

Abstract
Assessment of Alzheimer’s disease (AD)-related neurofibrillary pathology requires a procedure that permits a sufficient differentiation between initial, intermediate, and late stages. The gradual deposition of a hyperphosphorylated tau protein within select neuronal types in specific nuclei or areas is central to the disease process. The staging of AD-related neurofibrillary pathology originally described in 1991 was performed on unconventionally thick sections (100 μm) using a modern silver technique and reflected the progress of the disease process based chiefly on the topographic expansion of the lesions. To better meet the demands of routine laboratories this procedure is revised here by adapting tissue selection and processing to the needs of paraffin-embedded sections (5–15 μm) and by introducing a robust immunoreaction (AT8) for hyperphosphorylated tau protein that can be processed on an automated basis. It is anticipated that this revised methodological protocol will enable a more uniform application of the staging procedure.
Keywords: Alzheimer’s disease, Neurofibrillary changes, Immunocytochemistry, Hyperphosphorylated tau protein, Neuropathologic staging, Pretangles
Introduction
The development of intraneuronal lesions at selectively vulnerable brain sites is central to the pathological process in Alzheimer’s disease (AD) [42, 46, 55, 56, 58, 94]. The lesions consist chiefly of hyperphosphorylated tau protein and include pretangle material, neurofibrillary tangles (NFTs) in cell bodies, neuropil threads (NTs) in neuronal processes, and material in dystrophic nerve cell processes of neuritic plaques (NPs) [7, 19, 30].
The AD-related pathological process spans decades and, during this time, the distribution pattern of the lesions develops according to a predictable sequence [8, 21, 89, 93, for a broader discussion of the histopathological diagnosis of AD, see 54, 69, 76]. A staging system for the intraneuronal lesions introduced in 1991 differentiated initial, intermediate, and late phases of the disease process in both non-symptomatic and symptomatic individuals [21, 24–28, 39, 40, 43, 44, 63, 71, 75, 79–82, 84, 85, 87]. In 1997, this staging system was incorporated into the NIH-Reagan criteria for the neuropathological diagnosis of AD [64, 66, 83]. The Braak system was based upon assessment of two 100 μm sections processed according to the silver-iodate technique proposed by Gallyas [49–51, 67, 68, 78]. The first section included the hippocampal formation at uncal level, the anterior parahippocampal gyrus, and portions of the adjoining occipito-temporal gyrus. The second section, taken from the occipital neocortex, encompassed portions of the striate area, parastriate area, and peristriate region (Fig. 1). Distinctive differences in the topographical distribution pattern of the neurofibrillary lesions enabled the observer to assign a given autopsy case to one of six stages [21, 22]. This simple system had the advantage of being reproducible without having to rely on quantitative assessments or knowledge of patient-related data (age, gender, cognitive status). Furthermore, the result of the silver reactions in unconventionally thick sections provided a means of “reading” a given stage with the unaided eye.
Fig. 1.
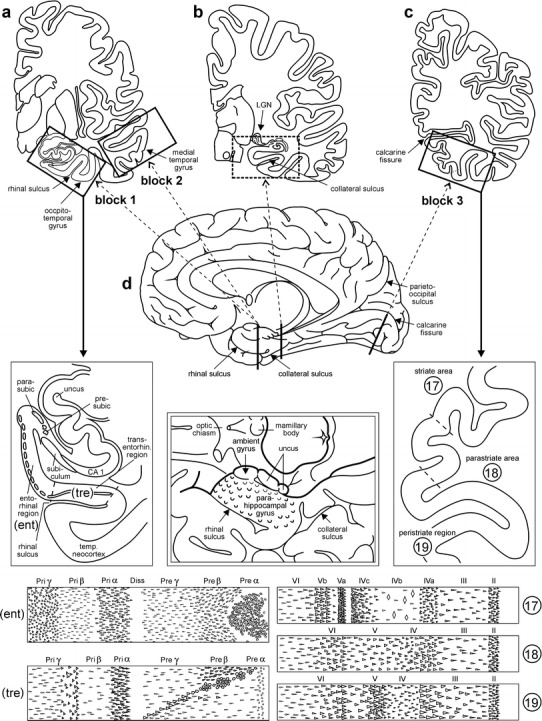
Scheme showing the location of the three blocks of tissue required for staging of AD-related neurofibrillary changes. The first block at the far left (a) includes anteromedial portions of the temporal lobe. It is cut at the mid-uncal or amygdala level (frontal section at the level of the mamillary bodies) and includes the parahippocampal and adjoining occipito-temporal gyri (see enlarged insert below a). The cutting line runs through the rhinal sulcus. The second block comes from the same level and includes part of the medial and superior temporal gyri (a). The third block at the far right (c) is removed from basal portions of the occipital lobe. The cut is oriented perpendicular to the calcarine fissure. The block includes the neocortex covering the lower bank of the calcarine fissure and the adjoining basal occipital gyri. It thus shows portions of the peristriate region as well as of the parastriate and striate areas (see enlarged insert below c). (b) This block provides the classical view of the hippocampal formation and is removed at the level of the lateral geniculate nucleus. It is routinely dissected for diverse diagnostic purposes of the hippocampal formation. The cutting line runs through the collateral sulcus (d). The parahippocampal gyrus at this latitude abuts posteriorly on the lingual gyrus and contains either posterior portions of the entorhinal and transentorhinal regions or lingual neocortex. Insofar as the first temporal block at mid-uncal level is essential for the evaluation of the transentorhinal and entorhinal regions (diagnosis of AD stages I–III), the classical hippocampus section is not absolutely required for staging. The middle drawing in the second row indicated by a double frame shows the anatomical landmarks of the entorhinal region seen basally. Note the wart-like elevations in anterior portions of the parahippocampal gyrus roughly outlining the extent of the entorhinal allocortex. The lower schemata highlight the lamination pattern of the areas that need to be evaluated for staging purposes. The various allocortical and neocortical laminae are indicated across the upper margins. 17, 18, 19 striate area, parastriate area, peristriate region. Abbreviations: CA1 first sector of the Ammon’s horn, ent entorhinal region, parasubic parasubiculum, presubic presubiculum, temp. neocortex temporal neocortex, tre transentorhinal region (mesocortex), transentorhin. transentorhinal
For routine diagnostic purposes, however, such a system is problematic because it calls for unusually thick sections cut from blocks embedded in an unconventional medium [polyethylene glycol (PEG)] [91]. In addition, the method requires that free floating sections be stained by experienced laboratory assistants using a non-automated silver technique. These features drastically limit the feasibility of the original staging protocol for routine diagnostic use in the majority of neuropathological laboratories [35]. At the same time, they account for the fact that the staging system has found broad acceptance in a research context while having been subjected to numerous modifications [9, 18, 36, 39, 40, 53, 59–62, 69–71, 75, 76, 83, 88]. Neuropathologists in routine diagnostic praxis as well as reference centers that maintain brain banks are interested in a uniform staging procedure so that the material submitted by various participating institutions can be used and evaluated according to the same criteria. Such a staging system must be reproducible, cost-effective, and easy.
In recent years, sensitive immunocytochemical methods have been developed, the application of which makes it possible to reliably detect not only incipient neurofibrillary pathology in mildly involved brain regions of non-symptomatic individuals but also, with disease progression, the full extent of the intraneuronal pathology in the end phase [19, 90]. Neurofibrillary changes of the Alzheimer type consist of stable proteins that are impervious to postmortal delay or suboptimal fixation conditions, and immunoreactions for demonstration of hyperphosphorylated tau protein can be carried out even on tissue that has been stored for decades in formaldehyde [1, 72].
Immunoreactions against hyperphosphorylated tau, however, cannot fully replace the Gallyas silver staining method (see Technical addendum), inasmuch as both techniques identify partially different structures. At the beginning of the intraneuronal changes, a soluble and non-argyrophilic material develops, filling the somata of involved nerve cells as well as dendritic processes and axons. Thereafter, the distal dendritic segments become snarled and develop dilated appendages. The soluble “pretangle” material is identifiable in immunoreactions for hyperphosphorylated tau protein but remains Gallyas-negative. It is the harbinger of an argyrophilic, insoluble, and non-biodegradable fibrillary material that emerges after cross-linkage and aggregation of the soluble pretangle material [7, 19, 96]. The Gallyas-positive neurofibrillary aggregations gradually fill the cytoplasm, sometimes infiltrating the proximal dendrites to form a neurofibrillary tangle, and appear in dendritic segments as NTs without involving the axon [19, 45, 95]. Following neuronal death, the abnormal material remains visible in the tissue as extraneuronal ghost tangles or tombstone tangles. With time, ghost tangles are no longer immunoreactive for hyperphosphorylated tau protein and their argyrophilia gradually becomes less pronounced [17, 19, 34]. In summary, the pretangle material can only be detected by immunocytochemistry and this fact has been taken into account in the revised staging procedure presented here (Fig. 2).
Fig. 2.
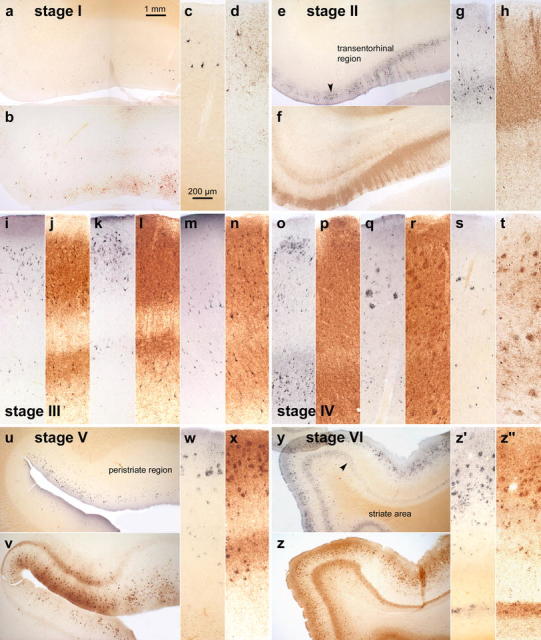
Comparison of Gallyas silver- and AT8-immunostaining of cortical neurofibrillary pathology as seen in adjacent serial 100 μm polyethylene glycol-embedded sections. The distribution pattern of the lesions throughout the various cortical fields that are necessary for staging purposes basically corresponds in both methods. It is possible with either technique to assess the progress of the neurofibrillary pathology. In the revised staging procedure, however, the greater emphasis on the presence of abnormal plexuses, which also include non-argyrophilic pretangle material in AT8-ir sections, facilitates rapid diagnostic assessment of the stages. a–d stage I: Mild involvement is confined to the transentorhinal region. Note that the plexus of AT8-ir nerve cell processes (b and d) is more conspicuous than that of argyrophilic neuropil threads (a and c). Sections originate from a non-demented 62-year-old male. e–h stage II: Lesional density increases and the pathology extends into the entorhinal region. Layer pre-α gradually sinks into a deeper position at the border between entorhinal and transentorhinal region (arrowhead). Note the greater breadth of the ir-plexus in comparison to silverstained nerve cell processes (compare f and h with e and g). Immunoreactions begin to show the deep entorhinal plexus (pri-α). The sections were obtained from a non-demented 78-year-old male. i–n stage III: The pathology in the outer and inner entorhinal (i, j) and transentorhinal (k, l) cellular layers worsens, and lesions extend into the adjoining neocortical association areas of the fusiform (occipito-temporal) gyrus (m, n). The sections originate from an 85-year-old female. o–t stage IV: The density of the lesions increases in both the entorhinal region (o, p) and fusiform gyrus (q–r) with a gradual decrease of the pallid lines (lamina dissecans in p and outer line of Baillarger in r). The neurofibrillary pathology now extends up to the medial temporal gyrus (s, t). Sections were taken from an 80-year-old female. u–x stage V: The lesions extend widely into the occipital lobe and appear in the peristriate region. Note the presence of a deep plexus in AT8-immunoreactions (v, x). Sections were obtained from a 66-year-old demented female. y-z′ stage VI: Lesions are visible even in the parastriate and striate areas of the occipital neocortex. Note the clear-cut line in layer V of the striate area (z′ and z″). The sections originate from a demented 75-year-old male. Scale bar in a applies to all overviews and that in c to all micrographs of cortical areas
In view of the progress that has been made in the demonstration of the neurofibrillary changes of the Alzheimer type, it seemed expedient to revise the 1991 staging procedure by introducing immunoreactions for visualization of hyperphosphorylated tau and by adapting the tissue selection and processing to the demands of the routine diagnostic laboratory. The goal remains the same, namely to stage the AD-related neurofibrillary pathology in six stages, as previously, with emphasis this time on the plexuses formed of both pretangle and tangle material, but using paraffin sections immunostained for hyperphosphorylated tau and processed on an automated basis. To illustrate the advantages and disadvantages of both methods, required brain regions with lesions representing AD stages I–VI have been digitally photographed both in silver- and immunostained 100 μm PEG sections and in 7 μm paraffin sections immunostained for hyperphosphorylated tau protein (AT8–antibody). The revised procedure is intended to facilitate the uniform application of the staging procedure, which now can be performed with greater efficiency than previously.
Processing
Fixation and macroscopic preparation
Brains obtained at autopsy should be fixed by immersion in 10% formalin (4% aqueous solution of HCHO) for one week or longer. Partially remove the meninges to uncover the rhinal sulcus, collateral sulcus, and calcarine fissure (Fig. 1a–d).
Whereas the original staging procedure requires evaluation of thick silver-stained sections from two relatively large blocks of cortical tissue, the revised version uses immunostained paraffin sections microtomed from three blocks of conventional size that fit routine tissue cassettes. Figure 1d shows the cutting lines for removal of the three blocks and, in addition, those for the classical view of the hippocampal formation. Alternatively, the tissue on one side of a cut can be used for conventional paraffin embedding (thin sections), and that on the other side for PEG embedding (thick sections).
The first block includes anteromedial portions of the temporal lobe cut at the mid-uncal or amygdala level (frontal section through the temporal lobe at the level of the mamillary bodies) encompassing anterior portions of both the parahippocampal gyrus and adjoining occipito-temporal gyrus. The cutting line runs through the rhinal sulcus (Fig. 1a, d). The sections from this block contain central portions of the entorhinal region and the adjoining transentorhinal region, the latter of which is concealed in the depths of the rhinal sulcus [23, 98]. This block is essential for assessment of neurofibrillary AD stages I–III.
The second block simplifies assessment of stage IV. It is obtained from the same slice as the first block and includes portions of the medial and superior temporal gyri (Fig. 1a, d). As an alternative to the first two blocks, the entire slice through the temporal lobe can be used, provided slides of sufficient size are available. Reduction of this slice to two blocks is recommended to avoid exceeding the size of conventional tissue cassettes.
The third block is removed halfway between the occipital pole and the junction of the parieto-occipital sulcus with the calcarine fissure. The cut is oriented perpendicular to the calcarine fissure (Fig. 1c, d). Again, the size of the block has been reduced to fit standard tissue cassettes. Care has to be taken that the block includes part of the lower bank of the calcarine fissure and the adjoining basal occipital gyri encompassing portions of the neocortex, i.e., the peristriate region, parastriate field, and a clearly definable primary field, the striate area (Brodmann field 17 with the macroscopically identifiable line of Gennari). This block is indispensable for recognition of the neurofibrillary AD stages V and VI.
Immunocytochemistry
Mounted paraffin sections of 5–15 μm thickness are de-waxed and re-hydrated.
The monoclonal antibody AT8 (Innogenetics, Belgium) is one of several commercially available specific antibodies that show robust immunoreactivity for hyperphosphorylated tau protein, and a recently published immunocytochemical trial using this antibody has yielded reproducible results [1]. AT8 does not cross-react with normal tau epitopes or require special pre-treatments, and it is exceptionally reliable in human autopsy material regardless of the length of the fixation time in formaldehyde and/or the condition of the preserved tissue [16, 19, 57, 77]. When performed on paraffin sections (5–15 μm), AT8-immunoreactions permit counter-staining for other structures of interest, provided that diaminobenzidine is used as a chromogen. Homogeneous immunoreactions can also be achieved using PEG sections (50–150 μm) (Fig. 2). The sections are incubated for 40 h at 4°C with the AT8 antibody (1:2,000) and thereafter processed for 2 h with the second biotinylated antibody (anti-mouse IgG). Reactions are visualized with the ABC-complex (Vectastain) and 3,3-diaminobenzidine (Sigma).
Prolonged fixation of brain tissue in a formaldehyde solution may cause metachromatic precipitations (Buscaino bodies or mucocytes) [73]. Components of this material partially react with silver methods and also may interfere with immunoreactions. The precipitations can be removed with pyridine or a tenside solution [1 unit volume Tween 20 (Merck-Schuchardt 822184) and 9 unit volumes de-ionized water] at 80°C for 30 min or both. The sections are then rinsed thoroughly under running tap water and transferred to de-ionized water.
Comparison between Gallyas silver- and AT8-immunostained thick (100 μm) sections
Figure 2 is designed to facilitate a direct comparison between selected cortical areas in 100 μm thick PEG-embedded sections. The first section of each pair has been silverstained according to a modified version of the technique originally proposed by Gallyas [22, 31, 49–51, 67, 68, 78], whereas the second serial section (i.e., back-to-back sections from the identical tissue block) underwent staining with the antibody AT8.
Intraneuronal neurofibrillary tangles are visualized with equal clarity by both methods (Fig. 2). Further, the plexuses of argyrophilic NTs are visible not only in the Gallyas sections but also in the AT8-immunostained sections—in the latter, however, the plexuses can be seen to include additional pathologically altered neuronal components (dendrites, axons) that contain non-argyrophilic “pretangle” material (see Fig. 2f, h). Such immunostained plexuses appear much more compact than those depicted by the silver stain, and their obvious advantage is that they can be recognized immediately with the naked eye (see Fig. 3). This applies particularly to the AT8-immunoreactive plexuses located in the deep cortical layers (see Fig. 2v, x, z, z″), whereas in Gallyas sections such macroscopic recognition is not always possible (compare Fig. 2v with Fig. 2u).
Fig. 3.
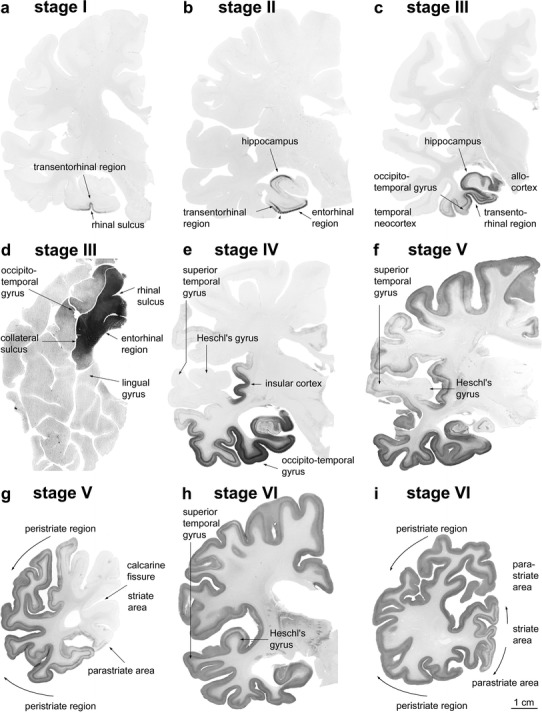
Stages I–VI of cortical neurofibrillary pathology in 100 μm polyethylene glycol-embedded hemisphere sections immunostained for hyperphosphorylated tau (AT8, Innogenetics). a stage I: Involvement is slight and all but confined to the transentorhinal region (part of the temporal mesocortex), located on the medial surface of the rhinal sulcus. The section originates from a non-demented 80-year-old female. b stage II: Additional immunoreactivity occurs in layer pre-α or layer II of the entorhinal region. The layer gradually sinks into a deeper position in the transentorhinal region (arrow). The border between the entorhinal and transentorhinal regions is clearly recognizable in these early stages (arrowhead). Furthermore, the lesions make headway into the hippocampus (arrow). The section was obtained from a non-demented 80-year-old male. c stage III: The lesions in the hippocampal formation worsen. Entorhinal layers pre-α and, additionally, pri-α of the deep layers become strongly involved. Lesions extend through the transentorhinal region into the adjoining high order sensory association areas of the temporal neocortex. The lesions generally do not extend beyond the occipito-temporal gyrus laterally (arrow) and lingual gyrus posteriorly. The section originates from a 90-year-old female. d stage III: A flat section through the entire basal surface of the temporal lobe reveals the severe involvement of the entorhinal cortex (anterior portions of the parahippocampal gyrus) at stage III and shows the tendency of the pathology to extend from there into the adjacent neocortex, i.e., occipito-temporal gyrus laterally (arrow) and lingual gyrus posteriorly (arrow). e stage IV: The third and fourth sectors of the Ammon’s horn and a large portion of the insular cortex (arrow) become affected. The involvement of the neocortical high order sensory association cortex of the temporal lobe now extends up to the medial temporal gyrus and stops short of the superior temporal gyrus (arrow). The primary fields of the neocortex (see transverse gyrus of Heschl) and, to a large extent, also the premotor and first order sensory association areas of the neocortex remain intact. This section was taken from an 82-year-old demented female. f stage V: In addition to the presence of AD-related lesions in all of the regions involved in stage IV, pathological changes appear in the superior temporal gyrus and even encroach to a mild degree upon the premotor and first order sensory association areas of the neocortex. g stage V: In the occipital lobe, the peristriate region shows varying degrees of affection, and lesions occasionally can even be seen in the parastriate area. Stage V sections were obtained from a 90-year-old female with dementia. h–i stage VI: Strong immunoreactivity can be detected even in the first order sensory association areas (e.g., the parastriate area) and the primary areas of the neocortex (e.g., the striate area) of the occipital neocortex. Compare the superior temporal gyrus and transverse gyrus of Heschl at stage V with the same structures at stage VI. Both stage VI sections originate from a severely demented 70-year-old female Alzheimer patient. Scale bar applies to all thick sections
Nonetheless, the distribution pattern of the immunoreactive cortical alterations throughout the various fields that are crucial for staging purposes corresponds to that of the argyrophilic lesions (Fig. 2) and, as such, it allows the observer to trace the progress of the neurofibrillary pathology in both silverstained (Fig. 2) and immunostained sections alike (Fig. 3). The greater emphasis on the abnormal plexuses in AT8-immunoreactive sections, however, facilitates the immediate diagnostic assessment of the stages, as, for instance, is readily evident even in the scaled down photographs of the hemisphere sections shown in Fig. 3. These immunopositive plexuses are still visible macroscopically in paraffin sections (5–15 μm), and it is helpful, initially, without using the microscope, to view all three slides against a light background to assign them preliminarily to a given stage.
The final diagnosis is essentially based on recognition of the topographical distribution pattern of the neurofibrillary pathology and calls for a precise knowledge of which regions in the cerebral cortex, in which sequence, develop the AD-related neurofibrillary lesions. This decision can be made with almost the same degree of accuracy regardless of whether immunostained or silverstained sections are employed, although, based on experience, there is a slight tendency to assign a higher stage to the immunostained slides. The frequency of stage I cases, for example, is somewhat higher in AT8-immunostained sections because in the incipient phases of the disease process AT8-immunopositive nerve cells appear that still lack argyrophilic material. Thus, it is advisable to perform the staging procedure using either the Gallyas or AT8 technique but not both methods.
The staging system
AD-related neurofibrillary changes occur at predisposed cortical and subcortical sites. The distribution pattern and developmental sequence of the lesions are predictable and permit identification of six stages, which can be subsumed under three more general units: I–II, III–IV, V–VI [4–6, 21, 28, 32, 37, 38, 47, 65, 66, 83]. Initial diagnosis as to whether the bulk of the abnormal tau protein is detectable in the transentorhinal and entorhinal regions (stages I–II), in the limbic allocortex and adjoining neocortex (stages III–IV), or in the neocortex, including the secondary and primary fields (stages V–VI), simplifies the subsequent task of differentiation.
Cases without cortical AD-related neurofibrillary pathology
The transentorhinal region as well as the entorhinal region and hippocampal formation remain devoid of AT8-immunoreactive nerve cells (Fig. 4a, b).
Fig. 4.
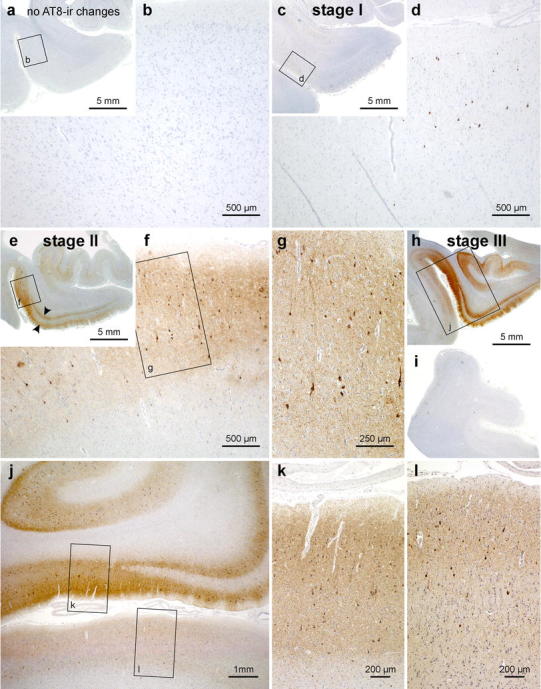
Progress of cortical neurofibrillary pathology, as seen in paraffin sections immunostained for hyperphosphorylated tau (AT8, Innogenetics). a, b Control case displaying no AT8-ir intraneuronal changes. Note that even the transentorhinal region is devoid of immunoreactivity. c, d stage I: The first AT8-ir pyramidal cells often are more easily detected in thick sections than in paraffin material. Closer inspection of the predilection site (transentorhinal region in d, framed area in c), however, reveals the initial lesions. The meshwork of ir-neurites is as yet not well developed. e–g stage II: Many AT8-ir projection neurons are seen in the transentorhinal region accompanied by a well-developed plexus of ir-neurites (f, g). The pathology also extends into the entorhinal layers pre-α and pri-α (arrowheads in e). h–l stage III: The transentorhinal and entorhinal regions are more severely involved than in the preceeding stage, and the pathology now extends into the adjoining temporal neocortex of the occipito-temporal and lingual gyri (h, j). The middle temporal gyrus remains uninvolved (i). Scale bar in a, c, e, h is also valid for i and Fig. 5 a, e, f, j, k, o below
Major characteristics of stages I–II
Stage I: Lesions develop in the transentorhinal region (Figs. 2a–c, 3a, 4c, d) [23]
Subcortical nuclei (i.e., locus coeruleus, magnocellular nuclei of the basal forebrain) occasionally show the earliest alterations in the absence of cortical involvement [29]. The transentorhinal region is the first site in the cerebral cortex to become involved. AT8-immunoreactive (ir) projection cells contain hyperphosphorylated tau in both the cell body and all of its neuronal processes (Fig. 4c, d). Late phases of the stage show abundant AT8-ir neurons that permit recognition of the descent of the superficial entorhinal cellular layer (pre-α, i.e., the outer layer-α of the external principal lamina) from its uppermost position at the entorhinal border to its deepest position at the transition towards the adjoining temporal neocortex (Figs. 1, 3a) [23]. The entorhinal region proper remains uninvolved or minimally involved.
Stage II: Lesions extend into the entorhinal region (Figs. 2e, f, 3b, 4e)
From the transentorhinal region, the lesions encroach upon the entorhinal region, particularly its superficial cellular layer, pre-α (Figs. 1, 3b, 4e–g). The deep layer, pri-α, gradually becomes visible (Figs. 2f, 3b, 4e), shows sharply defined upper and lower boundaries, and is separated from pre-α by the broad, wedge-shaped lamina dissecans (myelinated fiber plexus) [23, 98]. AT8-ir pyramidal cells appear in sectors 1 and 2 of the hippocampal Ammon’s horn (CA1/CA2) (Fig. 3b). Dilations develop transiently in apical dendrites that pass through the stratum lacunosum moleculare of CA1 [20]. Scattered NPs appear in CA1. Fine networks of AT8-ir neurites form in both the stratum radiatum and stratum oriens.
Major characteristics of stages III–IV
Stage III: Lesions extend into the neocortex of the fusiform and lingual gyri (Figs. 2i–n, 3c, d, 4h)
The lesions in stage II sites become more severe. The outer entorhinal cellular layers (pre-α, pre-β, pre-γ) and most of the molecular layer become filled with intermeshing AT8-ir neurites, whereas the pale lamina dissecans contains a few radially oriented neurites. The deep layer pri-α is heavily affected and gradually thins in the transentorhinal region as it approaches the temporal neocortex (Figs. 3c, 4h). CA1 appears band-like, and transient dendritic changes in CA1 reach their culmination point [20]. CA2 is filled with large and strongly AT8-ir pyramidal cells. A moderate number of mossy cells with characteristic dendritic excrescences appear in CA3 and CA4 [97]. The granule cells of the fascia dentata remain uninvolved. AT8-ir sections showing the classical view of the hippocampal formation (Figs. 1b, d) facilitate recognition of lesions in the fascia dentata and Ammon’s horn [92].
From the transentorhinal region, the lesions encroach upon the neocortex of the fusiform and lingual gyri, and then diminish markedly beyond this point (Figs. 3d, 4j). An AT8-ir plexus fills the cellular layers of the temporal neocortex (Figs. 2n, 4l). The outer line of Baillarger is barely developed and gradually becomes recognizable only with increasing distance from the transentorhinal region. A few NPs develop in the outer layers II–IV.
Stage IV: The disease process progresses more widely into neocortical association areas (Figs. 2o–t, 3e, 5a)
Fig. 5.
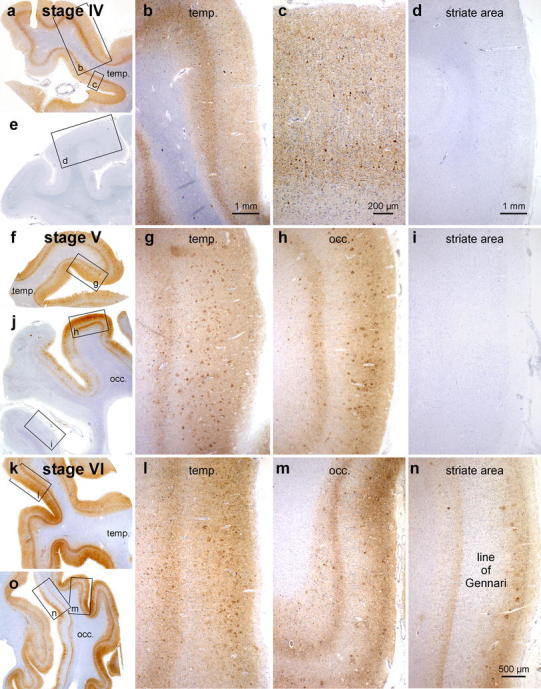
Progress of cortical neurofibrillary pathology, as seen in paraffin sections immunostained for hyperphosphorylated tau (AT8, Innogenetics). a–e stage IV: The disease process extends into the high order sensory association neocortex of the temporal lobe (temp.) and includes the medial temporal gyrus (a–c). The peristriate region as well as the parastriate field and striate area of the occipital lobe (occ.) still lack the neurofibrillary pathology (e, d). f–j stage V: Large numbers of neuritic plaques appear in the neocortex (g, h). Pathological changes now encroach to a mild degree upon premotor areas and first order sensory association fields. In the occipital lobe (j), it is chiefly the peristriate region (h) that shows varying degrees of affection, and lesions occasionally even develop in the parastriate area. The striate area remains uninvolved (i, j). k–o stage VI: Drastic affection of the neocortex is seen at stage VI with involvement of almost all areas. Strong immunoreactivity can be recognized even in premotor areas and first order sensory association areas (e.g., the parastriate area m), as well as in primary neocortical areas (e.g., the striate area n). The borderline between the striate and the parastriate areas is drawn easily and—owing to the sudden cessation of the line of Gennari (plexus of myelinated fibers)—usually can be detected with the unaided eye. A key feature of stage VI is the involvement of the striate area (n), characterized by a dense neuritic mesh in layer V with sharply drawn upper and lower boundaries. Note that the myelin-rich line of Gennari (layer IVb n) is virtually devoid of neurofibrillary pathology. Scale bar in n also applies to g, h, i, l, m
Lesional density increases in sites affected in stage III. A few AT8-ir pyramidal cells appear in the subiculum. The density of the neuritic plexuses of the entorhinal and transentorhinal regions increases and causes a corresponding blurring of the lamina dissecans. The deep plexus spans all of the deep layers: pri-α, pri-β, and pri-γ, and from there penetrates widely into the white substance. This aspect of maximum involvement undergoes little change until the end-phase of AD. Thus, the pathological features of the entorhinal and transentorhinal regions must not be taken into account for further differentiation of stages V and VI. CA1/CA2 are recognizable as dense bands. The varicose dendritic segments vanish from CA1 without leaving behind any remnants. Large numbers of mossy cells in CA3 and CA4 become AT8-ir. A few AT8-ir granule cells appear in the fascia dentata.
In stage IV, the pathology extends broadly into the mature neocortex. A slight decrease in the immunoreactivity of neocortical NPs can be recognized starting at the border facing the transentorhinal region. Dense neuritic plexuses develop up to the middle temporal convolution (Figs. 2s, t, 4a–c), and a rapid decrease in the severity of the lesions occurs at the transition to the superior temporal gyrus (Fig. 3e). The occipital neocortex is unaffected (Fig. 5e, d) or contains blotch-like local accumulations of AT8-ir pyramidal cells and/or NPs in the peristriate region (Brodmann area 19).
Major characteristics of stages V–VI:
Stage V: The neocortical pathology extends fanlike in frontal, superolateral, and occipital directions, and reaches the peristriate region (Figs. 2u–x, 3f, g, 5f, j)
From sites involved at stage IV, the lesions appear in hitherto uninvolved areas and extend widely into the first temporal convolution (Fig. 3f) as well as into high order association areas of the frontal, parietal, and occipital neocortex (peristriate region, Figs. 2v–x, 3g). Initially, unevenly and loosely distributed NPs appear in layers II and III, followed by large numbers of AT8-ir pyramidal cells in layers IIIa, b and V. The lower border of the outer neuritic plexus in layers II-IIIab blurs at its transition to the uninvolved layers IIIc and IV (outer line of Baillarger, Fig. 5g). In stage V, the deep plexus of layer V is narrow and tends not to extend into layer VI and the white matter (Fig. 5g, h). The same pattern (only less pronounced) is seen in secondary areas of the neocortex, where uneven accumulations of NPs predominate. Affection of layer V is weak (Fig. 5j). The primary visual field (striate area) contains only isolated signs of the pathology consisting of NPs (Fig. 5i, j). Isolated AT8-ir neurons also can be seen in layer IIIab (Fig. 5i, j).
Stage VI: The pathology reaches the secondary and primary neocortical areas and, in the occipital lobe, extends into the striate area (Figs. 2y–z″, 3h, i)
Most areas of the neocortex show severe affection and nearly all layers are filled with AT8-ir neurites. As such, the outer line of Baillarger—a pallid stripe in stage V—begins to blur (Fig. 5l). Layer V still appears as a recognizable band but continues into the neuritic plexus of layer VI. The underlying white substance contains AT8-ir axons. A decrease in immunoreactivity of NPs is seen in many neocortical areas and is most pronounced in the basal temporal fields. In the occipital lobe, the pathology breaches the parastriate and striate areas (Figs. 2y–z″, 3i, 5m–o). Large numbers of NPs and AT8-ir nerve cells appear in layers II and IIIab. Baillarger’s outer line or the line of Gennari maintains a light appearance, interrupted only by radially oriented AT8-ir neurites. A sharply drawn AT8-ir plexus follows in layer V (Figs. 2z, z″, 5n, o).
Discussion
By applying the silver technique proposed by Bielschowsky [11–15] Alzheimer [2, 3] became the first to describe the NFTs that develop in the course of the disease that bears his name. This staining technique has been in use for decades but has been subjected to numerous modifications [10, 33, 36, 48, 52, 74, 99]. In systematic studies, Gallyas [49, 50] replaced the critical steps of the Bielschowsky technique by means of more manageable reactions and developed a reliable method for selectively demonstrating AD-related neurofibrillary changes. Following its adaptation for use on 100 μm thick sections, this method was standardized for a procedure to stage the development of the cortical AD-related neurofibrillary pathology that gradually found international acceptance [64, 66, 83]. Nonetheless, one of the unavoidable pitfalls associated with using silverstaining techniques is that different laboratories produce results of widely varying quality [1]. Subsequently, the 1991 staging protocol, too, underwent a series of permutations, among them the application of various types of silver impregnations, the analysis of cortical sites that are fundamentally less well suited for the procedure, and the use of tissue sections, the thickness of which differed from that originally proposed [8, 9, 18, 21, 22, 36, 39–41, 53, 54, 59–61, 69–71, 75, 76, 78, 83]. A radical reduction of the different stages also has been suggested [62].
Since AD is an ongoing and not a static process, every staging procedure is, de facto, an artificial construct. It is the extent of brain involvement rather than qualitative changes in the neurofibrillary pathology that increases with disease progression and, as such, the concept of six neuropathological stages (and only six stages) is not entirely amenable to pathological states of a “transitional” nature that do not fulfill the criteria for one of the six neurofibrillary stages described above.
Here, a revised version of the 1991 staging procedure is presented that can be performed on paraffin sections of conventional thickness, which have been immunostained with the AT8 antibody and processed on an automated basis, thereby fulfilling the demands of the routine laboratory. The simplicity and uniformity of any staging system is the prerequisite for effective comparisons of results among laboratories and for reliable as well as reproducible classification of a disease process [35, 86].
Technical addendum
The previous staging protocol relied upon an advanced but inexpensive silver technique that exploits the physical development of the nucleation sites and in so doing permits careful control of the entire staining procedure [49–51]. Insoluble fibrillary AD-related material can be visualized virtually in the absence of distracting background staining [66, 67, 78]. The technique can be applied to routinely fixed autopsy material, even when the material has been stored for decades in formaldehyde solutions. It facilitates processing of large numbers and/or large sections (e.g., hemisphere sections). A homogeneous staining that permeates the entire thickness of a section is achieved even in 50–150 μm sections [22]. Thin paraffin sections (5–15 μm) can also be used and counter-stained for easy identification of cytoarchitectonic units or specific nuclei [31]. Neurofibrillary changes of the Alzheimer type (NFTs, NTs, NPs) appear in black and, thus, contrast well against an almost unstained background. Connective tissue, glial filaments, normal components of the neuronal cytoskeleton, Pick bodies, Lewy bodies/neurites, and corpora amylacea remain unstained. Abnormal tau-protein in argyrophilic grain disease (AGD), in progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and Niemann Pick type C (NPC) can be visualized as well [46, 78].
The staging procedure originally required sectioning at a thickness of 100 μm. Silverstained or immunostained sections of such thickness are optimal for the demands of low power (stereo) microscopy and greatly facilitate recognition of the laminar and areal distribution pattern of the lesions (see hemisphere sections in Fig. 3). Sections of this thickness can be gained from non-embedded brain tissue with the aid of a vibratome or a freezing microtome. Alternatively, the tissue blocks can be embedded in PEG [91] and sectioned with a sliding microtome. Application of PEG (400 and 1,000: Merck-Schuchardt 807 485 and 807 488) is rapid, simple, and causes little shrinkage [22].
The blocks are transferred from 96% ethanol to PEG 400 and their surfaces covered with blotting paper. Blocks are placed on a rotating table and after having sunk to the bottom (this can take several days), they are transferred to fresh PEG 400 for an additional day. Then, transfer to PEG 1000 at 54°C for 1 day. Embed in fresh PEG 1000, mount, and section at 50–150 μm. Transfer sections to 70% ethanol to remove the embedding medium. Store sections in formaldehyde solutions. Prior to staining, transfer sections to de-ionized water. It is important to note that the Gallyas silver technique displays only highly aggregated fibrillary material, whereas the AT8-immunoreaction also visualizes the non-argyrophilic material that initially develops within involved neurons (pretangle material).
Acknowledgments
The skillful assistance (tissue processing and staining) of Dr. R.A. Kauppinen (Kuopio), Mr. M. Bouzrou (Frankfurt/Main), and (illustrations) Ms. I. Szász (Frankfurt/Main) is gratefully acknowledged.
Footnotes
This study was made possible by funding from the German Research Council (Deutsche Forschungsgemeinschaft) and BrainNet Europe II (European Commission LSHM-CT-2004-503039). This publication reflects only the viewpoint of the authors, the European Community is not responsible for its use or contents.
References
- 1.Alafuzoff I, Pikkarainen M, Al-Sarraj S, Arzberger T, Bell J, Bodi I, Bogdanovic N, Budka H, Bugiani O, Ferrer I, Gelpi E, Gaiccone G, Graeber MB, Hauw JJ, Kamphorst W, King A, Kopp N, Korkolopolou P, Kovacs GG, Meyronet D, Marchi P, Patsouris E, Preusser M, Ravid R, Roggendorf W, Seilhean D, Streichneberger N, Thal DR, BNE consortium, Kretzschmar H (2006) Inter-laboratory comparison of assessments of AD-related lesions. A study of the BrainNet Europe consortium. J Neuropathol Exp Neurol 65 (in press) [DOI] [PubMed]
- 2.Alzheimer A. Über einen eigenartigen schweren Erkrankungsprozeß der Hirnrinde. Neurolog Centralbl. 1906;23:1129–1136. [Google Scholar]
- 3.Alzheimer A. Über eigenartige Krankheitsfälle des späteren Alters. Z ges Neurol Psychiatr. 1911;4:356–385. [Google Scholar]
- 4.Arnold SE, Hyman BT, Flory J, Damasio AR, van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex. 1991;1:103–116. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- 5.Arriagada PV, Growdon J, Hedley-Whyte E, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992a;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 6.Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology. 1992b;42:1681–1688. doi: 10.1212/wnl.42.9.1681. [DOI] [PubMed] [Google Scholar]
- 7.Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G, Seitelberger F, Grundke-Iqbal I, Wisniewski HM. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res. 1989;477:90–99. doi: 10.1016/0006-8993(89)91396-6. [DOI] [PubMed] [Google Scholar]
- 8.Bancher C, Braak H, Fischer P, Jellinger KA. Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer’s and Parkinson’s disease patients. Neurosci Lett. 1993;162:179–182. doi: 10.1016/0304-3940(93)90590-H. [DOI] [PubMed] [Google Scholar]
- 9.Bancher C, Paulus W, Paukner K, Jellinger K. Neuropathologic diagnosis of Alzheimer’s disease: consensus between practicing neuropathologists? Alzheimer Dis Assoc Disord. 1997;11:207–219. [PubMed] [Google Scholar]
- 10.Beech RH, Davenport HA. The Bielschowsky staining technique. A study of the factors influencing its specificity for nerve fibers. Stain Technol. 1933;8:11–30. [Google Scholar]
- 11.Bielschowsky M. Die Silberimprägnation der Axenzylinder. Neurol Centralb. 1902;13:579–584. [Google Scholar]
- 12.Bielschowsky M. Die Silberimprägnation der Neurofibrillen. Neurol Centralb. 1903;21:997–1006. [Google Scholar]
- 13.Bielschowsky M. Die Silberimprägnation der Neurofibrillen. Einige Bemerkungen zu der von mir angegebenen Methode und den von ihr gelieferten Bildern. J Psychol Neurol. 1904;3:169–189. [Google Scholar]
- 14.Bielschowsky M. Die Darstellung der Axenzylinder peripherischer Nervenfasern und der Axenzylinder zentraler markhaltiger Nervenfasern. Ein Nachtrag zu der von mir angegebenen Imprägnationsmethode der Neurofibrillen. J Psychol Neurol. 1905;4:228–231. [Google Scholar]
- 15.Bielschowsky M. Eine Modifikation meines Silberimprägnations-verfahrens zur Darstellung der Neurofibrillen. J Psychol Neurol. 1909;12:135–137. [Google Scholar]
- 16.Biernat J, Mandelkow EM, Schröter E, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M, Mandelkow E. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serin-proline motifs upstream of the microtubule binding region. EMBO J. 1992;11:1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bobinski M, Wegiel J, Tarnawski M, de Leon MJ, Reisberg B, Miller DC, Wisniewski HM. Duration of neurofibrillary changes in the hippocampal pyramidal neurons. Brain Res. 1998;799:156–158. doi: 10.1016/S0006-8993(98)00441-7. [DOI] [PubMed] [Google Scholar]
- 18.Bowler JV, Munoz DG, Merskey H, Haschinski V. Fallacies in the pathological confirmation of the diagnosis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1998;64:18–24. doi: 10.1136/jnnp.64.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braak E, Braak H, Mandelkow EM. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994;87:554–567. doi: 10.1007/BF00293315. [DOI] [PubMed] [Google Scholar]
- 20.Braak E, Braak H. Alzheimer’s disease: transiently developing dendritic changes in pyramidal cells of sector CA1 of the Ammon’s horn. Acta Neuropathol. 1997a;93:323–325. doi: 10.1007/s004010050622. [DOI] [PubMed] [Google Scholar]
- 21.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991a;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 22.Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991b;1:213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- 23.Braak H, Braak E. The human entorhinal cortex. Normal morphology and lamina-specific pathology in various diseases. Neurosci Res. 1992;15:6–31. doi: 10.1016/0168-0102(92)90014-4. [DOI] [PubMed] [Google Scholar]
- 24.Braak H, Braak E. Pathology of Alzheimer’s disease. In: Calne DB, editor. Neurodegenerative diseases. Philadelphia: Saunders; 1994. pp. 585–613. [Google Scholar]
- 25.Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–284. doi: 10.1016/0197-4580(95)00021-6. [DOI] [PubMed] [Google Scholar]
- 26.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997b;18:351–357. doi: 10.1016/S0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 27.Braak H, Braak E. Diagnostic criteria for neuropathologic assessment of Alzheimer’s disease. Neurobiol Aging. 1997c;18(Suppl 4):85–88. doi: 10.1016/S0197-4580(97)00062-6. [DOI] [PubMed] [Google Scholar]
- 28.Braak H, Braak E (1999) Temporal sequence of Alzheimer’s disease-related pathology. In: Peters A, Morrison JH (eds) Cerebral cortex, vol 14 Plenum Press, New York, pp 475–512
- 29.Braak H, Del Tredici K. Alzheimer’s disease: intraneuronal alterations precede insoluble amyloid-β formation. Neurobiol Aging. 2004;25:713–718. doi: 10.1016/j.neurobiolaging.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 30.Braak H, Braak E, Grundke-Iqbal I, Iqbal K. Occurrence of neuropil threads in the senile human brain and in Alzheimer’s disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett. 1986;65:351–355. doi: 10.1016/0304-3940(86)90288-0. [DOI] [PubMed] [Google Scholar]
- 31.Braak H, Braak E, Ohm TG, Bohl J. Silver impregnation of Alzheimer’s neurofibrillary changes counterstained for basophilic material and lipofuscin pigment. Stain Technol. 1988;63:197–200. doi: 10.3109/10520298809107184. [DOI] [PubMed] [Google Scholar]
- 32.Braak H, Del Tredici K, Schultz C, Braak E (2000) Vulnerability of select neuronal types to Alzheimer’s disease. In: Khachaturian ZS, Mesulam MM (eds) Alzheimer’s disease. A compendium of current theories. Ann NY Acad Sci 924:53–61 [DOI] [PubMed]
- 33.Churukian CJ, Kazee AM, Lapham LW, Eskin TA. Microwave modification of Bielschowsky silver impregnation method for diagnosis of Alzheimer’s disease. J Histotechnol. 1992;15:299–302. [Google Scholar]
- 34.Cras P, Smith MA, Richey PL, Siedlak SL, Mulvihill P, Perry G. Extracellular neurofibrillary tangles reflect neuronal loss and provide further evidence of extensive protein cross-linking in Alzheimer’s disease. Acta Neuropathol. 1995;89:291–295. doi: 10.1007/BF00309621. [DOI] [PubMed] [Google Scholar]
- 35.Coleman PD. Research uses of neuropathological data. Neurobiol Aging. 1997;18(Suppl 4):97–98. doi: 10.1016/S0197-4580(97)00059-6. [DOI] [PubMed] [Google Scholar]
- 36.Cullen KM, Halliday GM, Cartwright H, Kril JJ. Improved selectivity and sensitivity in the visualization of neurofibrillary tangles, plaques and neuropil threads. Neurodegeneration. 1996;5:177–187. doi: 10.1006/neur.1996.0025. [DOI] [PubMed] [Google Scholar]
- 37.Delacourte A, David JP, Dergeant N, Buée L, Wattez A, Vermersch P, Ghozali F, Fallet-Bianco C, Pasquier F, Lebert F, Petit H, Di Menza C. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology. 1999;52:1158–1165. doi: 10.1212/wnl.52.6.1158. [DOI] [PubMed] [Google Scholar]
- 38.Dickson DW. The value of cross sectional neuroanatomical studies as a conceptual framework for prospective clinicopathological studies. Neurobiol Aging. 1997;18:382–386. doi: 10.1016/S0197-4580(97)00053-5. [DOI] [PubMed] [Google Scholar]
- 39.Duyckaerts C, Hauw JJ. Diagnosis and staging of Alzheimer’s disease. Neurobiol Aging. 1997a;18(Suppl 4):33–42. doi: 10.1016/S0197-4580(97)00067-5. [DOI] [PubMed] [Google Scholar]
- 40.Duyckaerts C, Hauw JJ. Prevalence, incidence and duration of Braak’s stages in the general population: can we know? Neurobiol Aging. 1997b;18(Suppl 4):362–369. doi: 10.1016/S0197-4580(97)00047-X. [DOI] [PubMed] [Google Scholar]
- 41.Duyckaerts C, He Y, Seilhean D, Delaère P, Piette F, Braak H, Hauw JJ. Diagnosis and staging of Alzheimer’s disease in a prospective study involving aged individuals. Neurobiol Aging. 1994;15(Suppl 1):140–141. [Google Scholar]
- 42.Duyckaerts C, Delaère P, He Y, Camilleri S, Braak H, Piette F, Hauw JJ. The relative merits of tau- and amyloid markers in the neuropathology of Alzheimer’s disease. In: Bergener M, Finkel SI, editors. Treating Alzheimer’s and other dementias. Heidelberg Berlin New York: Springer; 1995. pp. 81–89. [Google Scholar]
- 43.Duyckaerts C, Bennecib M, Grignon Y, Piette F, Hauw JJ. Is the topography of Alzheimer’s disease lesions a clue to their pathogenesis? Bull Acad Natl Med. 1996;180:1703–1714. [PubMed] [Google Scholar]
- 44.Duyckaerts C, Bennecib M, Grignon Y, Uchihara T, He Y, Piette F, Hauw JJ. Modeling the relation between neurofibrillary tangles and intellectual status. Neurobiol Aging. 1997;18(Suppl. 4):267–273. doi: 10.1016/S0197-4580(97)80306-5. [DOI] [PubMed] [Google Scholar]
- 45.Esiri MM, Hyman BT, Beyreuther K, Masters C. Aging and dementia. In: Graham DL, Lantos PI, editors. Greenfield’s neuropathology. London: Arnold; 1997. pp. 153–234. [Google Scholar]
- 46.Feany MB, Dickson DW. Neurodegenerative disorders with extensive tau pathology: a comparative study and review. Ann Neurol. 1996;40:139–148. doi: 10.1002/ana.410400204. [DOI] [PubMed] [Google Scholar]
- 47.Fewster PH, Griffin-Brooks S, MacGregor J, Ojalvo-Rose E, Ball MJ. A topographical pathway by which histopathological lesions disseminate through the brain of patients with Alzheimer’s disease. Dementia. 1991;2:121–132. [Google Scholar]
- 48.Flowers D, Harasty J, Halliday G, Kril J. Microwave modification of the methenamine silver technique for the demonstration of Alzheimer-type pathology. J Histotechnol. 1996;19:33–38. [Google Scholar]
- 49.Gallyas F. Silver staining of Alzheimer’s neurofibrillary changes by means of physical development. Acta Morph Acad Sci Hung. 1971;19:1–8. [PubMed] [Google Scholar]
- 50.Gallyas F. Light insensitive physical developers. Stain Technol. 1979;54:173–176. doi: 10.3109/10520297909112679. [DOI] [PubMed] [Google Scholar]
- 51.Gallyas F, Wolff JR. Metal-catalyzed oxidation renders silver intensification selective. Application for the histochemistry of diaminobenzidine and neurofibrillary changes. J Histochem Cytochem. 1986;34:1667–1672. doi: 10.1177/34.12.3537114. [DOI] [PubMed] [Google Scholar]
- 52.Garvey W, Fathi A, Bigelow F, Jimenez CL, Carpenter BF. Rapid, reliable and economical silver stain for neurofibrillary tangles and senile plaques. J Histotechnol. 1991;14:39–42. [Google Scholar]
- 53.Geddes JW, Tekirian TL, Soultanian NS, Ashford JW, Davis DG, Markesbery WR. Comparison of neuropathologic criteria for the diagnosis of Alzheimer’s disease. Neurobiol Aging. 1997;18(Suppl 4):99–105. doi: 10.1016/S0197-4580(97)00063-8. [DOI] [PubMed] [Google Scholar]
- 54.Gertz HJ, Xuereb JH, Huppert FA, Brayne C, McGee MA, Paykel ES, Harrington C, Mukaetova-Ladinska E, Arendt T, Wischik CM. Examination of the validity of the hierarchical model of neuropathological staging in normal aging and Alzheimer’s disease. Acta Neuropathol. 1998;95:154–158. doi: 10.1007/s004010050780. [DOI] [PubMed] [Google Scholar]
- 55.Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci. 1993;16:460–465. doi: 10.1016/0166-2236(93)90078-Z. [DOI] [PubMed] [Google Scholar]
- 56.Goedert M. Filamentous nerve cell inclusions in neurodegenerative diseases: tauopathies and α-synucleinopathies. Phil Trans R Soc Lond B Biol Sci. 1999;354:1101–1108. doi: 10.1098/rstb.1999.0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167–169. doi: 10.1016/0304-3940(95)11484-E. [DOI] [PubMed] [Google Scholar]
- 58.Goedert M, Trojanowski JQ, Lee VMY. The neurofibrillary pathology of Alzheimer’s disease. In: Rosenberg RN, editor. The molecular and genetic basis of neurological disease. 2nd. Boston: Butterworth-Heinemann; 1997. pp. 613–627. [Google Scholar]
- 59.Gold G, Bouras C, Kövari E, Canuto A, González Glaría B, Malky A, Hof PR, Michel JP, Giannakopoulos P. Clinical validity of Braak neuropathological staging in the oldest-old. Acta Neuropathol. 2000;99:579–582. doi: 10.1007/s004010051163. [DOI] [PubMed] [Google Scholar]
- 60.Grober E, Dickson D, Sliwinski MJ, Buschke H, Katz M, Crystal H, Lipton RB. Memory and mental status correlates of modified Braak staging. Neurobiol Aging. 1999;20:573–579. doi: 10.1016/S0197-4580(99)00063-9. [DOI] [PubMed] [Google Scholar]
- 61.Halliday G, Ng T, Rodriguez M, Harding A, Blumbergs P, Evans W, Fabian V, Fryer V, Gonzales M, Harper C, Kalnins R, Masters CL, McLean C, Milder DG, Pamphlett R, Scott G, Tannenberg A., Kril J. Consensus neuropathological diagnosis of common dementia syndromes: testing and standardizing the use of multiple diagnostic criteria. Acta Neuropathol. 2002;104:72–78. doi: 10.1007/s00401-002-0529-5. [DOI] [PubMed] [Google Scholar]
- 62.Harding AJ, Kril JJ, Halliday GM. Practical measures to simplify the Braak tangle staging method for routine pathological screening. Acta Neuropathol. 2000;99:199–208. doi: 10.1007/PL00007425. [DOI] [PubMed] [Google Scholar]
- 63.Hyman BT. The neuropathological diagnosis of Alzheimer’s disease: clinical-pathological studies. Neurobiol Aging. 1997;18(Suppl. 4):27–32. doi: 10.1016/S0197-4580(97)00066-3. [DOI] [PubMed] [Google Scholar]
- 64.Hyman BT. New neuropathological criteria for Alzheimer’s disease. Arch Neurol. 1998;55:1174–1176. doi: 10.1001/archneur.55.9.1174. [DOI] [PubMed] [Google Scholar]
- 65.Hyman BT, Gomez-Isla T. Alzheimer’s disease is a laminar, regional, and neural system specific diasease, not a global brain disease. Neurobiol Aging. 1994;15:353–354. doi: 10.1016/0197-4580(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 66.Hyman BT, Trojanowski JQ. Editorial on consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute working group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:1095–1097. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 67.Iqbal K, Braak E, Braak H, Zaidi T, Grundke-Iqbal I. A silver impregnation method for labeling both Alzheimer paired helical filaments and their polypeptides separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Neurobiol Aging. 1991;12:357–361. doi: 10.1016/0197-4580(91)90022-C. [DOI] [PubMed] [Google Scholar]
- 68.Iqbal K, Braak H, Braak E, Grundke-Iqbal I. Silver labeling of Alzheimer neurofibrillary changes and brain β amyloid. J Histotechnol. 1993;16:335–342. [Google Scholar]
- 69.Jellinger KA. The neuropathological diagnosis of Alzheimer’s disease. J Neural Transm. 1998;53(Suppl):97–118. doi: 10.1007/978-3-7091-6467-9_9. [DOI] [PubMed] [Google Scholar]
- 70.Jellinger K. Clinical validity of Braak staging in the oldest-old. Acta Neuropathol. 2000;99:583–584. doi: 10.1007/s004010051164. [DOI] [PubMed] [Google Scholar]
- 71.Jellinger KA, Bancher C. Proposals for re-evaluation of current autopsy criteria for the diagnosis of Alzheimer’s disease. Neurobiol Aging. 1997;18(Suppl 4):55–65. doi: 10.1016/S0197-4580(97)00071-7. [DOI] [PubMed] [Google Scholar]
- 72.Kauppinen T, Martikainen P, Alafuzoff I (2006) Human postmortem brain tissue and 2 mm tissue microarrays. Appl Immunohistochem Mol Morphol (in press) [DOI] [PubMed]
- 73.Kreutzberg GW, Blakemore WF, Graeber MB. Cellular pathology of the central nervous system. In: Graham DI, Lantos PL, editors. Greenfield’s neuropathology. London: Arnold; 1997. pp. 85–156. [Google Scholar]
- 74.Lamy C, Duyckaerts C, Delaère P, Payan C, Fermanian J, Poulain V, Hauw JJ. Comparison of seven staining methods for senile plaques and neurofibrillary tangles in a prospective series of 15 elderly patients. Neuropathol Appl Neurobiol. 1989;15:563–578. doi: 10.1111/j.1365-2990.1989.tb01255.x. [DOI] [PubMed] [Google Scholar]
- 75.Markesbery WR. Neuropathological criteria for the diagnosis of Alzheimer’s disease. Neurobiol Aging. 1997;18(Suppl 4):13–19. doi: 10.1016/S0197-4580(97)00064-X. [DOI] [PubMed] [Google Scholar]
- 76.McKeel DW, Price JL, Miller JP, Grant EA, Xiong C, Berg L, Morris JC. Neuropathologic criteria for diagnosing Alzheimer disease in persons with pure dementia of the Alzheimer type. J Neuropathol Exp Neurol. 2004;63:1028–1037. doi: 10.1093/jnen/63.10.1028. [DOI] [PubMed] [Google Scholar]
- 77.Mercken M, Vandermeeren M, Lübke U, Six J, Boons J, Van de Voorde A, Martin J-J, Gheuens J. Monoclonal antibodies with selective specifity for Alzheimer tau are directed against phosphatase-sensitive epitopes. Acta Neuropathol. 1992;84:265–272. doi: 10.1007/BF00227819. [DOI] [PubMed] [Google Scholar]
- 78.Munoz DG. Stains for the differential diagnosis of degenerative diseases. Biotechn Histochem. 1999;74:311–320. doi: 10.3109/10520299909034668. [DOI] [PubMed] [Google Scholar]
- 79.Nagy Zs, Vatter-Bittner B, Braak H, Braak E, Yilmazer D, Schultz C, Hanke J. Staging of Alzheimer-type pathology: an interrater–intrarater study. Dementia. 1997;8:248–251. doi: 10.1159/000106639. [DOI] [PubMed] [Google Scholar]
- 80.Nagy S, Yilmazer-Hanke DM, Braak H, Braak E, Schultz C, Hanke J. Assessment of the pathological stages of Alzheimer’s disease in thin paraffin sections: a comparative study. Dement Geriatr Cogn Disord. 1998;9:140–144. doi: 10.1159/000017038. [DOI] [PubMed] [Google Scholar]
- 81.Nagy S, Hindley NJ, Braak H, Braak E, Yilmazer-Hanke DM, Schultz C, Barnetson L, Jobst KA, Smith AD. Relationship between clinical and radiological diagnostic criteria for Alzheimer’s disease and the extent of neuropathology as reflected by “stages”: a prospective study. Dement Geriatr Cogn Disord. 1999a;10:109–114. doi: 10.1159/000017110. [DOI] [PubMed] [Google Scholar]
- 82.Nagy S, Hindley NJ, Braak H, Braak E, Yilmazer-Hanke DM, Schultz C, Barnetson L, King EMF, Jobst KA, Smith AD. The progression of Alzheimer’s disease from limbic regions to the neocortex: clinical, radiological and pathological relationships. Dement Geriatr Cogn Disord. 1999b;10:115–120. doi: 10.1159/000017111. [DOI] [PubMed] [Google Scholar]
- 83.Newell KL, Hyman BT, Growdon JH, Hedley-Whyte ET. Application of the National Institute on Aging (NIA)-Reagan Institute criteria for the neuropathological diagnosis of Alzheimer’s disease. J Neuropathol Exp Neurol. 1999;58:1147–1155. doi: 10.1097/00005072-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 84.Ohm TG, Müller H, Braak H, Bohl J. Close-meshed prevalence rates of different stages as a tool to uncover the rate of Alzheimer’s disease-related neurofibrillary changes. Neuroscience. 1995;64:209–217. doi: 10.1016/0306-4522(95)90397-P. [DOI] [PubMed] [Google Scholar]
- 85.Ohm TG, Glöckner F, Distl R, Treiber-Held S, Meske V, Schönheit B. Plasticity and the spread of Alzheimer’s disease-like changes. Neurochem Res. 2003;28:1715–1723. doi: 10.1023/A:1026017206925. [DOI] [PubMed] [Google Scholar]
- 86.Paulus W, Bancher C, Jellinger K. Interrater reliability in the neuropathologic diagnosis of Alzheimer’s disease. Neurology. 1992;42:329–332. doi: 10.1212/wnl.42.2.329. [DOI] [PubMed] [Google Scholar]
- 87.Petersen RC, Parisi JE, Dickson DW, Johnson KA, Knopman DS, Boeve BF, Jicha GA, Ivnik RJ, Smith GE, Tangalos EG, Braak H, Kokmen E. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63:665–672. doi: 10.1001/archneur.63.5.665. [DOI] [PubMed] [Google Scholar]
- 88.Price JL. Diagnostic criteria for Alzheimer’s disease. Neurobiol Aging. 1997;18(Suppl 4):67–70. doi: 10.1016/S0197-4580(97)00072-9. [DOI] [PubMed] [Google Scholar]
- 89.Schönheit B, Zarski R, Ohm TG. Spatial and temporal relationships between plaques and tangles in Alzheimer-pathology. Neurobiol Aging. 2004;25:697–711. doi: 10.1016/j.neurobiolaging.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 90.Schmidt ML, Lee VMY, Trojanowski JQ. Comparative epitope analysis of neuronal cytoskeletal proteins in Alzheimer’s disease senile plaque neurites and neuropil threads. Lab Invest. 1991;64:352–357. [PubMed] [Google Scholar]
- 91.Smithson KG, MacVicar BA, Hatton GI. Polyethylene glycol embedding: a technique compatible with immunocytochemistry, enzyme histochemistry, histofluorescence and intracellular staining. J Neurosci Methods. 1983;7:27–41. doi: 10.1016/0165-0270(83)90016-X. [DOI] [PubMed] [Google Scholar]
- 92.Thal DR, Holzer M, Rüb U, Waldmann G, Günzel S, Zedlick D, Schober R. Alzheimer-related τ-pathology in the perforant path target zone and in the hippocampal stratum oriens and radiatum correlates with onset and degree of dementia. Exp Neurol. 2000;163:98–110. doi: 10.1006/exnr.2000.7380. [DOI] [PubMed] [Google Scholar]
- 93.Thal DR, Del Tredici K, Braak H (2004) Neurodegeneration in normal brain aging and disease. SAGE 23, p 26 [DOI] [PubMed]
- 94.Tolnay M, Probst A. Review: tau protein pathology in Alzheimer’s disease and related disorders. Neuropathol Appl Neurobiol. 1999;25:171–187. doi: 10.1046/j.1365-2990.1999.00182.x. [DOI] [PubMed] [Google Scholar]
- 95.Trojanowski JQ, Shin RW, Schmidt ML, Lee VMY. Relationship between plaques, tangles, and dystrophic processes in Alzheimer’s disease. Neurobiol Aging. 1995;16:335–345. doi: 10.1016/0197-4580(94)00176-2. [DOI] [PubMed] [Google Scholar]
- 96.Uchihara T, Nakamura A, Yamazaki M, Moris O. Evolution from pretangle neurons to neurofibrillary tangles monitored by thiazin red combined with Gallyas method and double immunofluorescence. Acta Neuropathol. 2001;101:535–539. doi: 10.1007/s004010000306. [DOI] [PubMed] [Google Scholar]
- 97.van Hoesen GW, Hyman BT. Hippocampal formation: anatomy and the patterns of pathology in Alzheimer’s disease. Prog Brain Res. 1990;83:445–457. doi: 10.1016/s0079-6123(08)61268-6. [DOI] [PubMed] [Google Scholar]
- 98.van Hoesen GW, Hyman BT, Damasio AR. Entorhinal cortex pathology in Alzheimer’s disease. Hippocampus. 1991;1:1–8. doi: 10.1002/hipo.450010102. [DOI] [PubMed] [Google Scholar]
- 99.Yamamoto T, Hirano A. A comparative study of modified Bielschowsky, Bodian, and thioflavin S stains on Alzheimer’s neurofibrillary tangles. Neuropathol Appl Neurobiol. 1986;12:3–9. doi: 10.1111/j.1365-2990.1986.tb00677.x. [DOI] [PubMed] [Google Scholar]