottom J., Stallcup W.B., Hamlyn P.J., Lieberman A.R., Anderson P.N. NG2 proteoglycan expression in the peripheral nervous system: upregulation following injury and comparison with CNS lesions. Mol Cell Neurosci. 2004;25:572–584. doi: 10.1016/j.mcn.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 72.Li Z., Wermuth P.J., Benn B.S., Lisanti M.P., Jimenez S.A. Caveolin-1 deficiency induces spontaneous endothelial-to-mesenchymal transition in murine pulmonary endothelial cells in vitro. Am J Pathol. 2013;182:325–331. doi: 10.1016/j.ajpath.2012.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morgan W.H., Lind C.R., Kain S., Fatehee N., Bala A., Yu D.Y. Retinal vein pulsation is in phase with intracranial pressure and not intraocular pressure. Invest Ophthalmol Vis Sci. 2012;53:4676–4681. doi: 10.1167/iovs.12-9837. [DOI] [PubMed] [Google Scholar]
- 74.Morgan W.H. Central venous pulsations: new findings, clinical importance and relation to cerebrospinal fluid pressure. J Glaucoma. 2013;22(Suppl 5):S15–S16. doi: 10.1097/IJG.0b013e31829349d7. [DOI] [PubMed] [Google Scholar]
- 75.Legler U., Jonas J.B. Frequency of spontaneous pulsations of the central retinal vein in glaucoma. J Glaucoma. 2009;18:210–212. doi: 10.1097/IJG.0b013e318182edd2. [DOI] [PubMed] [Google Scholar]
- 76.Yu J., Bergaya S., Murata T., Alp I.F., Bauer M.P., Lin M.I., Drab M., Kurzchalia T.V., Stan R.V., Sessa W.C. Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest. 2006;116:1284–1291. doi: 10.1172/JCI27100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sinha B., Köster D., Ruez R., Gonnord P., Bastiani M., Abankwa D., Stan R.V., Butler-Browne G., Vedie B., Johannes L., Morone N., Parton R.G., Raposo G., Sens P., Lamaze C., Nassoy P. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell. 2011;144:402–413. doi: 10.1016/j.cell.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
WT and Cav-1 KO vessels have TJ morphology. Electron micrographs reveal abundant caveolae in WT vascular endothelial cells (arrows) and absent caveolae in Cav-1 KO vessels. Junctions (boxed regions) have similar gross appearance in both Cav-1 KO and WT retinas. BM, basement membrane; EC, endothelial cells; P, pericytes. Scale bar = 500 nm.
Phosphorylation status of ZO-1 and occludin was not altered in Cav-1 KO retinas. A: Lysates from WT and Cav-1 KO retinas were immunoprecipitated using phosphotyrosine 4G10 antibody and blotted for ZO-1 and phosphotyrosine (4G10). Multiple proteins exhibited enhanced tyrosine phosphorylation in Cav-1 KO retinas (arrows) but ZO-1 was not recovered in phosphotyrosine immunoprecipitates from either KO or WT retinas. B: Occludin phosphorylation identified by the reduced migration on SDS-PAGE gels. Upon treatment with alkaline phosphatase, the slower migrating bands collapsed and occludin ran as a single 60-kDa band. No changes in occludin phosphorylation were detected between Cav-1 KO and WT retinas. AP, alkaline phosphatase; B, beads; FT, flow-through; IN, input; pY, phosphotyrosine.
Sucrose gradient analysis revealed similar fractionation pattern of claudin-5 in the Cav-1 KO and WT retinas. A: Western blotting shows that claudin-5 fractionation pattern in the Cav-1 KO retinas does not differ from that in the WT retinas. B: Densitometry analysis of the Western blotting results.
eNOS hyperactivity in Cav-1 KO retinas contributes to arterial dilation. WT and Cav-1 KO mice were treated with 30 mg/kg body weight L-NAME or PBS for 2 hours and retinas were stained using anti-CD31 antibody (red). Artery diameters were measured to 500 μm from the ONH, or to the first bifurcation before 500 μm, measuring only the common artery trunk. A–D: Representative images show that PBS-injected Cav-1 KO arteries (C) are more dilated than WT arteries (A). In response to L-NAME treatment, arteries in WT (B) and Cav-1 KO (D) retinas constricted to a similar extent. E: Quantification of artery diameters. Five to six arteries from each retina were measured, averaged, and collected as one sample. Data are expressed as means ± SEM. n = 4 per group. ∗∗P < 0.01 versus WT PBS. ††††P < 0.0001 versus WT L-NAME. ‡‡‡P < 0.001 versus Cav-1 KO PBS. Two-way analysis of variance with Newman–Keuls post hoc analysis. Scale bar = 50 μm.
Cav-1–dependent arterial dilation is not corrected by eNOS deletion. Arteries from WT (A), Cav-1 KO (C), eNOS KO (D) and Cav-1 and eNOS DKO (B) mice were measured according to the criteria described for Supplemental Figure S4 and quantified (E). In contrast to the transient pharmacological inhibition of NOS activity shown in Supplemental Figure S4, chronic genetic deletion of eNOS did not correct artery diameter. In fact, eNOS KO retinas also had enlarged diameters, suggesting a compensatory mechanism to dilate arteries when eNOS is constitutively deleted. Data are expressed as means ± SEM. n = 7 or 8 per genotype. ∗P < 0.05 versus WT. One-way analysis of variance with Newman–Keuls post hoc analysis was used. Scale bar = 50 μm.
NG2 deficiency is not dependent on eNOS activity. Cav-1 and eNOS DKO retinas were stained with mural cell markers, NG2 (green) and PDGFRβ (blue) and endothelial marker, CD31 (red). Compared with WT, DKO veins have obvious NG2 deficiency, as also observed in Cav-1 KO retinas (Figure 7). As in the Cav-1 KO mice, another mural cell marker (PDGFRβ) and an endothelial marker (CD31) did not differ between DKO and WT retinas, suggesting that such NG2 reduction is not due to eNOS activity but rather depends on Cav-1, reinforcing the possibility that Cav-1 might play an important role in NG2 biology (eg, trafficking, membrane targeting, or polarization). Scale bar = 50 μm.
Total NG2 transcript levels in Cav-1 KO retinas. Gene expression was measured by RT-qPCR, normalized to housekeeping gene RPL19 and presented as fold change over control (WT). The results did not reach our statistical threshold (P = 0.11 Student’s t-test); however, given that NG2 is reduced on a subset of retinal vessels (the venous system, and notably the branch veins), NG2 transcripts from total retinal mRNA may not reveal vessel-specific reductions. Data are expressed as means ± SEM. n = 10 (WT) or 14 (Cav-1 KO).
Mural cell phenotypic changes observed in Cav-1 KO branch retinal veins are also apparent in Cav-1 and eNOS DKO mice but not in eNOS KO mice. Retinas were stained with the mural cell markers NG2 (blue) and αSMA (red) and the endothelial marker CD31 (green). DKO branch retinal veins exhibited reduced NG2 and enhanced αSMA immunoreactivity distal to the ONH, as well as spindly mural cell morphology proximal to the ONH, as was observed for Cav-1 KO mice. Scale bar = 50 μm (A–H); 10 μm (I–P).