

Abstract
Pulmonary hypertension (PH) is a disease of diverse etiology. Although primary PH can develop in the absence of prior disease, PH more commonly develops in conjunction with other pulmonary pathologies. We previously reported a mouse model in which PH occurs as a sequela of Pneumocystis infection in the context of transient CD4 depletion. Here, we report that instead of the expected Th2 pathways, the Th1 cytokine IFN-γ is essential for the development of PH, as wild-type mice developed PH but IFN-γ knockout mice did not. Because gene expression analysis showed few strain differences that were not immune-function related, we focused on those responses as potential pathologic mechanisms. In addition to dependence on IFN-γ, we found that when CD4 cells were continuously depleted, but infection was limited by antibiotic treatment, PH did not occur, confirming that CD4 T cells are required for PH development. Also, although CD8 T-cells are implicated in the pathology of Pneumocystis pneumonia, they did not have a role in the onset of PH. Finally, we found differences in immune cell phenotypes that correlated with PH, including elevated CD204 expression in lung CD11c+ cells, but their role remains unclear. Overall, we demonstrate that a transient, localized, immune response requiring IFN-γ and CD4-T cells can disrupt pulmonary vascular function and promote lingering PH.
Pulmonary hypertension (PH) is a devastating disease with complex etiology and, in all likelihood, diverse mechanisms of pathology. A recent reclassification of the types of PH involves five major divisions, including forms associated with specific causative agents (such as drugs), hypoxia, and infectious agents (such as schistosomes).1 A common feature of many of these agents is that they initiate local inflammation, which may act as a trigger for the development of PH,2–4 even if the inflammation does not persist after the manifestation of PH. However, there does not seem to be any single inflammatory mediator responsible for the inflammatory initiation of PH. For example, several immune cell types (T cells, B cells, and macrophages) and inflammatory cytokines (TGF-β, IL-1β, IL-6, RANTES, and IL-13) have been implicated in various forms of PH.5–9
In the T helper 1 and T helper 2 (Th1 and Th2) paradigm, CD4+ Th2 cells drive an immune response characterized by the production of cytokines such as IL-4, IL-5, and IL-13, as well as by secreted antibody (in particular, IgE).10 In several studies using animal models, a strong case has been made for a role of Th2 immune responses as instigators of PH. For example, a Th2 response associated with sensitization to an antigen and subsequent challenge with that antigen can result in muscularization of smaller pulmonary arteries, and this response is associated with CD4+ cells and IL-13.11 The protein resistin-like alpha (Retnla; alias cysteine-rich secreted protein FIZZ1) can be induced by hypoxia (which is associated with vascular remodeling12), but it is also induced in Th2 immune responses; in some cases, the Th2-associated molecule Retn1a appears to have a strong association with vascular remodeling and resultant PH,13,14 which may be related to its induction by hypoxia, itself a potent stimulator of vascular remodeling.14 An interesting mouse model of PH associated with repeated inhalation of spores of the fungus Stachybotrys chartarum also is associated with the Th2 cytokines IL-4 and IL-5, but not the Th1 cytokine IFN-γ.15 Finally, in what is probably one of the best-known examples of PH in conjunction with an infectious agent, schistosomiasis-induced PH appears to be associated with the Th2 cytokine IL-13.16 Furthermore, IL-13 is implicated in several other forms of PH.17
In contrast, Th1 immune responses, which are characterized by the secretion of cytokines such as IFN-γ and TNF-α and activation of phagocytic macrophages, appear to have little connection with the development of vascular remodeling and PH. Despite a few reports of elevated TNF-α in conjunction with clinical syndromes that include PH,7,18 there is very little association of PH with the canonical Th1 cytokine, IFN-γ, although there is one report of IFN-γ having a synergistic effect with other cytokines on in vitro pulmonary vascular cell remodeling.19 Indeed, this lack of effect is illustrated by the fact that IFN-γ has been used in clinical treatment of idiopathic pulmonary fibrosis,20 although with little effectiveness, even though ≤40% of patients with idiopathic pulmonary fibrosis also exhibit PH.21
Recently, we reported that PH developed in the aftermath of a resolved Pneumocystis pneumonia in mice, in the context of a transient depletion of CD4+ cells.22 At the time, it was unclear which immune responses are involved in the development of PH in that mouse model; although there were elevated levels of Retnla (FIZZ1) in the bronchoalveolar lavage fluid (BALF) of mice that developed PH, as well as some perivascular fibrosis, IL-4 signaling was not required for these developments.22 In the present study, surprisingly, we found that the Th1 cytokine IFN-γ is absolutely required for the development of PH in this mouse model, and that perivascular fibrosis does not appear to be a cause of PH. Furthermore, having previously established that onset of PH in these mice is correlated with the resurgence of CD4+ T cells after depletion,22 with the present study we have demonstrated that CD4+ cells are also absolutely required for the development of PH.
Materials and Methods
Animals
Most mice used in these experiments were raised in the Animal Research Facility Center at Montana State University, from stock originally obtained from the Jackson Laboratory (Bar Harbor, ME). The IFN-γ–knockout mice were on a BALB/c background (stock no. 002286). STAT6 knockout mice on a BALB/c background (stock no. 002828) and IL-12p40 knockout mice on a BALB/c background (stock no. 002694) were purchased directly from the Jackson Laboratory. SCID mice were raised from stock originally obtained from Charles River Laboratories International (stock no. 236; Wilmington, MA). In some cases, BALB/c and C57BL/6 mice were purchased directly from the NIH–National Cancer Institute Mouse Repository (Frederick, MD). All animals were housed in isolation rooms in high-efficiency particulate absorption–filtered ventilated cages, and received autoclaved mouse chow and acidified water.
Mouse Depletion and Infection Regimens
Pneumocystis murina (originally obtained from The Trudeau Institute, Saranac Lake, NY) was maintained by serial colonization of SCID source mice. To inoculate experimental mice, the lungs of infected source mice were homogenized, P. murina nuclei were enumerated, and 107 P. murina organisms were administered via intratracheal delivery to isoflurane-anesthetized mice, as described previously.23 Mice were depleted of specific cell populations by periodic intraperitoneal injections of antibody. For short-term depletion of CD4+ cells, 300 μg of GK1.5 (antibody grown from hybridoma originally obtained from ATCC, Manassas, VA) was injected at days −3, 0, +3, and +7, relative to the day of infection. With this procedure, resurgence of CD4+ cells occurs at 18 to 24 days after infection, initiating clearance of the P. murina infection.22 Wild-type BALB/c treated in this way are here referred to as BALB-STD, and IFN-γ knockout mice treated in this way are referred to as IFN-γ–STD (where STD stands for short-term depletion). When continuous depletion of CD4+ cells was required, this protocol was followed by twice weekly doses of 300 μg GK1.5.
To determine whether Pneumocystis-associated PH can occur in the absence of CD4+ T cells, groups of mice were continuously depleted of CD4+ cells, infected with P. murina, and the subsequent fungal infection was then cleared by antibiotic administration. This was done by switching the mice to a medicated chow containing 0.124% sulfamethoxazole and 0.025% trimethoprim (Sulfa-Trim formula 5TXS; Test Diet, Richmond, IN) (SMX-TMP) at 19 days after infection, so that clearance kinetics of P. murina were similar to those of the groups with short-term depletion. The animals were then assessed as described at 36 days after infection. In other experiments, a group of BALB-STD mice were also depleted continuously of CD8+ cells, with twice-weekly intraperitoneal injections of 300 μg of the depleting antibody TIB-210 [BALB-STD (−CD8)] (antibody from the TIB-210, hybridoma; ATCC).
Physiological Measurements and Tissue Sampling
Right ventricular pressure (RVP) was measured by transthoracic cannulation of pentobarbital-anesthetized animals, as described previously.22 After measurement, anesthetized mice were euthanized by exsanguination. Bronchoalveolar lavage was then performed by nicking the trachea and lavaging the lungs with 3 mL of PBS containing 3 mmol/L EDTA. A 100-μL aliquot of each lavage was spun onto a slide using a cytospin centrifuge, and stained with Dade Behring Diff-Quik (Siemens Healthcare Diagnostics, Newark, DE), and another small aliquot was taken for microscopic enumeration of cells in BALF, using a hemocytometer. The remaining lavage sample was centrifuged at 900 × g for 10 minutes. Supernatant aliquots were frozen at −80°C for later analysis of soluble mediators, and the pelleted cells were used for fluorescence-activated cell sorting analysis, as described below. At this time, the heart was also removed, and the relative RV mass was determined, as described previously.22 In brief, the atria were trimmed away from the heart, which was then weighed; next, the RV was cut away from the heart and weighed, as was the remaining left ventricle (LV) and septum (S). Relative RV mass (%) was calculated as [RV/(LV + S)] × 100.
Lung tissue was collected by first tying off the small right lobes of the lung with suture, cutting these lobes off, and then infusing the single large left lobe with phosphate-buffered formalin. This lobe was then immersed in the formalin solution, and later processed for histology. In some cases, the left lobe was instilled with OCT optimal cutting temperature compound (Sakura Finetek, Torrance, CA), quick frozen, and later used for cryostat sectioning and immunostaining. The small right lobes were immediately homogenized in PBS, and the homogenate was used for enumeration of P. murina burden, as described previously.22 Aliquots of the homogenate were either directly frozen in liquid nitrogen for later zymography or, before freezing, were supplemented with EDTA for collagen analysis or with complete protease inhibitors (P8340; Sigma-Aldrich, St. Louis, MO) and phosphatase inhibitors (P5726; Sigma-Aldrich) for Western blot analysis.
Flow Cytometry
BALF cells were resuspended in Fc block solution and stained for cell surface antigens, as described previously.22 A variety of antibody cocktails were used. Typically, two cocktails (each with three to six antibodies) were used to stain for lymphocyte markers (CD4, CD8, CD19, CD3, CD49b, and γδ-TCR) and a tetramer specific for natural killer T (NKT) cells. Natural killer (NK) cells were quantified based on CD3−, CD49b+ staining. A third cocktail was used for macrophage surface antigens (CD11c, CD11b, CD204, CD80, and CD23). Antibodies were from eBioscience (San Diego, CA), BioLegend, (San Diego, CA), and BD Biosciences (San Jose, CA). Separate acquisition schemes and set-up single stain tubes were used for macrophages and lymphocytes, because of the high autofluorescence of alveolar macrophages; acquisition was performed on either an LSR II or FACSCanto flow cytometer (BD Biosciences). Analysis was performed using FlowJo software version 9.6 (Tree Star, Ashland, OR). Percentages of cell subset numbers were combined with cell count and differential count data to obtain actual cell number estimates.
Biochemical Assays
Soluble collagen was measured in lung homogenates using a Sircol dye kit (Biocolor, Carrickfergus, UK), as described previously.22 Concentrations of IFN-γ, IL-13, CCL2, IL-12p40, and granulocyte-macrophage colony-stimulating factor (GM-CSF) in BALF were determined using commercial enzyme-linked immunosorbent assay kits (eBioscience).
Gene Expression Analysis
Total RNA was collected from the lungs of BALB-STD and IFN-γ–STD mice, as well as from immunocompetent BALB/c mice that were P. murina–infected but not depleted of CD4 and from control uninfected BALB/c mice, at day 38 after infection, using a Qiagen (Valencia, CA) RNeasy maxi kit procedure. RNA was evaluated using an RNA 6000 NanoChip assay on a 2100 Bioanalyzer instrument (Agilent Technologies, Santa Clara, CA); only RNA with an RNA integrity number of >7.5 was used for analysis. Total RNA was amplified, biotin-labeled, and hybridized to GeneChip Mouse 430A 2.0 genome arrays (Affymetrix, Santa Clara, CA), using 500 ng total RNA and an Ambion MessageAmp premier RNA amplification kit (AM1792; Life Technologies, Carlsbad, CA). The labeled cRNA was purified, fragmented, and hybridized to the arrays at 45°C for 16 hours with constant rotational mixing at 60 rpm. Washing and staining of the arrays was performed using a GeneChip fluidics station 450 (Affymetrix). Arrays were scanned using a GeneChip scanner 7G and GCOS software version 1.4 (Affymetrix).
Microarray data were analyzed using FlexArray software version 1.5 (http://www.gqinnovationcenter.com/services/bioinformatics/flexarray/index.aspx?l=e, last accessed September 24, 2010). Files in Affymetrix CEL format were imported and normalized using GC robust multiarray normalization (GC-RMA). Three data filters were applied: i) signal intensities of at least 50 in one biological replicate; ii) at least one condition called present by the GCOS software; and iii) a fold change of at least 2 in any comparison. Statistical significance between conditions was determined using analysis of variance, resulting in a final gene list of 406 genes. The log2 values were imported into Genesis software version 1.7.524 for hierarchical clustering. Grouping genes into simplified categories was performed using the generic Gene Ontology GO slim set from the GO Consortium (http://www.geneontology.org/GO.slims.shtml, last accessed November 21, 2012). Total lung gene expression data of P. murina–infected wild-type and IFN-γ–knockout mice are available at Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo, accession number GSE51750).
Statistical Analysis
Preliminary group analysis was performed using Numbers software version '09 (Apple, Cupertino, CA). Final statistical analysis with analysis of variance followed by Tukey’s post hoc tests was performed using GraphPad Prism 6 software (Graph Pad Software, La Jolla, CA).
Results
IFN-γ Is Necessary for PH Development
Because our previous studies with IL-4 receptor knockout mice suggested that Th2 immune mechanisms may not be required for development of PH in our model, we repeated these experiments in mice deficient in the transcription factor STAT6, the cellular pathway through which many Th2 responses are mediated. As with the IL-4 receptor knockout mice, the STAT6 signaling knockout mice were not protected from development of PH in this experimental model (Figure 1). Although RVP was slightly reduced, compared with wild-type BALB-STD mice (Figure 1A), RV mass was not significantly different (Figure 1B).
Figure 1.
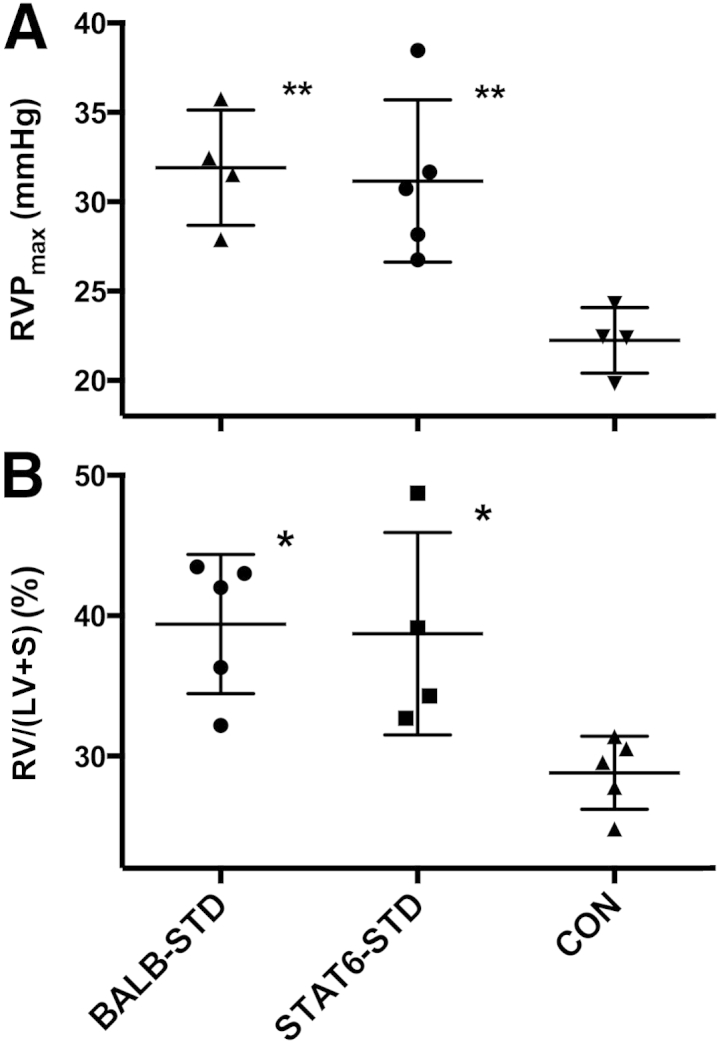
Under short-term CD4 depletion and P. murina infection, STAT6 knockout mice (STAT6-STD) are not protected from developing PH. A: RVP did not differ between BALB-STD and STAT6-STD mice. B: RV hypertrophy was present in both CD4-depleted strains. Data are representative of two independent experiments and are expressed as means ± SEM. n = 4 or 5. ∗P ≤ 0.05, ∗∗P ≤ 0.01 versus control (CON).
For comparison, we performed similar experiments with Th1-deficient mouse strains in Th1-related molecules. The cytokine IL-12 is important in driving development of Th1 responses, and (as we show below) the p40 subunit of this molecule is significantly elevated in the BALF of BALB-STD mice. Although the p40 subunit of IL-12 can be either antagonistic or supportive of Th1 actions,25 results with IL-12p40 knockout mice did not differ from results with BALB-STD mice (data not shown). In contrast, mice deficient in IFN-γ clearly did not develop PH when subjected to the same experimental procedure as wild-type BALB/c mice (Figure 2).
Figure 2.
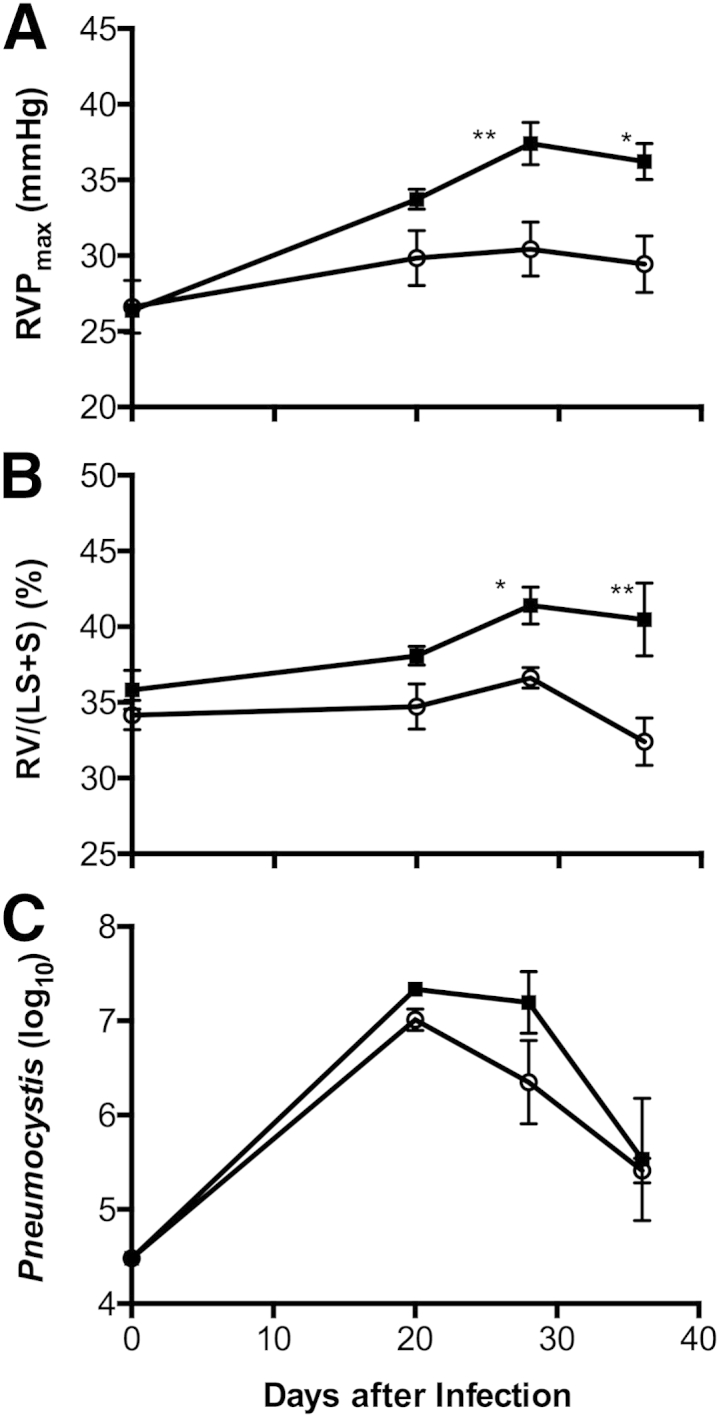
Under short-term CD4 depletion and P. murina infection, PH develops in wild-type BALB/c mice (BALB-STD, squares) but not in IFN-γ knockout mice (IFN-γ–STD, circles). RVP became elevated (A), and persistent RV hypertrophy developed (B), even as the P. murina infection was cleared (C). A Pneumocystis count of approximately 25,000 (4.4 on a log10 scale) is the limit of detection for this procedure. Data are representative of three independent experiments and are expressed as means ± SEM. n = 5. ∗P ≤ 0.05, ∗∗P ≤ 0.01.
Next, we characterized the onset of PH in BALB-STD mice. An increase in RVP coincided with the beginning of resurgence of CD4+ cells at 20 days after inoculation (Figure 3) and continued through the 36-day measurement period; there was no significant change in RVP in IFN-γ–STD mice (Figure 2A). The change in RVP was mirrored by significant RV hypertrophy in BALB-STD mice, but (as expected) there was no change in RV mass in IFN-γ–STD mice (Figure 2B). In contrast to these physiological responses, there was no significant difference in the kinetics of clearance of P. murina; in both BALB-STD and IFN-γ–STD mice, P. murina levels were highest at 20 days after infection and had declined to residual levels by 36 days after infection (Figure 2C).
Figure 3.

Under short-term CD4 depletion and P. murina infection, the influx of pulmonary inflammatory cells differs distinctly between BALB-STD mice (which develop PH) and IFN-γ–STD mice (which do not develop PH). Cell types counted included total lung cells; macrophages (Macs), polymorphonuclear cells (PMN), eosinophils (EOS), natural killer (NK) cells, and γδ T cells as percentage in BALF; and B cells, CD4+ T cells, CD8+ T cells, and CD4+CD8+ cells. Important differences are described in the Results section. Data are representative of three independent experiments and are expressed as means ± SEM. n = 5. ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001, and ∗∗∗∗P ≤ 0.0001 versus control.
Cellular and Cytokine Inflammatory Responses Are Distinctly Different in BALB-STD and IFN-γ–STD Mice
Because the onset of Pneumocystis-associated PH is tied to the immune response, and to CD4+ T cells in particular, we selected three time points (20, 28, and 36 days after infection) to more closely examine the development from baseline of a differential immune response in BALB-STD mice (which exhibit PH) and in IFN-γ–STD mice (which do not). As expected, given the broad and complex role of IFN-γ in the immune response, we observed many significant differences in the immune response in these two strains of mice (Figure 3). As the immune response proceeded, there was a greater alveolar influx of total cells in the IFN-γ–STD mice, although the types of cells predominant in each strain differed distinctly. In the IFN-γ–STD mice, there were significantly more macrophages and γδ T cells, and marginally greater numbers of neutrophils and eosinophils at the peak of the inflammatory response (20 to 28 days after infection). In contrast, the BALB-STD mice had relatively more NK cells in the BALF at this time. There were no consistent differences in the numbers of NKT cells (data not shown). Resolution of inflammation was more rapid in the IFN-γ–STD mice than in the BALB-STD mice. We had previously found that inflammation was nearly resolved in BALB-STD mice by 50 days.22 In the present study, at 36 days there was still a persistent lymphocytic inflammation in BALB-STD mice, but inflammation had already begun to recede in IFN-γ–STD mice. Numbers of B cells, CD4+ T cells, and double-positive (CD4+CD8+) T cells were significantly higher in BALB-STD mice at 36 days (Figure 3). Notably, there were greater numbers of alveolar CD8+ T cells than CD4+ T cells in both strains of mice, although the absolute number of CD8+ cells was higher in IFN-γ–STD mice at 28 days.
Also as expected, there were substantial differences in the pulmonary inflammatory cytokine environment during Pneumocystis infection in these two mouse strains. IFN-γ was markedly elevated in BALB-STD mice at 20 and 28 days after infection, but rapidly dropped to baseline levels by 36 days (Figure 4A). IL-12p40 is another cytokine that may have Th1-type actions, but is reported to have other immunomodulatory functions as well.26,27 Levels of IL-12p40 increased at 20 days after infection in both strains; IL-12p40 remained elevated in WT mice through 36 days after infection, but had declined to baseline levels in IFN-γ–STD mice by day 36 (Figure 4B). The baseline level of pulmonary IL-13 was higher in IFN-γ–STD mice than in BALB-STD mice, and in the latter, IL-13 fell to significantly lower levels at 30 to 36 days after infection (Figure 4C); a slight increase in IL-13 in BALB-STD mice at 20 days after infection did not reach statistical significance. It is also notable that levels of the cytokine GM-CSF, important in development and recruitment of macrophages, were significantly higher in the IFN-γ–STD mice over the course of the measurement period (Figure 4D), which may in part explain the higher numbers of alveolar macrophages noted above. Levels of CCL-2, another cytokine implicated in the pulmonary recruitment of macrophages, did not differ significantly between the two mouse strains (Figure 4E).
Figure 4.
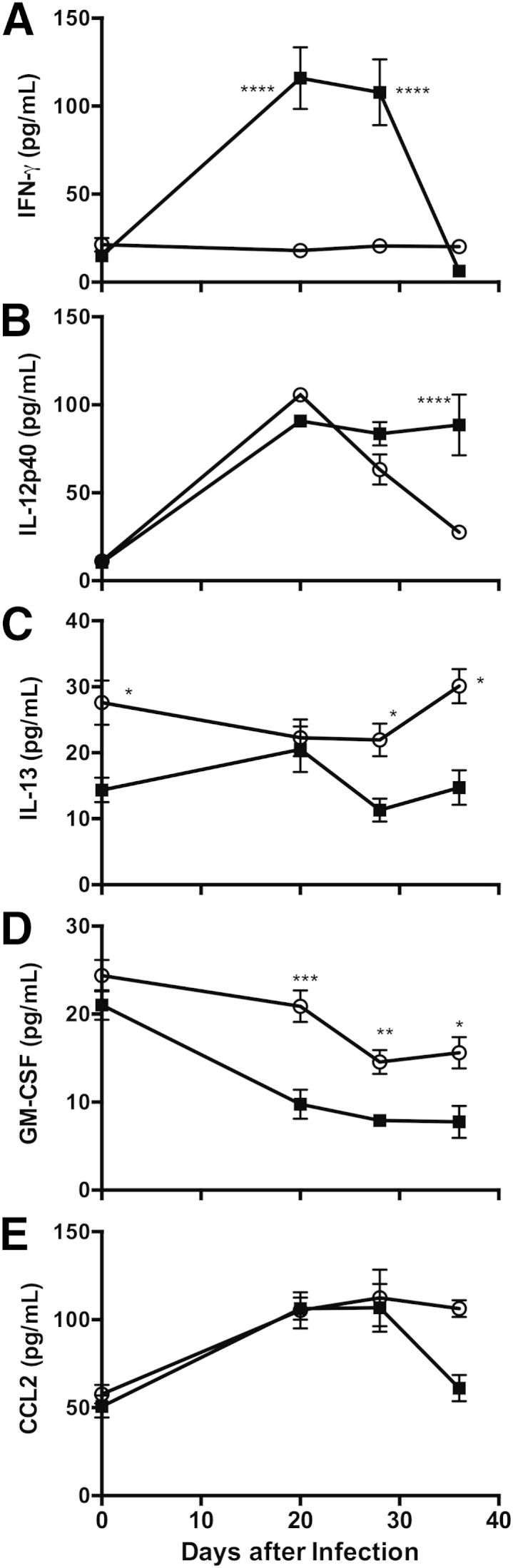
IFN-γ (A) and IL-12p40 (B) levels were elevated at times in BALF from BALB-STD mice, compared with IFN-γ–STD mice (which do not develop PH), whereas levels of IL-13 (C) and GM-CSF (D) were significantly lower. E: Levels of CCL2 did not differ significantly between the two mouse strains. Data are representative of three independent experiments and are expressed as means ± SEM. n = 5. ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001, and ∗∗∗∗P ≤ 0.0001 versus control.
Perivascular Fibrosis Is Not Correlated with PH
We previously observed areas of perivascular fibrosis in lungs of mice with PH.22 In the present study, we similarly observed residual areas of perivascular inflammation, with some indication of fibrosis, as shown by collagen deposition (Figure 5). However, this response was similar in BALB-STD, IFN-γ–STD, and STAT6-STD mice, compared with controls (Figure 5, A–D). Because pressure regulation in the pulmonary blood flow may be related more to the caliber of smaller arterioles, we also looked at arterioles near alveolar ducts. Less residual inflammation was observed near these smaller vessels, with no indication of perivascular fibrosis (Figure 5, E–H).
Figure 5.

In representative images, perivascular inflammation lingers around larger pulmonary arteries adjacent to airways (arrowheads), and collagen deposition (blue) in these areas is similar in BALB-STD (A), IFN-γ–STD (B), and STAT6-STD (C) mice but is less than that in the control (D). Although slight inflammation without collagen deposition remains around smaller arterioles near alveolar ducts (arrows) in P. murina–infected mice, it is similar in BALB-STD (E), IFN-γ–STD (F), and STAT6-STD (G) mice but is absent in the control (H). Original magnification, ×200. Scale bar = 50 μm (B and F).
Gene Expression Analysis Shows a Mostly Differential Immune Response
Analysis of total lung RNA of BALB-STD, IFN-γ–STD, and immunocompetent BALB/c Pneumocystis-infected mice was made in comparison with RNA from BALB/c control untreated mice. Using the criteria described above, from the array of 14,000 possible genes, we identified 211, 360, and 111 genes, respectively in those three strains, that demonstrated a twofold or greater change in expression. To facilitate evaluation of the expressed genes from a larger perspective, we categorized the genes into a defined subset of Gene Ontology groups (generic GO Slim). As expected, the immune system process category had one of the largest numbers of modulated genes, although large numbers of modulated genes were found also in other categories, including signal transduction, response to stress, and anatomical structure development (Supplemental Table S1).
Because an immune response occurred in each of the three groups, but PH was persistent only in the BALB-STD group, we compared the three lists to determine what unique genes, if any, are associated with PH. The BALB-STD group had only two up-regulated genes and one down-regulated gene unique to that group (Supplemental Table S2); furthermore, these genes were only marginally greater than twofold modulated, and their values differed only slightly from the other two groups. We therefore expanded our analysis to include genes that, although either up-regulated or down-regulated in two or more groups, exhibited much greater regulation (at least twofold) in one group relative to the other two. This analysis allowed for a greater number of genes to be considered (a total of 45) (Supplemental Table S2). Most of these genes also reflect the different immune response in the two strains. For example, BALB-STD mice exhibited higher expression of genes related to certain antibody subtypes (eg, IgG3), but the IFN-γ–STD mice had higher expression of CD8 and CD3 antigens and some chemokines (CXCR6, CCL9). The IFN–STD mice also exhibited higher gene expression of the matrix metallopeptidase MMP12; this was an intriguing finding, but we could not detect any actual difference in matrix metallopeptidase activity via zymography (data not shown).
CD4+ T Cells Are Required for Development of PH
To demonstrate that the connection between CD4+ T cells and the onset of PH in Pneumocystis-infected mice with short-term depletion of CD4 is causal, and not simply correlational, BALB/c mice were depleted of CD4+ cells for the entire duration of an experiment. Because this would normally result in fatal Pneumocystis pneumonia, at 20 days after infection the mice were switched to lab chow containing an antibiotic cocktail (SMX/TMP), to clear the Pneumocystis infection. When the experiment was terminated at 40 days after infection, Pneumocystis-infected mice with continuous CD4 depletion and SMX/TMP treatment did not exhibit any significant signs of PH, in that neither RVP nor RV mass differed significantly from control untreated mice (Figure 6, A and B). BALB-STD mice were the only group with significantly elevated CD4+ cells and macrophages (Figure 6, C and E), but CD8+ cells were elevated in both of the Pneumocystis-infected groups (Figure 6D). As has been reviewed,28 significant production of anti-Pneumocystis antibody does not occur in the absence of CD4+ T cells (Figure 6F).
Figure 6.

Mice continuously depleted of CD4+ cells but treated with SMX/TMP do not develop PH. Antibiotic treatment commenced at 20 days after infection in P. murina–infected (PC+SMX) or uninfected (SMX only) mice. At 40 days after infection, only the BALB-STD mice exhibited PH (A) and RV hypertrophy (B). Numbers of CD4+ cells (C), CD8+ cells (D), macrophages (E), and anti-Pneumocystis antibody (F) were measured in BALF. Data are representative of two independent experiments and are expressed as means ± SEM. n = 5. ∗P ≤ 0.05, ∗∗P ≤ 0.01, and ∗∗∗P ≤ 0.001 as compared to PC+SMX. OD, optical density 405 nm.
CD8+ T Cells Are Not Implicated in Development of PH
In contrast to CD4+ depletion, when CD8+ cells were depleted for the entire duration of the experiment, there was no significant difference in either RVP or RV hypertrophy, compared with BALB-STD mice (Figure 7, A and B). This was despite very little difference in the relative numbers of other inflammatory cells when CD8+ cells were depleted (Figure 7, C, E, and F).
Figure 7.

CD8+ cells do not contribute to the development of PH. Two groups of mice were transiently depleted of CD4+ cells, and one group was also continuously depleted of CD8+ cells [BALB-STD (−CD8)]. There was no significant difference in PH (A), RV mass (B). Numbers of CD4+ cells (C), CD8+ cells (D), macrophages (E), and B cells (F) were measured in BALF. Data are representative of two independent experiments and are expressed as means ± SEM. n = 5. ∗∗P ≤ 0.01 as compared to the BALB-STD (−CD8) group.
Certain Immune Cell Phenotypes Correlate with PH
Although our results highlighted the necessity of CD4+ T cells in the development of PH, we also examined whether other immune cells, perhaps in response to CD4+ cell actions, exhibit any phenotype unique to the BALB-STD group and correlating with PH occurrence. The one group of cells that demonstrated consistent changes was pulmonary CD11c+ cells (which in the lung includes both alveolar macrophages and dendritic cells, as well as some interstitial cells). In all experimental groups that exhibited PH, CD11c+ cells had highly up-regulated expression of the scavenger receptor CD204 (SR-A) (Figure 8). This up-regulation was not present in CD11c+ cells in IFN-γ–STD mice, nor in mice subjected to continuous CD4 depletion (with antibiotic treatment), neither of which exhibited PH. In contrast, when CD8+ cells were depleted, there was up-regulation in CD11c+ CD204 expression, correlating with PH in this group. This pattern was also evident in CD11c+ cells obtained from digestion of the lung (data not shown), which included interstitial cells as well as alveolar cells.
Figure 8.
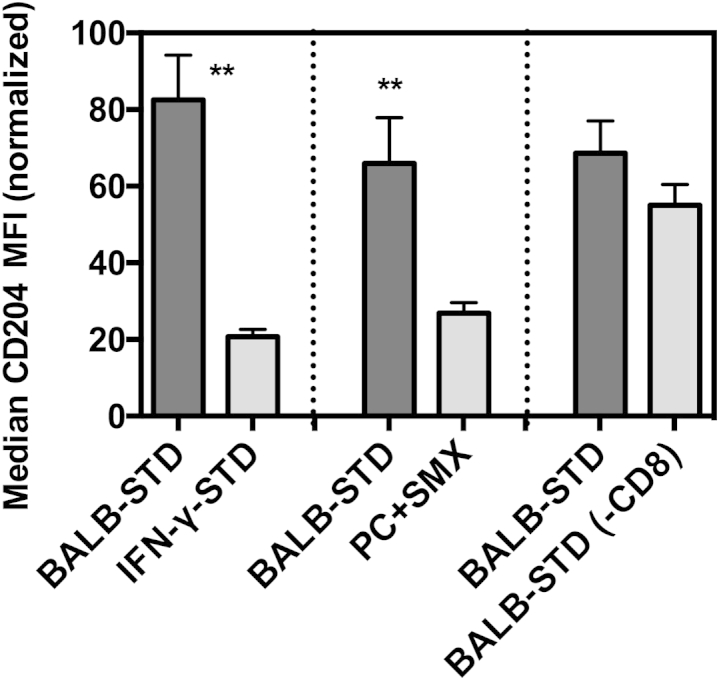
Up-regulated CD204 expression on lung CD11c+ cells correlates with PH. Flow cytometric assessment of median fluorescence intensity (MFI) of Alexa Fluor 647–conjugated anti-mouse CD204 on gated CD11c+ BALF cells. Shown are comparisons of BALB-STD with IFN-γ–STD (Figure 3), PC+SMX (Figure 6), and BALB-STD (−CD8) (Figure 7). Statistical tests were performed on raw MFI values within each experiment, but values were normalized (100% is the highest MFI of BALB-STD group, and 0% is the mean CD204 MFI of control uninfected mice) for the composite graph. Data are representative of two independent experiments and are expressed as means ± SEM. ∗∗P ≤ 0.01.
Discussion
Although PH presents as a well-defined set of distinct symptoms, a number of different mechanistic pathways lead to this disease. We present here a unique developmental path of PH, in which an acute immune response to an infectious organism leads to a chronic impairment after the acute illness has been eliminated. We also show that it is the context and quality of the immune response that determines whether it will direct the onset of PH. The reconstitution of immune function that occurred during an ongoing Pneumocystis infection after a transient period of partial immunosuppression, although successful in eliminating the pathogen, was aberrant in such a way as to cause long-term deleterious effects on pulmonary vascular function.
In our previous study,22 the onset of PH in these mice was correlated with the resurgence of CD4+ cells, and not with the burden of Pneumocystis. Here, we confirm that CD4+ cells are required for the development of PH. Although CD8+ T cells are known to be instrumental in pathology accrued during terminal Pneumocystis pneumonia in the absence of CD4+ cells,29 CD8+ cells are not implicated in the development of PH in our experimental model, despite the presence of large numbers of CD8+ cells in the lung during the onset of PH. It is quite likely that the CD4+ cells are not the proximal cause of changes to the pulmonary vasculature, but instead determine the actions of other cells, potentially other immune effector cells, that more directly locally mediate whatever the vascular changes may underlie PH, as discussed below. Indeed, our gene array analysis confirmed that most of the observed changes in pulmonary gene expression are immune system related.
An immune-associated mechanism proposed in our previous study is that perivascular fibrosis induced by inflammation during clearance of Pneumocystis may be the mechanistic link between the immune response and the increased resistance in pulmonary blood flow leading to RV hypertrophy. At that time, we speculated that Th2-type immune responses typically mediated through IL-4 and/or IL-13 that are permissive to the development of fibrosis may be instrumental in the development of PH. However, just as we previously showed that IL-4 signaling is not required for PH, we show here that other Th2-associated pathways that signal through STAT6 (IL-13 for example) were not required for the development of PH. In fact, after using several different mouse strains with genetic knockouts of immune system components, we were surprised to find that the only factor we examined that was necessary for PH was IFN-γ, a Th1-type cytokine. Because slight amounts of perivascular fibrosis occurred in both IFN-γ–STD and wild-type mice in these experiments, we concluded that this was probably a general response to the extended period of inflammation coincident with the Pneumocystis infection in the context of transient CD4 depletion, and thus was not a strictly Th2-associated phenomenon. This also indicates that, although there are compelling arguments that PH can be associated with increased adventitial and/or smooth muscle stiffness (a possible outcome of perivascular fibrosis),30 this was not a significant factor in the PH we observed in our experimental model.
The finding that IFN-γ is required for the onset of PH in our mouse model provided us with an experimental method by which we could elucidate differences in both the immune response and other downstream pulmonary changes instrumental in the development of PH. We observed several obvious and consistent differences in the cellular inflammatory response between IFN-γ–STD and BALB-STD mice, such as increased numbers of macrophages and γδ T cells in IFN-γ–STD mice and increased numbers of NK and double-positive (CD4+CD8+) lymphocytes in wild-type mice. Although none of these cell types is known to have a clear mechanistic connection to changes in the pulmonary vasculature, some warrant further investigation. The CD4+CD8+ cells are unusual, but they would presumably be depleted with anti-CD8 injections; furthermore, because this does not ameliorate PH, they probably are not mechanistically involved. NK cells have recently been implicated as potentially important effector cells in clearance of Pneumocystis; they secrete IFN-γ, and seem to be regulated in those actions by CD4+ cells,31 which makes them intriguing candidates for further investigation. Also, although there are slightly fewer macrophage-type cells in BALB-STD mice than in IFN-γ–STD mice, we demonstrated clear phenotypic differences in lung CD11c+ cells that correlate with the development of PH, such as elevated expression of the scavenger receptor CD204. Furthermore, because IFN-γ is known to promote production of reactive oxygen species and reactive nitrogen species by macrophages, it will be crucial to explore whether the immune response we observe may be causing vascular dysregulation through oxidative mechanisms. An interesting parallel area of investigation in the literature posits just such a mechanism as involved in situations in which failure of transplanted tissue grafts occur because of failed blood supply to the graft. These investigators showed that changes in the production of NO by inducible NOS (iNOS) were disrupting the function of endothelial NOS (eNOS), reducing the ability of the arteries in these grafts to vasodilate, resulting in decreased blood flow and tissue damage. Most importantly, as in the present study, these effects were dependent on both CD4+ T cells and IFN-γ.32 Investigations are currently under way in our research group to determine whether these mechanisms are involved in our model of immune response-associated PH.
Regarding other downstream changes in the lung, our gene array data revealed very few significant changes in pulmonary gene regulation that were not immune-response related, and those gene products identified do not as yet have any obvious connection to PH. This paucity of results stands in contrast to gene expression studies in other experimental PH models. For example, hypoxic induction of PH in mice was accompanied by a much greater diversity of gene expression changes, including those with more direct involvement with vascular remodeling.33 Furthermore, gene expression analysis of tissues taken from patients with primary PH also reveals a more diverse set of regulated genes than we found.34 This may indicate that the vascular dysfunction that we describe is completely dependent on a lingering immune response, however small, and that this response may eventually recede, given enough time. It is also possible that other, nonimmune changes occurred that were highly local in nature (ie, confined only to perivascular regions in areas where significant inflammation occurred). In such a case, any changes in nonimmune gene RNA expression might be diluted out by sampling the entire lung tissue and therefore would not be readily apparent in our assay. This type of highly local response would probably instead result in dysregulation of vascular function, probably at the level of vascular endothelium or smooth muscle, and would not necessarily be reflected by overt vascular remodeling at the histological level. This is in contrast to forms of PH, both in humans and in animal models, that are characterized by noticeable vascular remodeling, including medial hypertrophy and endothelial proliferation in the pulmonary vasculature.35,36 However, the observation here and elsewhere37 that PH can occur through vascular dysregulation in the absence of significant histological remodeling reinforces the notion that there are many independent paths to persistent PH, and raises the question whether sustained periods of immune-induced vascular dysregulation may eventually lead to other forms of vascular remodeling.
CD4-mediated pathology is believed to be important in several clinical conditions, including allograft rejection,38 autoimmune disease,39 and immune reconstruction inflammatory syndrome (IRIS).40,41 The role of CD4+ cells in IRIS is especially complex. Resurgence of CD4+ cells with effector phenotypes and directed against residual pathogen antigens (often with elevated serum IFN-γ) is associated with many outbreaks of IRIS.42 In contrast, late-onset IRIS-associated Graves disease is associated with increased proportions of naïve CD4+ cells.43 Other investigators have proposed that resurgent CD4+ cells initiate IRIS symptoms primarily through activation of accumulated innate immune cells, such as macrophages.44 Although there is evidence that some of these phenomena occur in our model of Pneumocystis-associated PH (presence of antigen, modified CD4+ phenotypes, modified macrophage phenotype), it remains to be determined which CD4 actions facilitate the development of PH. The larger question is the relevance of this model to PH in HIV-infected individuals (HIV-PH). Given that the incidence of HIV-PH is similar before and after highly active antiretroviral therapy treatment,45 and that no significant correlation has yet been discovered between HIV-PH and AIDS-related opportunistic diseases, it does not seem that the pathological processes we demonstrated in mice are the primary cause of human HIV-PH. Nonetheless, the possibility exists that these processes may be synergistic with other mechanisms proposed for HIV-PH. For example, endothelial dysfunction and increased pulmonary vascular tone, possibly through increased stimulation of endothelin-1, have been suggested as a mechanism of HIV-PH.46 If the inflammatory processes we describe here occur in some AIDS patients, the possibility exists for enhanced endothelial dysfunction through additive effects of these mechanisms. Elucidation of the specific mechanisms of vascular dysfunction in our model will be required to determine the extent of this relevance to human disease, and this is the goal of our ongoing efforts.
Acknowledgments
We thank Katie Rowse, Anfin Erickson, Ann Harmsen, Abigail Leary, and Erin Dobrinen for technical assistance, Tammy Marcotte and the staff of the MSU Animal Resources Center for mouse care and breeding, and Nicole Meissner and Allen Harmsen for manuscript suggestions.
Footnotes
Supported by NIH grants (R01-HL096464 and P20-GM103500), an M.J. Murdock Charitable Trust equipment grant, and the Montana State University Agricultural Experimental Station.
Supplemental Data
References
- 1.Simonneau G., Robbins I.M., Beghetti M., Channick R.N., Delcroix M., Denton C.P., Elliott C.G., Gaine S.P., Gladwin M.T., Jing Z.C., Krowka M.J., Langleben D., Nakanishi N., Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S43–S54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Pullamsetti S.S., Savai R., Janssen W., Dahal B.K., Seeger W., Grimminger F., Ghofrani H.A., Weissmann N., Schermuly R.T. Inflammation, immunological reaction and role of infection in pulmonary hypertension. Clin Microbiol Infect. 2011;17:7–14. doi: 10.1111/j.1469-0691.2010.03285.x. [DOI] [PubMed] [Google Scholar]
- 3.Voelkel N.F., Tuder R.M. Cellular and molecular mechanisms in the pathogenesis of severe pulmonary hypertension. Eur Respir J. 1995;8:2129–2138. doi: 10.1183/09031936.95.08122129. [DOI] [PubMed] [Google Scholar]
- 4.Tuder R.M., Voelkel N.F. Pulmonary hypertension and inflammation. J Lab Clin Med. 1998;132:16–24. doi: 10.1016/s0022-2143(98)90020-8. [DOI] [PubMed] [Google Scholar]
- 5.Larsen K.O., Yndestad A., Sjaastad I., Løberg E.M., Goverud I.L., Halvorsen B., Jia J., Andreassen A.K., Husberg C., Jonasson S., Lipp M., Christensen G., Aukrust P., Skjønsberg O.H. Lack of CCR7 induces pulmonary hypertension involving perivascular leukocyte infiltration and inflammation. Am J Physiol Lung Cell Mol Physiol. 2011;301:L50–L59. doi: 10.1152/ajplung.00048.2010. [DOI] [PubMed] [Google Scholar]
- 6.Ulrich S., Taraseviciene-Stewart L., Huber L.C., Speich R., Voelkel N. Peripheral blood B lymphocytes derived from patients with idiopathic pulmonary arterial hypertension express a different RNA pattern compared with healthy controls: a cross sectional study. Respir Res. 2008;9:20. doi: 10.1186/1465-9921-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kubo K., Hanaoka M., Hayano T., Miyahara T., Hachiya T., Hayasaka M., Koizumi T., Fujimoto K., Kobayashi T., Honda T. Inflammatory cytokines in BAL fluid and pulmonary hemodynamics in high-altitude pulmonary edema. Respir Physiol. 1998;111:301–310. doi: 10.1016/s0034-5687(98)00006-1. [DOI] [PubMed] [Google Scholar]
- 8.Dorfmüller P., Perros F., Balabanian K., Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22:358–363. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- 9.Pinto R.F., Higuchi Mde L., Aiello V.D. Decreased numbers of T-lymphocytes and predominance of recently recruited macrophages in the walls of peripheral pulmonary arteries from 26 patients with pulmonary hypertension secondary to congenital cardiac shunts. Cardiovasc Pathol. 2004;13:268–275. doi: 10.1016/j.carpath.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Annunziato F., Romagnani S. Heterogeneity of human effector CD4+ T cells. Arthritis Res Ther. 2009;11:257. doi: 10.1186/ar2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daley E., Emson C., Guignabert C., de Waal Malefyt R., Louten J., Kurup V.P., Hogaboam C., Taraseviciene-Stewart L., Voelkel N.F., Rabinovitch M., Grunig E., Grunig G. Pulmonary arterial remodeling induced by a Th2 immune response. J Exp Med. 2008;205:361–372. doi: 10.1084/jem.20071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johns R.A. Th2 inflammation, hypoxia-induced mitogenic factor/FIZZ1, and pulmonary hypertension and vascular remodeling in schistosomiasis. Am J Respir Crit Care Med. 2010;181:203–205. doi: 10.1164/rccm.200912-1827ED. [DOI] [PubMed] [Google Scholar]
- 13.Yamaji-Kegan K., Su Q., Angelini D.J., Myers A.C., Cheadle C., Johns R.A. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) increases lung inflammation and activates pulmonary microvascular endothelial cells via an IL-4-dependent mechanism. J Immunol. 2010;185:5539–5548. doi: 10.4049/jimmunol.0904021. [DOI] [PubMed] [Google Scholar]
- 14.Angelini D.J., Su Q., Yamaji-Kegan K., Fan C., Skinner J.T., Champion H.C., Crow M.T., Johns R.A. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) induces the vascular and hemodynamic changes of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2009;296:L582–L593. doi: 10.1152/ajplung.90526.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagayoshi M., Tada Y., West J., Ochiai E., Watanabe A., Toyotome T., Tanabe N., Takiguchi Y., Shigeta A., Yasuda T., Shibuya K., Kamei K., Tatsumi K. Inhalation of Stachybotrys chartarum evokes pulmonary arterial remodeling in mice, attenuated by Rho-kinase inhibitor. Mycopathologia. 2011;172:5–15. doi: 10.1007/s11046-011-9400-3. [DOI] [PubMed] [Google Scholar]
- 16.Graham B.B., Mentink-Kane M.M., El-Haddad H., Purnell S., Zhang L., Zaiman A., Redente E.F., Riches D.W.H., Hassoun P.M., Bandeira A., Champion H.C., Butrous G., Wynn T.A., Tuder R.M. Schistosomiasis-induced experimental pulmonary hypertension: role of interleukin-13 signaling. Am J Pathol. 2010;177:1549–1561. doi: 10.2353/ajpath.2010.100063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christmann R.B., Hayes E., Pendergrass S., Padilla C., Farina G., Affandi A.J., Whitfield M.L., Farber H.W., Lafyatis R. Interferon and alternative activation of monocyte/macrophages in systemic sclerosis-associated pulmonary arterial hypertension. Arthritis Rheum. 2011;63:1718–1728. doi: 10.1002/art.30318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feinberg L., Temple D., de Marchena E., Patarca R., Mitrani A. Soluble immune mediators in POEMS syndrome with pulmonary hypertension: case report and review of the literature. Crit Rev Oncog. 1999;10:293–302. [PubMed] [Google Scholar]
- 19.Wort S.J., Ito M., Chou P.C., Mc Master S.K., Badiger R., Jazrawi E., de Souza P., Evans T.W., Mitchell J.A., Pinhu L., Ito K., Adcock I.M. Synergistic induction of endothelin-1 by tumor necrosis factor alpha and interferon gamma is due to enhanced NF-kappaB binding and histone acetylation at specific kappaB sites. J Biol Chem. 2009;284:24297–24305. doi: 10.1074/jbc.M109.032524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouros D. Interferon gamma for idiopathic pulmonary fibrosis. Lancet. 2009;374:180–182. doi: 10.1016/S0140-6736(09)60689-9. [DOI] [PubMed] [Google Scholar]
- 21.Castria D., Refini R.M., Bargagli E., Mezzasalma F., Pierli C., Rottoli P. Pulmonary hypertension in idiopathic pulmonary fibrosis: prevalence and clinical progress. Int J Immunopathol Pharmacol. 2012;25:681–689. doi: 10.1177/039463201202500314. [DOI] [PubMed] [Google Scholar]
- 22.Swain S.D., Han S., Harmsen A., Shampeny K., Harmsen A.G. Pulmonary hypertension can be a sequela of prior Pneumocystis pneumonia. Am J Pathol. 2007;171:790–799. doi: 10.2353/ajpath.2007.070178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swain S.D., Lee S.J., Nussenzweig M.C., Harmsen A.G. Absence of the macrophage mannose receptor in mice does not increase susceptibility to Pneumocystis carinii infection in vivo. Infect Immun. 2003;71:6213–6221. doi: 10.1128/IAI.71.11.6213-6221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sturn A., Quackenbush J., Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. 2002;18:207–208. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
- 25.Cooper A.M., Khader S.A. IL-12p40: an inherently agonistic cytokine. Trends in Immunol. 2007;28:33–38. doi: 10.1016/j.it.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Wang S.Z., Bao Y.X., Rosenberger C.L., Tesfaigzi Y., Stark J.M., Harrod K.S. IL-12p40 and IL-18 modulate inflammatory and immune responses to respiratory syncytial virus infection. J Immunol. 2004;173:4040–4049. doi: 10.4049/jimmunol.173.6.4040. [DOI] [PubMed] [Google Scholar]
- 27.Onari Y., Yokoyama A., Haruta Y., Nakashima T., Iwamoto H., Hattori N., Kohno N. IL-12p40 is essential for the down-regulation of airway hyperresponsiveness in a mouse model of bronchial asthma with prolonged antigen exposure. Clin Exp Allergy. 2009;39:290–298. doi: 10.1111/j.1365-2222.2008.03131.x. [DOI] [PubMed] [Google Scholar]
- 28.Garvy B.A., Gigliotti F. Antibody-mediated immunity to Pneumocystis in the lungs. In: Fidel P.L., Huffnagle G.B., editors. Fungal Immunology: From an Organ Perspective. Springer; New York: 2005. pp. 291–302. [Google Scholar]
- 29.Wright T.W., Gigliotti F., Finkelstein J.N., McBride J.T., An C.L., Harmsen A.G. Immune-mediated inflammation directly impairs pulmonary function, contributing to the pathogenesis of Pneumocystis carinii pneumonia. J Clin Invest. 1999;104:1307–1317. doi: 10.1172/JCI6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeffery T.K., Morrell N.W. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog Cardiovasc Dis. 2002;45:173–202. doi: 10.1053/pcad.2002.130041. [DOI] [PubMed] [Google Scholar]
- 31.Kelly M.N., Zheng M., Ruan S., Kolls J., D’Souza A., Shellito J.E. Memory CD4+ T cells are required for optimal NK Cell effector functions against the opportunistic fungal pathogen Pneumocystis murina. J Immunol. 2013;190:285–295. doi: 10.4049/jimmunol.1200861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koh K.P., Wang Y., Yi T., Shiao S.L., Lorber M.I., Sessa W.C., Tellides G., Pober J.S. T cell-mediated vascular dysfunction of human allografts results from IFN-gamma dysregulation of NO synthase. J Clin Invest. 2004;114:846–856. doi: 10.1172/JCI21767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gharib S.A., Luchtel D.L., Madtes D.K., Glenny R.W. Global gene annotation analysis and transcriptional profiling identify key biological modules in hypoxic pulmonary hypertension. Physiol Genomics. 2005;22:14–23. doi: 10.1152/physiolgenomics.00265.2004. [DOI] [PubMed] [Google Scholar]
- 34.Geraci M.W., Moore M., Gesell T., Yeager M.E., Alger L., Golpon H., Gao B., Loyd J.E., Tuder R.M., Voelkel N.F. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ Res. 2001;88:555–562. doi: 10.1161/01.res.88.6.555. [DOI] [PubMed] [Google Scholar]
- 35.Tuder R.M., Marecki J.C., Richter A., Fijalkowska I., Flores S. Pathology of pulmonary hypertension. Clin Chest Med. 2007;28:23–42. doi: 10.1016/j.ccm.2006.11.010. vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Durmowicz A.G., Stenmark K.R. Mechanisms of structural remodeling in chronic pulmonary hypertension. Pediatr Rev. 1999;20:e91–e102. [PubMed] [Google Scholar]
- 37.Hyvelin J.M., Howell K., Nichol A., Costello C.M., Preston R.J., McLoughlin P. Inhibition of Rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ Res. 2005;97:185–191. doi: 10.1161/01.RES.0000174287.17953.83. [DOI] [PubMed] [Google Scholar]
- 38.Atalar K., Afzali B., Lord G., Lombardi G. Relative roles of Th1 and Th17 effector cells in allograft rejection. Curr Opin Organ Transplant. 2009;14:23–29. doi: 10.1097/MOT.0b013e32831b70c2. [DOI] [PubMed] [Google Scholar]
- 39.Bolon B. Cellular and molecular mechanisms of autoimmune disease. Toxicol Pathol. 2012;40:216–229. doi: 10.1177/0192623311428481. [DOI] [PubMed] [Google Scholar]
- 40.Mahnke Y.D., Greenwald J.H., DerSimonian R., Roby G., Antonelli L.R.V., Sher A., Roederer M., Sereti I. Selective expansion of polyfunctional pathogen-specific CD4(+) T cells in HIV-1-infected patients with immune reconstitution inflammatory syndrome. Blood. 2012;119:3105–3112. doi: 10.1182/blood-2011-09-380840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson H., de Jong B.C., Peterson K., Jaye A., Kampmann B., Ota M.O.C., Sutherland J.S. Skewing of the CD4(+) T-cell pool toward monofunctional antigen-specific responses in patients with immune reconstitution inflammatory syndrome in The Gambia. Clin Infect Dis. 2013;57:594–603. doi: 10.1093/cid/cit285. [DOI] [PubMed] [Google Scholar]
- 42.Antonelli L.R.V., Mahnke Y., Hodge J.N., Porter B.O., Barber D.L., DerSimonian R., Greenwald J.H., Roby G., Mican J., Sher A., Roederer M., Sereti I. Elevated frequencies of highly activated CD4+ T cells in HIV+ patients developing immune reconstitution inflammatory syndrome. Blood. 2010;116:3818–3827. doi: 10.1182/blood-2010-05-285080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheikh V., DerSimonian R., Richterman A.G., Porter B.O., Natarajan V., Burbelo P.D., Rupert A., Santich B.H., Kardava L., Mican J.M., Moir S., Sereti I. Graves’ disease as immune reconstitution disease in HIV-positive patients is associated with naive and primary thymic emigrant CD4 T-cell recovery. AIDS. 2013 doi: 10.1097/QAD.0000000000000006. [Epub ahead of press] [DOI] [PubMed] [Google Scholar]
- 44.Barber D.L., Andrade B.B., Sereti I., Sher A. Immune reconstitution inflammatory syndrome: the trouble with immunity when you had none. Nat Rev Microbiol. 2012;10:150–156. doi: 10.1038/nrmicro2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sitbon O., Lascoux-Combe C., Delfraissy J.F., Yeni P.G., Raffi F., De Zuttere D., Gressin V., Clerson P., Sereni D., Simonneau G. Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era. Am J Respir Crit Care Med. 2008;177:108–113. doi: 10.1164/rccm.200704-541OC. [DOI] [PubMed] [Google Scholar]
- 46.Kanmogne G.D., Primeaux C., Grammas P. Induction of apoptosis and endothelin-1 secretion in primary human lung endothelial cells by HIV-1 gp120 proteins. Biochem Biophys Res Commun. 2005;333:1107–1115. doi: 10.1016/j.bbrc.2005.05.198. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.