

Abstract
Encapsulation of cisplatin (CDDP) into nanoparticles (NPs) with high drug loading and encapsulation efficiency has been difficult due to the poor solubility of CDDP. However, this barrier has been overcome with a reverse microemulsion method appropriating CDDP’s poor solubility to our advantage promoting the synthesis of a pure cisplatin nanoparticle with a high drug loading capacity (approximately 80.8wt%). Actively targeted CDDP NPs exhibited significant accumulation in human A375M melanoma tumor cells in vivo. In addition, CDDP NPs achieved potent anti-tumor efficacy through the neighboring effect at a dose of 1 mg/kg when injected weekly via IV without inducing nephrotoxicity. The neighboring effect regards an observation made in vivo when the tumor cells that took up CDDP NPs released active drug following apoptosis. Via diffusion, surrounding cells that were previously unaffected showed intake of the released drug and their apoptosis soon followed. This observation was also made in vitro when A375M melanoma tumor cells incubated with CDDP NPs exhibited release of active drug and induced apoptosis on untreated neighboring cells. However, the neighboring effect was unique to rapidly proliferating tumor cells. Liver functional parameters and H&E staining of liver tissue in vivo failed to detect any difference between CDDP NP treated and control groups in terms of tissue health. By simultaneously promoting an increase in cytotoxicity and a lesser degree of side effects over free CDDP, CDDP NPs show great therapeutic potential with lower doses of drug while enhancing anti-cancer effectiveness.
Keywords: Cisplatin, Chemotherapy, Nanomedicine, Drug delivery, Neighboring effect
The use of cisplatin (CDDP) as a cytotoxic drug was pioneered by Rosenberg while studying the effects of electrical fields on the growth of bacteria.1 Investigating the properties of platinum compounds, Rosenberg discovered that DNA damage induced by the crosslinking of CDDP and DNA initiates DNA repair mechanisms and triggers cell apoptosis when repair proves unsuccessful. Utilizing this interaction, CDDP has become a first-line therapy against a wide spectrum of solid neoplasms, including bladder, ovarian, colorectal and melanoma cancers.2, 3 However, drug resistance and related systemic toxicities (e.g. nephro- and neuro- toxicities) limit the clinical use of CDDP.4, 5
Formulating small molecule drugs into nanoparticles (NPs), such as liposomal or polymeric formulations allows for a significant reduction of adverse side effects while maintaining anti-tumor efficacy. Therefore, this class of nanomedicine is currently established as the cutting edge method in treating a variety of cancers.6, 7 With modification, NPs are able to avoid undesired uptake by the reticuloendothelial system (RES) and improve circulation of their encapsulated drugs in the blood compared to free drug.8 Thus, drug efficacy can be greatly increased without a subsequent increase in collateral damage to healthy tissues.
Similarly, uptake of NPs by tumor cells can be mediated by tumor targeting ligands, such as aptamer,9 RGD peptide and anisamide (AA).10–12 The accumulation of nano-sized formulations in tumors is also highly dependent on the enhanced permeability and retention (EPR) effect due to the disorganized and tortuous tumor endothelium.13 Nonetheless, the accessibility of NPs into tumor cells primarily depends on the properties of the NPs, especially size. NPs with a diameter less than 50 nm can penetrate deeper into poorly permeable, hypo-vascular tumors with greater efficiency than larger NPs.14, 15
However, the poor solubility of inorganic CDDP in both water and oil significantly limits the development of NPs with high drug loading and encapsulation efficacy. In our previous study, lipid-coated CDDP (LPC) NPs composed entirely of CDDP and outer leaflet lipids were successfully synthesized and characterized with high drug loading capacity. Compared to other lipid-based cisplatin nanocapsules,16–18 LPC NPs were much smaller and more homogeneous in size.
Herein, we evaluated the anticancer efficacy of LPC NPs on A375M melanoma xenograft tumors. Furthermore, the in vitro release profile of LPC NPs in cells incubated in a medium with 50% fetal bovine serum evaluated. Also, the diffusion and distance dependent neighboring effect of LPC NPs was additionally examined both in vitro and in vivo. Finally, the biodistribution and safety profile of LPC NPs was determined.
RESULTS AND DISCUSSIONS
Physiochemical characterizations of LPC NPs
While the major side effects of CDDP can be minimized through the usage of NPs for drug delivery, the poor solubility of CDDP has hampered the development of a successful nanoparticulate formulation. In the present study, we have successfully synthesized lipid-coated, platinum-filled drug formulations (LPC NPs) characterized with a core of CDDP and 80wt% of drug loading. LPC NPs were negatively stained with uranyl acetate for transmission electron microscopy (TEM). The images revealed the core/membrane nanostructure of NPs with a size of approximately 20 nm in diameter (Figure 1a). DLS results (Figure 1b) further indicated that the hydrodynamic diameter of NPs was approximately 30 nm, slightly larger than the diameter observed in TEM images. The drug loading capacity of LPC NPs determined using inductively coupled plasma mass spectrometry (ICP-MS) was 80.8wt%. Other liposomal formulations of CDDP based drugs, such as SPI-77 (6.7wt%) and Lipoplatin (10wt%), which are either in phase II clinical trials or clinically approved respectively, cannot achieve such high drug loading.19
Figure 1. LPC NPs were characterized with a small size and narrow dispersity.
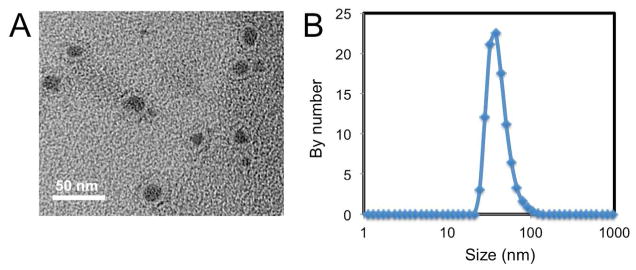
(a) Characterization of LPC NPs using TEM. LPC NPs were negatively stained with uranyl acetate. Scale bar represents 50 nm. (b) Characterization of LPC NPs using dynamic light scattering (DLS).
LPC NPs delivered CDDP efficiently into A375M cells and show significant efficacy
To test the anticancer efficacy of LPC NPs, we first evaluated the cytotoxicity of LPC NPs in A375M melanoma cancer cells. As shown in Figure 2a, the LPC NPs showed a nearly ten-fold lower IC50 than free drug (1.2 v.s. 10.2 μM) regarding the growth inhibition in A375M cells. As a control, empty liposome vesicles did not induce any cytotoxicity (data not shown). Figure 2b and c quantitatively presented cellular uptake of NPs measured using ICP-MS. As indicated, LPC NPs delivered CDDP efficiently into A375M cells with a 6.5 fold increase in internalized drug over free CDDP. In vitro studies illustrated that LPC NPs efficiently transported CDDP into cells and resulted in a significantly lower IC50 over free CDDP.
Figure 2. LPC NPs exhibited high toxicity and strong transport ability of CDDP.
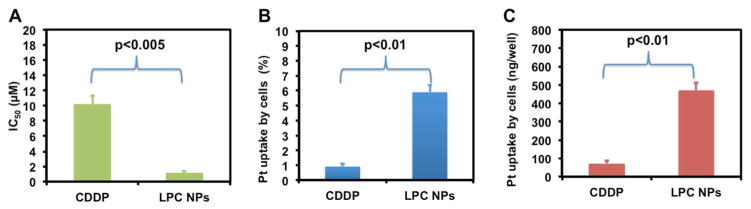
(a) IC50 of CDDP and LPC NPs in A375M cells. (b) The amount of cell uptake of CDDP and LPC NPs in A375M cells quantified using ICP-MS. Data is expressed as % uptake. (c) The amount of the Pt drug associated with cells after incubation with 100 μM CDDP or LPC NPs in 24 well plates. Each bar represents the mean ± SEM of 3 independent experiments. The analysis of variance is completed using a one-way ANOVA.
LPC NPs showed high accumulation of CDDP in A375M xenograft bearing mice and significant anti-tumor efficacy at a low dose
The biodistribution of free CDDP and LPC NPs in tumor-bearing mice was compared. Twenty-four hours post-IV injection, 10.5% of the injected dose per gram of LPC NPs accumulated in the tumors, which was significantly higher than the 1.2% of the injected dose per gram of free CDDP (Figure 3a). To determine the efficacy of LPC NPs in treating A375M tumors, the drugs were administered weekly by IV injection at a dose of 1.0 mg/kg Pt. LPC NPs inhibited the growth of A375M tumors significantly without reducing the body weight of the treated animals (Figure 3 b and c). However, free CDDP at the same dose and dosing schedule was ineffective, possibly due to a low tumor accumulation.
Figure 3. LPC NPs showed high accumulation in A375M tumor cells and impeded the growth of tumors at 1.0 mg/kg of Pt.
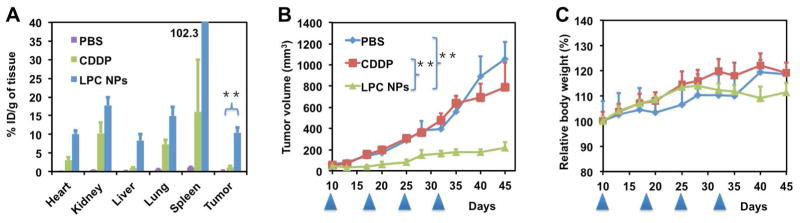
(a) Pt distribution in A375M tumor bearing mice administered with CDDP and LPC NPs. One mg/kg of Pt was administered weekly via IV injection; (b–c) effects of CDDP and LPC NPs on tumor growth and body weight respectively of A375M tumor bearing mice. The arrowheads indicate the time of injection. The results are displayed as mean ± SEM (error bars) of five animals per group. The analysis of variance is computed using a one-way ANOVA. ** indicates p < 0.01.
In vivo, the small size of LPC NPs facilitated the accumulation of LPC NPs in tumor cells through the EPR effect. Therefore, LPC NPs achieved an accumulation of 10.5% injected dose (ID)/g in A375M tumor cells and exhibited significant anticancer therapeutic effect at a low dose and generous dosing schedule while free CDDP was ineffective at the same dose. LPC NPs are therefore capable of inducing considerable anti-tumor efficacy at a significantly lower dose than free CDDP and can be applied to treat a wide range of cancers.
LPC NPs induced discernible apoptosis in A375M tumors
After confirming that LPC NPs showed significant antitumor efficacy, an additional experiment was used to evaluate their efficacy in treating large tumors. Mice bearing A375M melanoma tumors of approximately 600 mm3 were dosed with IV administrations of LPC NPs at a dose of 3.0 mg/kg Pt once a week, for a period of two weeks. One week after the final injection, the mice were sacrificed and the tumors were assayed using TUNEL, a marker of apoptosis. As shown in Figure 4, about 90% of tumor cells were apoptotic, resulting in a 60% reduction in tumor volume (data not shown). Since it is highly unlikely that NPs can reach 90% of tumor cells, additional mechanisms must be contributing to the tumor reduction. The majority of apoptosis may actually be induced by the small fraction of cells that took up the NPs in a pattern known as the neighboring effect. This phenomenon is characterized as the uptake of NPs by tumor cells that become in situ drug depots and release active drugs to induce apoptosis in surrounding cells. Therefore, the neighboring effect is a distance and diffusion dependent effect.
Figure 4. LPC NPs induced apoptosis in 90% of tumor cells.
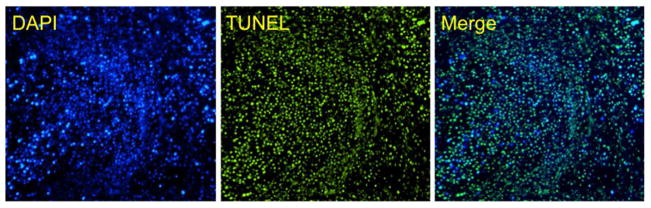
Effects of LPC NPs on A375M tumor cell apoptosis using TUNEL assay. The tumors were treated once a week for two weeks with IV injections containing 3.0 mg/kg of Pt.
Neighboring effect contributed to significant in vivo apoptosis
In support of our hypothesis in vivo, we investigated the intracellular distribution of LPC NPs in tumors and tested the apoptosis of tumor cells using TUNEL and CDDP-DNA adduct antibody. To determine the mechanism of the neighboring effect, we used a lipophilic dye, 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) to label LPC NPs. DiI was entrapped in an asymmetric bilayer. Results indicated that only 5.3% of the tumor cells took up the NPs and yet, 26.7% of cells underwent apoptosis (Figure 5a). It is possible that the amount of NPs in some TUNEL positive cells was too low to be detected because of the detection limitations of the technique. Therefore, we used a “nearest neighbor” analysis to eliminate this possibility. The number of apoptotic cells was measured as a function of the distance to the nearest DiI positive cells. In groups treated with LPC NPs, the large number of green cells (TUNEL positive) close to red cells (DiI positive) gradually decayed to a small number of green cells far from red cells (Figure 5c). The data therefore indicated that the neighboring effect is indeed facilitated by diffusion and varies with distance from the depot cell.
Figure 5. Neighboring effect was studied using TUNEL assay and detection of CDDP-DNA adduct.

LPC NPs were labeled with DiI dye (red). The mice were sacrificed twenty-four hours after receiving a single IV injection of LPC NPs at a dose of 1.0 mg/kg Pt. (a) The distribution of NPs was tracked by DiI dye, and the apoptotic tumor cells were detected by the TUNEL assay; (b) the formation of CDDP-DNA in tumor cells detected by CDDP-DNA antibody. (c) The number of TUNEL positive cells measured as a function of the distance to its nearest DiI positive cell; (d) the number of CDDP-DNA adduct positive cells measured as a function of the distance to its nearest DiI positive cell.
In addition, an antibody specific to the Pt-DNA adduct was used in an assay for a nearest neighbor analysis to determine if Pt-DNA adduct was the cause of cell death.20 As shown in Figure 5b, the formation of CDDP-DNA adducts was confirmed. It was consistently observed that a relatively small number of DiI-positive cells were able to induce the formation of the CDDP-DNA adduct in a large number of surrounding cells (Figure 5c and d). Therefore, formation of CDDP-DNA adduct is directly attributed to the release of CDDP in vivo. This data further provides strong evidence for the neighboring effect by suggesting that active Pt drugs released from dead or dying depot cells were diffused into previously unaffected cells.
In Vitro and intracellular release of drugs from NPs and cytotoxicity assays
To test the neighboring effect in vitro, intracellular release of CDDP from LPC NP was investigated. The kinetics regarding the release of platinum-based drugs from LPC NPs was evaluated in 50% FBS medium at 37°C. As shown in Figure 7a, LPC NPs exhibited a sustained release of Pt over time with a half-life of 3.0 h.
Figure 7. LPC NPs showed a controlled release pattern in medium and in cells.
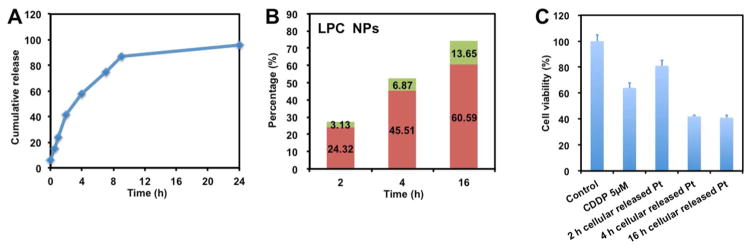
(a) In vitro release kinetics of encapsulated platinum in 50% FBS medium at 37°C and the cellular release of Pt from LPC NPs treated cells. (b) Percentages of Pt in the released medium that were pelletable (green) and unpelletable (red) are shown. (c) The cytotoxic activity of released drugs from NP treated cells at different time points. Cells were treated with 5 μM CDDP for comparison. Each bar represents the mean ± SEM of 3 independent experiments.
We also labeled the NPs using fluorescent NBD-PE lipid and incubated them with cells. Some of the nanoparticles were co-localized with lysosomes as indicated by yellow spots (Figure S1). However, a large number of the NPs taken into the tumor cells did not co-localize with lysosomes.
We further tested the neighboring effect in vitro using the procedure shown in Figure 6. By culturing untreated cells with medium from LPC NPs treated cells, the activity of released CDDP was tested. Cells were first incubated with LPC NPs for 2, 4 or 16 h and subsequently washed and cultured. At different time points, the released, NPs and free drugs in the medium were separated by centrifugation at 16,000g for 20 min. After centrifugation, we observed that the LPC NPs exhibited cellular release and that free drugs composed a major fraction of the medium (Figure 7b). To test the activity of drugs released from cells which previously entrapped NPs, the medium collected at different time points was transferred and incubated with untreated cells. After 48 h, the viability of the tumor cells was assayed using MTS. As shown in Figure 7c, the medium containing more drugs was more toxic.
Figure 6.
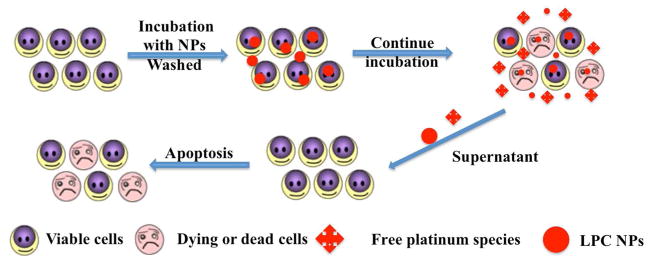
The procedures used to validate the neighboring effect in vitro.
Study of the neighboring effect in vitro
In addition, we further investigated the neighboring effect using a common protocol. A375M-GFP cells that stably expressed green-fluorescence protein (green) were treated with 50 μM of LPC NPs for 4 h, washed, and mixed with untreated A375M cells at a 1:10 ratio. Cells were incubated for an additional 24 or 48 h. Then, cell apoptosis was examined with Alexa Fluor 568-labeled Annexin V (red), an apoptosis marker.
In Figure 8a and b, many cells that were near the green, NP-treated cells were undergoing apoptosis. Groups treated with LPC NPs exhibited a pronounced effect while cells treated with CDDP showed only minimal signs of the neighboring effect. The apoptosis results were further quantified using flow cytometry. Cells were analyzed at 24 (upper panels) or 48 (lower panels) h (Figure 8a). Untreated cells served as the control; after 24 or 48 h, A375M-GFP cells survived, while cells treated with CDDP died and disappeared at both time points. Furthermore, at 24 or 48 h the CDDP-treated cells did not induce significant apoptosis in the unlabeled and untreated cells. At 48 h, less than 6% of untreated cells were apoptotic. In contrast, cells treated with LPC NPs induced a higher percent of apoptotic cells, which were not directly exposed to CDDP. At 48 h, few green cells were left in both cases, but 70% apoptotic cells appeared in the untreated cell population for LPC NPs. It demonstrated that CDDP released from dead or dying cells was able to induce apoptosis on untreated tumor cells. These results confirm that the neighboring effect as characterized by the release of active drug from dead or dying cells after NP internalization and subsequent apoptosis in previously unaffected cells was validated both in vivo and in vitro. The cells transfected with NPs do in fact, serve as drug depots and affect the untreated cells in a manner dependent on distance and diffusion. Though the detailed mechanism behind the transport of the drugs from the depot cells to other cells is still unknown, we will continue to investigate the mechanism of the neighboring effect.
Figure 8. The neighboring effect demonstrated by co-culturing CDDP transfected A375M-GFP cells and A375M cells at a 1:10 ratio.

A375M-GFP cells were treated with LPC NPs (50 μM) for 4 h. After 24 or 48 h of co-culturing, the cell nuclei were stained with Hoechst 33342 (blue). (a–b) Apoptotic cells were stained with Alexa Fluor 568-labeled Annexin V (red) for fluorescence microscopy and flow cytometry analysis.
Safety evaluations LPC NPs are safe and no neighboring effect is observed in major organs
Although the neighboring effect displayed profound effects against rapidly proliferating tumor cells, its potential toxicity toward normal organs is of concern. Therefore, mechanism of the neighboring effect in normal tissues was studied. Since the liver was characterized as the major organ affecting clearance of NPs, the functional parameters aspartate transaminase (AST) and aspartate aminotransferase (ALT) of liver cells treated with free CDDP or LPC NPs were studied. The data indicated that the AST and ALT functional parameters from mice treated with CDDP and LPC NPs fell within the normal range (Figure S2). Furthermore, the comparison between H&E stained liver cells treated with LPC NPs and PBS displayed negligible differences in morphology (Figure 9). Therefore, LPC NPs only posed a minimal threat to normal liver function, which was probably due to the strong repair ability of cisplatin-induced DNA damage in the liver.21–23
Figure 9. HE staining showed LPC NPs did not induce nephrotoxicity.

H&E staining of liver, spleen and kidney tissue from mice that received four doses of treatment (1 mg/kg each).
In addition, it was shown that Kupffer cells were responsible for harmlessly removing most of the NPs in the liver (Figure 10) while hepatocytes showed minimal LPC NPs uptake. Therefore, while the formation of the CDDP-DNA adduct was observed in some liver cells (Figure 11), subsequent apoptosis was not noted (Figure 12). This observation could be due to the successful repair of CDDP-DNA adducts which is has been reported in previous works.21–23 This pattern was also found in other critical organs such as the kidney, spleen, heart, and lung.
Figure 10. DiI-labeled LPC NPs (red) in liver were mainly taken up by Kupffer cells.
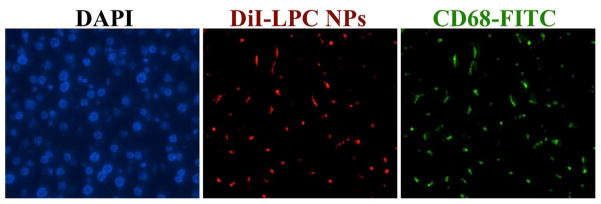
Kupffer cells were stained using CD68 antibody (green) and the hepatocyte nuclei were stained using DAPI (blue). The mice were sacrificed twenty-four hours after receiving a single IV injection of LPC NPs at a dose of 1.0 mg/kg Pt.
Figure 11. Although CDDP-DNA adducts were detected in kidney, liver and spleen, no neighboring effect is observed.

The distribution of DiI-labeled LPC NPs (red) and the detection of CDDP-DNA adduct (green) in kidney, liver and spleen. The mice were sacrificed twenty-four hours after receiving a single IV injection of LPC NPs at a dose of 1.0 mg/kg Pt.
Figure 12. No significant apoptosis was detected in organs from LPC NPs treated mice.

The detection of apoptotic cells in heart, liver, spleen, lung and kidney using TUNEL assay. The mice were sacrificed twenty-four hours after receiving a single IV injection of LPC NPs at a dose of 1.0 mg/kg Pt. Apoptotic cells were detected using TUNEL assay (green) and the cell nuclei were stained using DAPI (blue).
Because the spleen was responsible for significant NP uptake (Figure 3a), histological analysis of the spleen was also performed to exclude any spleen toxicity induced by the NPs (Figure 9). Although LPC NPs accumulated 6-fold higher in the spleen than in cisplatin-treated mice as shown in Figure 3a, the data in Figure 12 indicated that LPC NPs did not induce significant apoptosis spleen cells, which was consistent with other formulations.24, 25 It is believed that uptake was performed primarily by macrophages which can successfully internalize the NP to prevent cell apoptosis (Figure 12). Therefore, the repair of CDDP-DNA adduct was also observed in spleen.
In clinics, the use of CDDP is mainly limited by nephrotoxicity. To this end, the nephrotoxicity of free CDDP and LPC NPs was studied. It was observed that LPC NPs induced significantly less nephrotoxicity over free CDDP at the same dose. As shown in Figure 9, the morphology of kidneys treated with LPC NPs was similar to that treated with PBS. Therefore, no signs of nephrotoxicity were observed in kidneys from mice treated with LPC NPs while some nephrotoxicity was observed in mice treated with free CDDP. Glomeruloscelorsis, tubular cell atrophy, and cystic dilatation of renal tubes were observed in cells treated with free CDDP and indicated by rings, arrows, and squares, respectively. CDDP also induced significantly more apoptotic cells in kidney than LPC NPs (Figure 12). In addition, there were no toxicities in heart and lung for both CDDP and LPC NPs. Pathologic examination of other major organs (lung and heart) in mice that received long-term treatments (Figure S3) indicated that mice treated with LPC NPs suffered no organ damage.
These results indicated that while the neighboring effect was capable of inducing high levels of apoptosis in cancerous cells, its effects on healthy cells were nearly unobservable. A similar pattern was also observed in heart and lung cells in mice treated with LPC NPs. A key mechanism behind this observation is the formation of Pt-DNA adducts in both cancerous and healthy cells alike. However, the Pt-DNA adducts could be successfully repaired in healthy cells while they induced observable apoptosis in cancerous cells. The specificity of these NPs therefore allows a significant anti-tumor effect to be achieved at a low dose and generous dosing schedule.
CONCLUSIONS
The antitumor efficacy of LPC NPs was tested in vitro and in vivo. When administered into mice at a low weekly dose, LPC NPs effectively inhibited the growth of melanoma tumors while free CDDP proved ineffective at the same dose and dosing schedule. In addition, LPC NPs also exhibited the neighboring effect both in vivo and in vitro. The successful uptake of LPC NPs by the tumor cells and the release of active drug following apoptosis therefore furthers the effectiveness of the encapsulated drug. However, the neighboring effect was not induced in organ tissues due to their strong repair ability of the CDDP-DNA adduct. Thus, the tumor specific effect allows a magnification of anti-tumor efficacy at a low dose without pronounced side effects. Subsequently, both the therapeutic potential of CDDP and its safety toward normal tissues in vivo can be greatly optimized. Our studies have therefore distinguished the Pt drug delivery platform as an efficient and relatively safe candidate in the treatment of human melanoma tumors and a promising method for further explorations.
MATERIALS AND METHODS
Materials
All lipids were purchased from Avanti Polar Lipids (Alabaster, AL). DSPE-PEG-AA was synthesized in our lab as previously reported.10 CDDP, AgNO3 and other chemicals were obtained from Sigma-Aldrich (St Louis, MO) without further purification.
Cell lines
The human melanoma, A375M cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA). A375M-GFP was constructed by transfecting an A375M cell line with pEGFP-N1 plasmid. The episomal expression of the plasmid in the transfected cells was maintained by cultivating the cells in the media containing Neomycin. All cells were cultured in DMEM medium supplemented with 10% heat-inactivated, fetal bovine serum (FBS), 20 mM of L-glutamine, 100 U/ml of penicillin G sodium, and 100 mg/ml of streptomycin at 37 °C in an atmosphere of 5% CO2 and 95% air.
Preparations of LPC NPs
LPC NPs were synthesized according to our previous work.26 Briefly, 200 mM cis-[Pt(NH3)2(H2O)2](NO3)2 and 800 mM KCl in water were separately dispersed in a solution composed of Cyclohexane/Igepal CO-520 (71:29, V:V) and Cyclohexane/Triton-X100/Hexanol (75:15:10, V:V:V) (3:1) to form a well-dispersed, water-in-oil reverse micro-emulsion. One hundred μL DOPA (20 mM) was added to the CDDP precursor phase and the mixture was stirred. Then, the two emulsions were mixed for another 30 min while the reaction proceeded. After that, ethanol was added to the micro-emulsion and the particles were collected by centrifugation at 12,000 g. After being extensively washed with ethanol 2–3 times, the pellets were re-dispersed in 3.0 ml of chloroform and stored in a glass vial for further modification. Finally, 1.0 mL of LPC NPs core, 50 μL of 20 mM DOTAP, 50 μL of 20 mM Cholesterol and 50 μL of 10 mM DSPE–PEG-2000 or DSPE–PEG–AA were combined. After evaporating the chloroform, the residual lipids were dispersed in 1.0 mL of d-H2O. The particle size of LPC NPs was determined using a Malvern ZetaSizer Nano series (Westborough, MA). TEM images of LPC NPs were acquired using a JEOL 100CX II TEM (JEOL, Japan). The LPC NPs were negatively stained with 2% uranyl acetate.
Biodistribution
The mice were administered a single dose of 1.0 mg/kg Pt CDDP and LPC NPs. Each group contained five mice, which were sacrificed four hours following injection. Tissue samples were digested by concentrated nitric acid overnight at room temperature and processed according to the procedure reported previously the literature.27, 28 The concentration of Pt was measured using ICP-MS.
In vivo anticancer efficacy
Animals were maintained in the Center for Experimental Animals (an AAALAC accredited experimental animal facility) at the University of North Carolina. All procedures involving experimental animals were performed in accordance with the protocols approved by the University of North Carolina Institutional Animal Care and Use committee and conformed to the Guide for the Care and Use of Laboratory Animals (NIH publication No. 86–23, revised 1985). Female athymic nude mice, 5–6 weeks old and weighing 18–22 g were supplied by the University of North Carolina animal facility. 5×106 A375M cells were injected subcutaneously into the mice. After 10 days, the mice were randomly divided into four groups (4–6 mice per group). The mice were treated with weekly IV injections of CDDP and LPC NPs and saline as a control. A dose of 1.0 mg/kg Pt was administered. Thereafter, tumor growth and body weight were monitored. Tumor volume was calculated using the following formula: , with W being smaller than L. Finally, mice were sacrificed using a CO2 inhalation method. After the therapeutic experiment was done, blood samples were collected and allowed to clot for 2 h at room temperature. Serum was obtained through centrifugation for 20 min at 2,000 g. For liver and renal function experiments, the levels of aspartate aminotransferase, alanine aminotransferase, and blood urea nitrogen in the serum were measured. Major organs were collected after treatment and were formalin fixed and processed for routine H&E staining using standard methods. Images were collected using a Nikon light microscope (Nikon).
After the A375M tumor reached 600 mm3, the mice were treated with two weekly IV injections of LPC NPs at a dose of 3.0 mg/kg Pt. Seven days post the last injection, the mice were sacrificed and the tumors were assayed with TUNEL.
TUNEL assay
The tumors were fixed in 4.0% paraformaldehyde (PFA), paraffin-embedded, and sectioned at the UNC Lineberger Comprehensive Cancer Center Animal Histopathology Facility. To detect apoptotic cells in tumor tissues, a TUNEL assay, using a DeadEndTM Fluorometric TUNEL System (Promega, Madison, WI), was performed following the manufacturer’s protocols. Cell nuclei that were fluorescently stained with green were defined as TUNEL-positive nuclei. TUNEL-positive nuclei were monitored by using a fluorescence microscope (Nikon, Tokyo, Japan). The cell nuclei were stained with 4, 6-diaminidino-2-phenyl-indole (DAPI) Vectashield (Vector Laboratories, Inc., Burlingame, CA). TUNEL-positive cells in three slides of images taken at 40× magnification were counted to quantify apoptosis.
In vivo neighboring effect study
In order to study the neighboring effect, the LPC NPs were labeled with DiI dye (Sigma-Aldrich, St Louis, MO) and administered to nude mice bearing A375M tumors at a single dose of 1.0 mg/kg Pt. Each group contained three mice that were sacrificed 24 h post injection. The organs and tumor sections were prepared by the procedure described in the TUNEL assay in supporting information. The distribution of NPs (red) and TUNEL positive cells (green) were observed using a fluorescence microscope (Nikon, Tokyo, Japan). The distance between two cells was measured using the NIS-Elements Microscope Imaging Software (Nikon Corp., Tokyo, Japan).
In order to observe the distribution of LPC NPs in liver, the sections were incubated with a 1:250 dilution of CD68 primary antibody (Abcam, Cambridge, MA) at 4°C overnight followed by incubation with FITC-labeled secondary antibody (1:200, Santa Cruz, CA) for 1 h at room temperature. The sections were also stained by DAPI and covered with a coverslip. The sections were observed using a Nikon light microscope (Nikon Corp., Tokyo, Japan).
The CDDP-DNA adducts were detected using anti-CDDP modified DNA antibodies [CP9/19] (Abcam, Cambridge, MA). The sections were incubated with a 1:250 dilution of anti-CDDP modified DNA antibody [CP9/19] at 4°C overnight followed by incubation with FITC-labeled goat anti-(rat Ig) antibody (1:200, Santa Cruz, CA) for 1 h at room temperature. The sections were also stained by DAPI and covered with a coverslip. The sections were observed using a Nikon light microscope (Nikon Corp., Tokyo, Japan).
In vitro neighboring effect study
A375M-GFP cells (2 × 105) were seeded in 6-well plates (Corning Inc., Corning, NY) 20 h before the beginning of the experiments. The cells were first treated with CDDP and LPC NPs (50 μM Pt) at 37°C for 4 h and then trypsinized. The A375M-GFP cells were mixed with A375M cells at the ratio of 1:10 (total cell number: 2 × 105) and reseeded into 6-well plates. After culturing for 48 h, the cells were stained with Hoechst 33342 (Sigma, St Louis, MO) and Annexin V Alexa Fluor® 568 Conjugate (Invitrogen, Carlsbad, CA). Cells stained with Alexa Fluor® 568 Conjugate were observed with a fluorescence microscope (Nikon, Tokyo, Japan) and quantified using flow cytometry (Becton-Dickinson, Heidelberg, Germany). Results were processed using the Cellquest software (Becton-Dickinson).
Supplementary Material
Acknowledgments
This work was supported by NIH grants CA129835, CA129421, CA151652, CA151455 and CA149363. We thank Kelly Racette for her assistance in manuscript preparation and Chin-Ying Chung and Gavin Robertson for providing A375M and A375M-GFP cell line.
Footnotes
Supporting information available. Cell Toxicity Assay, Cellular Uptake, In Vitro Drug Release in 50% FBS and Cellular Release of Pt drug and Its Cell Toxicity are available in Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rosenber B, Vancamp L, Krigas T. Inhibition of Cell Division in Escherichia Coli by Electrolysis Products from a Platinum Electrode. Nature. 1965;205:698–699. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 2.Lebwohl D, Canetta R. Clinical Development of Platinum Complexes in Cancer Therapy: An Historical Perspective and an Update. Eur J Cancer. 1998;34:1522–1534. doi: 10.1016/s0959-8049(98)00224-x. [DOI] [PubMed] [Google Scholar]
- 3.Drayton RM, Catto JWF. Molecular Mechanisms of Cisplatin Resistance in Bladder Cancer. Expert Rev Anticanc. 2012;12:271–281. doi: 10.1586/era.11.201. [DOI] [PubMed] [Google Scholar]
- 4.Liang XJ, Meng H, Wang Y, He H, Meng J, Lu J, Wang PC, Zhao Y, Gao X, Sun B, et al. Metallofullerene Nanoparticles Circumvent Tumor Resistance to Cisplatin by Reactivating Endocytosis. Proc Natl Acad Sci US A. 2010;107:7449–7454. doi: 10.1073/pnas.0909707107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Go RS, Adjei AA. Review of the Comparative Pharmacology and Clinical Activity of Cisplatin and Carboplatin. J Clin Oncol. 1999;17:409–422. doi: 10.1200/JCO.1999.17.1.409. [DOI] [PubMed] [Google Scholar]
- 6.Farokhzad OC, Langer R. Nanomedicine: Developing Smarter Therapeutic and Diagnostic Modalities. Adv Drug Deliv Rev. 2006;58:1456–1459. doi: 10.1016/j.addr.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Davis ME, Chen Z, Shin DM. Nanoparticle Therapeutics: An Emerging Treatment Modality for Cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 8.Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic Polyethyleneglycols Effectively Prolong the Circulation Time of Liposomes. FEBS Lett. 1990;268:235–237. doi: 10.1016/0014-5793(90)81016-h. [DOI] [PubMed] [Google Scholar]
- 9.Farokhzad OC, Karp JM, Langer R. Nanoparticle–Aptamer Bioconjugates for Cancer Targeting. Expert Opin Drug Del. 2006;3:311–324. doi: 10.1517/17425247.3.3.311. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee R, Tyagi P, Li S, Huang L. Anisamide-Targeted Stealth Liposomes: A Potent Carrier for Targeting Doxorubicin to Human Prostate Cancer Cells. Int J Cancer. 2004;112:693–700. doi: 10.1002/ijc.20452. [DOI] [PubMed] [Google Scholar]
- 11.Li Z, Huang P, Zhang X, Lin J, Yang S, Liu B, Gao F, Xi P, Ren Q, Cui D. Rgd-Conjugated Dendrimer-Modified Gold Nanorods for in Vivo Tumor Targeting and Photothermal Therapy†. Mol Pharm. 2009;7:94–104. doi: 10.1021/mp9001415. [DOI] [PubMed] [Google Scholar]
- 12.Zhang CF, Jugold M, Woenne EC, Lammers T, Morgenstern B, Mueller MM, Zentgraf H, Bock M, Eisenhut M, Semmler W, et al. Specific Targeting of Tumor Angiogenesis by Rgd-Conjugated Ultrasmall Superparamagnetic Iron Oxide Particles Using a Clinical 1.5-T Magnetic Resonance Scanner. Cancer Res. 2007;67:1555–1562. doi: 10.1158/0008-5472.CAN-06-1668. [DOI] [PubMed] [Google Scholar]
- 13.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor Vascular Permeability and the Epr Effect in Macromolecular Therapeutics: A Review. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 14.Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, et al. Accumulation of Sub-100 Nm Polymeric Micelles in Poorly Permeable Tumours Depends on Size. Nat Nano. 2011;6:815–823. doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]
- 15.Chauhan VP, Stylianopoulos T, Martin JD, Popović Z, Chen O, Kamoun WS, Bawendi MG, Fukumura D, Jain RK. Normalization of Tumour Blood Vessels Improves the Delivery of Nanomedicines in a Size-Dependent Manner. Nat Nano. 2012;7:383–388. doi: 10.1038/nnano.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burger KN, Staffhorst RW, de Vijlder HC, Velinova MJ, Bomans PH, Frederik PM, de Kruijff B. Nanocapsules: Lipid-Coated Aggregates of Cisplatin with High Cytotoxicity. Nat Med. 2002;8:81–84. doi: 10.1038/nm0102-81. [DOI] [PubMed] [Google Scholar]
- 17.Hamelers IH, de Kroon AI. Nanocapsules: A Novel Lipid Formulation Platform for Platinum-Based Anti-Cancer Drugs. J Liposome Res. 2007;17:183–189. doi: 10.1080/08982100701530290. [DOI] [PubMed] [Google Scholar]
- 18.Khiati S, Luvino D, Oumzil K, Chauffert B, Camplo M, Barthelemy P. Nucleoside-Lipid-Based Nanoparticles for Cisplatin Delivery. ACS Nano. 2011;5:8649–8655. doi: 10.1021/nn202291k. [DOI] [PubMed] [Google Scholar]
- 19.Kieler-Ferguson HM, Fréchet JMJ, Szoka FC., Jr Clinical Developments of Chemotherapeutic Nanomedicines: Polymers and Liposomes for Delivery of Camptothecins and Platinum (Ii) Drugs. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2013;5:130–138. doi: 10.1002/wnan.1209. [DOI] [PubMed] [Google Scholar]
- 20.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted Delivery of Cisplatin to Prostate Cancer Cells by Aptamer Functionalized Pt(Iv) Prodrug-Plga-Peg Nanoparticles. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li X, Wang LK, Wang LW, Han XQ, Yang F, Gong ZJ. Cisplatin Protects against Acute Liver Failure by Inhibiting Nuclear Hmgb1 Release. Int J Mol Sci. 2013;14:11224–11237. doi: 10.3390/ijms140611224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Basu A, Krishnamurthy S. Cellular Responses to Cisplatin-Induced DNA Damage. J Nucleic Acids. 2010 doi: 10.4061/2010/201367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang TH, Lindsey-Boltz LA, Reardon JT, Sancar A. Circadian Control of Xpa and Excision Repair of Cisplatin-DNA Damage by Cryptochrome and Herc2 Ubiquitin Ligase. Proc Natl Acad Sci US A. 2010;107:4890–4895. doi: 10.1073/pnas.0915085107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newman MS, Colbern GT, Working PK, Engbers C, Amantea MA. Comparative Pharmacokinetics, Tissue Distribution, and Therapeutic Effectiveness of Cisplatin Encapsulated in Long-Circulating, Pegylated Liposomes (Spi-077) in Tumor-Bearing Mice. Cancer Chemoth Pharm. 1999;43:1–7. doi: 10.1007/s002800050855. [DOI] [PubMed] [Google Scholar]
- 25.Comenge J, Sotelo C, Romero F, Gallego O, Barnadas A, Parada TGC, Domínguez F, Puntes VF. Detoxifying Antitumoral Drugs Via Nanoconjugation: The Case of Gold Nanoparticles and Cisplatin. PLoS One. 2012;7:e47562. doi: 10.1371/journal.pone.0047562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo S, Miao L, Wang Y, Huang L. Pure Cisplatin Nanoparticle with Tunable Size and Surface Modification for Melanoma Cancer Therapy. 2013 in submission. [Google Scholar]
- 27.Graf N, Bielenberg DR, Kolishetti N, Muus C, Banyard J, Farokhzad OC, Lippard SJ. Alpha(V)Beta(3) Integrin-Targeted Plga-Peg Nanoparticles for Enhanced Anti-Tumor Efficacy of a Pt(Iv) Prodrug. ACS Nano. 2012;6:4530–4539. doi: 10.1021/nn301148e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mcgahan MC, Tyczkowska K. The Determination of Platinum in Biological-Materials by Electrothermal Atomic-Absorption Spectroscopy. Spectrochim Acta B. 1987;42:665–668. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.