

Abstract
MraY (phospho-MurNAc-pentapeptide translocase) is an integral membrane enzyme that catalyzes an essential step of bacterial cell wall biosynthesis: the transfer of the peptidoglycan precursor phospho-MurNAc-pentapeptide to the lipid carrier undecaprenyl phosphate. MraY has long been considered a promising target for the development of antibiotics, but the lack of a structure has hindered mechanistic understanding of this critical enzyme and the enzyme superfamily in general. The superfamily includes enzymes involved in bacterial lipopolysaccharide/teichoic acid formation and eukaryotic N-linked glycosylation, modifications that are central in many biological processes. We present the crystal structure of MraY from Aquifex aeolicus (MraYAA) at 3.3 Å resolution, which allows us to visualize the overall architecture, locate Mg2+ within the active site, and provide a structural basis of catalysis for this class of enzyme.
Bacteria maintain their cell shapes at different osmotic pressures by using the mesh-like layers of the cell wall to surround and stabilize the membrane. The cell wall of both Gram-negative and Gram-positive bacteria is composed of peptidoglycan, a cross-linked polymer of carbohydrates and amino acids. Because biosynthesis of peptidoglycan is a critical process for bacteria, it has been a major target for antibiotics (1, 2). Peptidoglycan biosynthesis involves three main stages. First, the peptidoglycan precursor UDP-N-acetylmuramoyl (MurNAc)–pentapeptide (l-Ala-γ-d-Glu-diaminopimelic acid/l-Lys-d-Ala-d-Ala) is synthesized in the cytosol. Second, this hydrophilic precursor is attached to a carrier lipid, and the lipid-linked precursor is flipped across the membrane to the periplasm.Third, peptidoglycan precursors are polymerized to form the cell wall.
MraY, or phospho-MurNAc-pentapeptide translocase, is an integral membrane protein responsible for the second stage of peptidoglycan biosynthesis. MraY catalyzes the transfer of phospho-MurNAc-pentapeptide from UDP-MurNAc-pentapeptide to the lipid carrier undecaprenyl phosphate (C55-P), yielding undecaprenyl-pyrophosphoryl-MurNAc-pentapeptide, also known as Lipid I (Fig. 1A). This MraY-mediated membrane step is Mg2+-dependent and essential for bacterial viability (3, 4). Pharmacologically, MraY has long been known as a promising target for the development of new antibiotics because it is the target of five different classes of natural product inhibitors with antibacterial activity as well as bacteriolytic protein E from bacteriophage phiX174 (1, 5–11).
Fig. 1. MraYAA is functional.
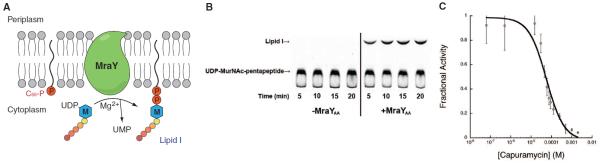
(A) Illustration of MraY-mediated translocation. The M-labeled hexagon represents MurNAc, and the five attached circles represent the pentapeptide (l-Ala-γ-d-Glu-diaminopimelic acid-d-Ala-d-Ala). MraY is shown in green, and phosphates are depicted as red circles. (B) Thin-layer chromatography-based translocase assay of detergent-solubilized MraYAA. The substrate UDP-MurNAc-pentapeptide and the product Lipid I are indicated with arrows. The identity of Lipid I was deduced from the similar retardation factor (Rf) values as published (13). The concentration of MraYAA was 100 μg/ml. (C) Capuramycin inhibition curve of detergent-solubilized MraYAA with IC50 = 56.4 ± 14.3 μM (Data are shown as means ± SD, n = 3 experiments). Capuramycin was preincubated with MraYAA for 20 min at 30°C before the reaction was initiated.
MraY belongs to a subfamily of the polyprenyl-phosphate N-acetyl hexosamine 1-phosphate transferase (PNPT) superfamily. The PNPT superfamily includes the MraY, WecA, TagO, WbcO, WbpL, and RgpG enzyme families, which are responsible for the synthesis of cell envelope polymers such as the O-antigen and teichoic acid in bacteria, and the GPT (UDP-GlcNAc:dolichol-P GlcNAc-1-P transferase) enzyme family, which is responsible for the committed step of N-linked glycosylation in eukaryotes (12). The structural basis of enzyme function has been elusive because no structure from this enzyme superfamily is available.
For structural and functional studies, recombinant MraY from Aquifex aeolicus (MraYAA) was expressed, purified, and crystallized (supplementary materials, materials and methods). Single anomalous dispersion phasing was performed by using a selenomethionine-substituted MraYAA crystal (fig. S1). The final model was refined with good geometry (table S1 and S2).
A thin-layer chromatography (TLC)–based translocase assay (13) shows that recombinant MraYAA can catalyze the transfer of phospho-MurNAc-pentapeptide to the carrier lipid C55-P (Fig. 1B). Endogenous Escherichia coli MraY does not contribute appreciably to the observed activity (fig. S2). We also used the translocase assay to show that MraYAA could be inhibited by the MraY-specific natural product inhibitor capuramycin with a median inhibitory concentration (IC50) value of about 56 μM (Fig. 1C) (7, 9).
MraYAA crystallizes as a dimer in the asymmetric unit. The twofold axis of the dimer is perpendicular to the putative membrane plane and parallel to one of the crystallographic twofold axes (Fig. 2A). At the center of the dimer interface is an oval-shaped tunnel. The tunnel is surrounded mostly by hydrophobic amino acids and is large enough to accommodate lipids (Fig. 2B). An unbiased electron density map shows that two elongated electron density peaks, too long to be detergent molecules, are intertwined within the dimer interface (fig. S3). The overall dimensions of the dimer are about 72 Å across the long axis and 55 Å across the short axis when viewed from the cytoplasmic side (Fig. 2B). Each protomer contains 10 transmembrane helices (TM1 to TM10), an interfacial helix (IH), a periplasmic β hairpin (PB), a periplasmic helix (PH), and five cytoplasmic loops (loop A to loop E) (Fig. 2C). Both the N- and C-termini of the structure are located on the periplasmic side, which is consistent with previous topological studies (14). TM9 breaks into two helical fragments (TM9a and TM9b), with TM9b displaying a substantial bend (~50° relative to the membrane normal) in the middle of the membrane. This bending causes TM9b to protrude ~20 Å into the lipid membrane away from the rest of structure (Fig. 2, A and B). This protrusion is buttressed by interactions with TM5. TM9b—together with TM3, TM4, and TM8—surrounds TM5 and thus generates a cleft around the inner leaflet membrane region of TM8 (Fig. 2B).
Fig. 2. Architecture and topology of MraY.
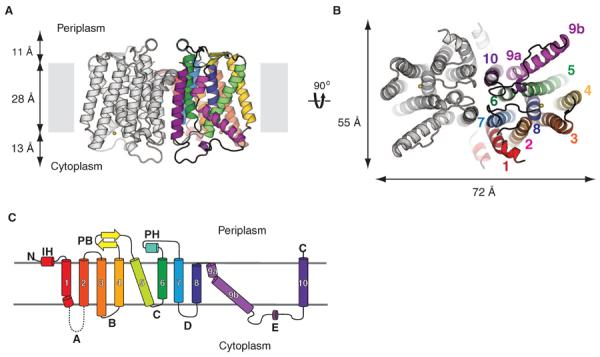
(A) View from within the membrane. Only transmembrane helices (TMs) from one protomer are colored. Loop E from only one protomer was present in the model, but loop E from both protomers are shown. The yellow sphere is Mg2+. (B) Cytoplasmic view. The yellow sphere is Mg2+. (C) Topology diagram of MraY protomer. Each TM is colored differently. TMs are given numbers, and cytoplasmic loops are given letters. Loop A is missing in the structure. The same colors are used for TMs in (A) through (C).
To test the oligomeric status of MraYAA, we performed cross-linking studies in both detergent micelles and lipid membranes. For detergent-solubilized MraYAA, we performed the experiment using the nonspecific cross-linker disuccinimidyl suberate (DSS), and for membrane-embedded MraYAA, we performed structure-guided disulfide cross-bridge experiments (fig. S4). The results show that MraYAA forms a dimer both in detergent micelles and in the membrane. Consistent with this observation, a recent bacterial two-hybrid study indicated that MraY enzymes in Caulobacter crescentus interact with each other (15).
To gain functional insight, we mapped sequence conservation onto the MraYAA structure (Fig. 3A and fig. S5). The highest conservation is localized around the cleft formed by the cytoplasmic and inner-leaflet membrane regions of TM3, TM4, TM8, and TM9b (Fig. 3A). Recent mutational studies of Bacillus subtilis MraY showed that 14 invariant polar/charged amino acid residues are essential as evidenced by both in vivo functional complementation assay and in vitro enzymatic assay (16). Most of these residues are localized in the cleft, suggesting that this region serves as the active site (Fig. 3B). On the basis of our mutational mapping, we predicted that three invariant aspartate residues (Asp117, Asp118, and Asp265) conserved throughout the entire PNPT family and two invariant histidine residues (His324 and His325) in the MraY family are catalytically important because they do not appear to be involved in maintaining the putative active site structure (Fig. 3B). We individually mutated each of these three invariant aspartate as well as three histidine residues (His324, His325, and His326) and found that mutation of any of the three aspartate residues and one histidine residue (His324) resulted in nearly complete loss of activity of MraYAA, whereas mutating a nonconserved residue (Ile83) away from the active site (on TM2) did not reduce catalytic activity (Fig. 3C). We observed that there is no appreciable difference in stability upon mutation. Thus, at least three acidic residues and one histidine residue (Asp117, Asp118, Asp265, and His324) in the active site are important for catalysis.
Fig. 3. Theactivesite of MraYAA.

(A) Conservation mapping on the MraYAA structure. Sequence conservation is colored on one protomer with a gradient from magenta (absolutely conserved) to cyan (least conserved) based on 28 MraY sequences (fig. S5). The structure is rotated ~45° toward the reader relative to Fig. 2A. The arrow indicates the active site cleft. (B) Mutation mapping on the MraYAA structure based on the studies of Al-Dabbagh et al. (16). Mutations leading to pronounced functional effects are shown in stick representation. Among these mutations, amino acid residues that appear to be important for catalysis and active site structural maintenance are colored orange and cyan, respectively. (C) Mutational studies of putative active-site residues of MraYAA using the same translocase assay as Fig. 1B. The specific activities of mutants were normalized to that of wild type (Data are shown as means ± SD, n = 3 experiments). (D) Active site Mn2+ (Mg2+)–binding site. Anomalous difference Fourier density for Mn2+, shown in green mesh contoured at 5.2σ, is cal culated from a data set collected from a crystal grown in the presence of Mn2+ without Ni2+ by using phases derived from the model without the metals. Another anomalous difference Fourier density peak for Mn2+, shown in red mesh contoured at 3.4σ, was calculated from a data set collected from a crystal soaked with Mn2+ in the presence of Ni2+ by using phases derived from the model without the metals.
It had been assumed that the residues corresponding to Asp117 and Asp118 in MraYAA are involved in Mg2+ coordination based on sequence similarity to the Mg2+-binding motif (DDXXD/N) of prenyl transferases (17), but this assumption was challenged recently (16). To identify Mg2+ within the active site of the structure, we performed anomalous scattering studies using Mn2+-substituted MraYAA crystals (experimental details are available in the supplementary materials, materials and methods). Anomalous difference Fourier density maps calculated from both Mn2+-soaked and Mn2+–co-crystallized crystals displayed peaks at the active site near Asp265 (Fig. 3D). The Mn2+ directly interacts only with Asp265 but not with Asp117 or Asp118. An FO-FC OMIT electron density map from the native data at 3.3 Å shows that there are unassigned density peaks within interacting distance of the Mg2+, which are probably involved in Mg2+ coordination (fig. S6).
From sequence conservation and topological studies, it was predicted that MraY contains five conserved cytoplasmic loops (fig. S5) (3). Substantial portions of the loops correspond to the inner-leaflet membrane region of TM helices in the structure. The fifth predicted loop is composed of TM9b and the loop connecting TM9b to TM10 (loop E) (Fig. 4A and fig. S5). This region shows subfamily-specific sequence conservation within the PNPT superfamily (18, 19), and it was predicted that it has a role in the specific recognition of sugars. This is consistent with a study of WecA, which showed that this region may interact with sugar nucleotides (18).
Fig. 4. Putative substrate-binding sites.
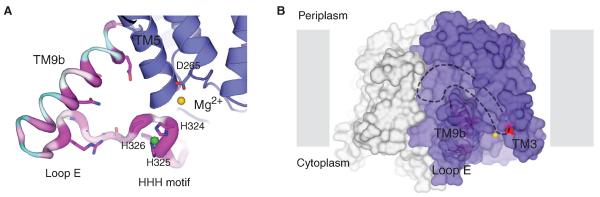
(A) Zoomed-in view of TM9b, loop E (the HHH motif). The structure is rotated 90° toward the reader relative to Fig. 3A. TM9b/loop E is shown in sausage representation, with the thicker region more conserved (magenta) and the thinner region less conserved (cyan). Side chain of highly conserved amino acids on TM9b and loop E as well as the catalytic residue D265 are shown in stick representation. Mg2+ and Ni2+ are shown as yellow and green spheres, respectively. (B) A hydrophobic groove connected to the active site is delineated with dashed lines on the MraYAA surface illustration. Mg2+ is shown as a yellow sphere, and D117 is colored red. Protomers are colored blue and gray.
Within the two protomers of the asymmetric unit, only one (chain A) shows well-resolved electron density for loop E (fig. S7). Loop E is ~23 amino acids long and is composed of several basic residues followed by a stretch of sequence (PXHHHXEXXG) that is highly conserved within the MraY family (hereafter termed “HHH motif”). The HHH motif includes a short stretch of helical segment followed by a loop (Fig. 4A). Two Ni2+ ions, verified through anomalous scattering studies, are coordinated by the short helical segment, one by the first two histidines (His324 and His325) and the second by the third histidine (His326) and Glu328 (fig. S8). The translocase assay with the concentrations of Mg2+ (100 mM) and Ni2+ (10 mM) used for crystallization resulted in full activity, suggesting that the structure of Ni2+-bound loop E reflects a physiologically relevant conformation (fig. S9).
TM9b is an amphipathic helix substantially tilted and protruding toward the lipid membrane and, together with loop E (HHH motif), abuts the active site, thus making the active site cleft deeper (Figs. 3 and 4A). Highly conserved polar and charged amino acids on TM9b and loop E (the HHH motif) are localized and pointed toward a region near the active site Mg2+, which is consistent with the previous notion that this region is the binding site for the hydrophilic substrate UDP-MurNAc-pentapeptide. An FO-FC OMIT map shows a τ-shaped density peak threaded and trapped in the gap between TM9b and loop E (fig. S10).
Bouhss and colleagues proposed that Asp117 (Asp98 in B. subtilis MraY) interacts with and deprotonates the phosphate moiety of C55-P. Asp117 is one of the three catalytically important acidic residues in the active site (16). The structure of MraYAA is in line with the prediction that Asp117 is the binding site for the phosphate group of C55-P because it is surrounded by the conserved residues Lys121, Lys133, and Asp265 as well as the Mg2+ ion (fig. S11). The surface representation of MraYAA reveals that there is an inverted-U–shaped groove surrounding TM9b that extends into the active site where Asp117 is located (Fig. 4B). Because C55-P is longer than the depth of the membrane, and is elastic owing to the polyisoprenyl group, the inverted-U–shaped groove within the membrane region of the enzyme could perhaps serve as the binding site for the hydrophobic substrate C55-P (Fig. 4B).
Supplementary Material
Acknowledgments
Data for this study were collected at beamlines NE-CAT ID 24-C and SER-CAT BM22/ID22 at the Advanced Photon Source. We thank C. Pemble and N. Nicely from the Duke x-ray crystallography facility for help with remote data collection; F. Murphy for help with data collection at ID 24-C; and Z. Johnson for critical reading of the manuscript. This work was supported by start-up funds from the Duke University Medical Center (S.-Y.L.), Duke University (J.H.), and the National Institutes of Health (NIH) (grants GM-51310 and AI-55588 to P.Z. and Lipid Maps glue grant GM-069338 to Z.G.). S.-Y.L. is a McKnight Scholar, Klingenstein fellow, Alfred P. Sloan Research fellow, Mallinckrodt Scholar, Whitehead Scholar, Basil O'Connor Starter Scholar, and NIH Director's New Innovator awardee. The authors declare no conflict of interest. The coordinates and structure factors are deposited in the Protein Data Bank (4J72). B.C.C. expressed, purified, and crystallized MraYAA. B.C.C. and S.-Y. L. collected the crystallographic data, solved the structure, and refined the model. B.C.C. performed the cross-linking studies and prepared all the protein for functional assays. J. Z. and R.A.G. performed the translocase assays (J.Z. performed all the mutational studies, and R.A.G. performed capuramycin and divalent metal experiments) under the guidance of P.Z. D.-Y.K. synthesized capuramycin under the guidance of J.H. Z.G. performed mass spectrometry characterization. B.C.C. and S.-Y.L. wrote the paper.
Footnotes
Supplementary Materials www.sciencemag.org/cgi/content/full/341/6149/1012/DC1
References and Notes
- 1.Winn M, Goss RJ, Kimura K, Bugg TD. Nat. Prod. Rep. 2010;27:279–304. doi: 10.1039/b816215h. [DOI] [PubMed] [Google Scholar]
- 2.Bugg TD, Braddick D, Dowson CG, Roper DI. Trends Biotechnol. 2011;29:167–173. doi: 10.1016/j.tibtech.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Bouhss A, Trunkfield AE, Bugg TD, Mengin-Lecreulx D. FEMS Microbiol. Rev. 2008;32:208–233. doi: 10.1111/j.1574-6976.2007.00089.x. [DOI] [PubMed] [Google Scholar]
- 4.Boyle DS, Donachie WD. J. Bacteriol. 1998;180:6429–6432. doi: 10.1128/jb.180.23.6429-6432.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mendel S, Holbourn JM, Schouten JA, Bugg TD. Microbiology. 2006;152:2959–2967. doi: 10.1099/mic.0.28776-0. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka S, Clemons WM., Jr. Mol. Microbiol. 2012;85:975–985. doi: 10.1111/j.1365-2958.2012.08153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koga T, et al. J. Antimicrob. Chemother. 2004;54:755–760. doi: 10.1093/jac/dkh417. [DOI] [PubMed] [Google Scholar]
- 8.Auberger N, et al. Org. Biomol. Chem. 2011;9:8301–8312. doi: 10.1039/c1ob06124k. [DOI] [PubMed] [Google Scholar]
- 9.Bugg TD, Lloyd AJ, Roper DI. Infect. Disord. Drug Targets. 2006;6:85–106. doi: 10.2174/187152606784112128. [DOI] [PubMed] [Google Scholar]
- 10.Dini C, Mra Y. Curr. Top. Med. Chem. 2005;5:1221–1236. doi: 10.2174/156802605774463042. [DOI] [PubMed] [Google Scholar]
- 11.Shapiro AB, Jahić H, Gao N, Hajec L, Rivin O. J. Biomol. Screen. 2012;17:662–672. doi: 10.1177/1087057112436885. [DOI] [PubMed] [Google Scholar]
- 12.Lehrman MA. Glycobiology. 1994;4:768–771. doi: 10.1093/glycob/4.6.768. [DOI] [PubMed] [Google Scholar]
- 13.Bouhss A, Crouvoisier M, Blanot D, Mengin-Lecreulx D. J. Biol. Chem. 2004;279:29974–29980. doi: 10.1074/jbc.M314165200. [DOI] [PubMed] [Google Scholar]
- 14.Bouhss A, Mengin-Lecreulx D, Le Beller D, Van Heijenoort J. Mol. Microbiol. 1999;34:576–585. doi: 10.1046/j.1365-2958.1999.01623.x. [DOI] [PubMed] [Google Scholar]
- 15.White CL, Kitich A, Gober JW. Mol. Microbiol. 2010;76:616–633. doi: 10.1111/j.1365-2958.2010.07108.x. [DOI] [PubMed] [Google Scholar]
- 16.Al-Dabbagh B, et al. Biochemistry. 2008;47:8919–8928. doi: 10.1021/bi8006274. [DOI] [PubMed] [Google Scholar]
- 17.Lloyd AJ, Brandish PE, Gilbey AM, Bugg TD. J. Bacteriol. 2004;186:1747–1757. doi: 10.1128/JB.186.6.1747-1757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amer AO, Valvano MA. Microbiology. 2001;147:3015–3025. doi: 10.1099/00221287-147-11-3015. [DOI] [PubMed] [Google Scholar]
- 19.Anderson MS, Eveland SS, Price NP. FEMS Microbiol. Lett. 2000;191:169–175. doi: 10.1111/j.1574-6968.2000.tb09335.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.