

Abstract
Generation of high-affinity antibodies in response to antigens/infectious agents is essential for developing long-lasting immune responses. B cell maturation and antibody responses to antigen stimulation require immunoglobulin (Ig) somatic hypermutation (SHM) and class-switch recombination (CSR) for high-affinity responses. Upon immunization with either the model antigen NP-CGG or heat-killed Pn14 derived from Streptococcus pneumoniae, knock-in (KI) mice hypomorphic for mTOR function had decreased ability to form germinal centers, develop high-affinity anti-NP or –Pn14 specific antibodies, and perform SHM/CSR. Hypomorphic mTOR mice also had a high mortality rate (40%) compared to WT (0%) littermates and had lower PspA specific antibody titers when immunized and challenged with live S. pneumoniae infection. Mice with mTOR deleted in their B cell lineage (KO) also produced fewer splenic germinal centers and decreased high-affinity antibody responses to NP-CGG than their WT littermates. CSR rates were lower in mTOR KI and KO mice, and pharmacologic inhibition of mTOR in WT B cells resulted in decreased rates of ex vivo CSR. RNA and protein levels of activation-induced cytidine deaminase (AID), a protein essential for SHM and CSR, were lower in B cells from both KI and B-cell specific KO mice, concomitant with increases in phosphorylated AKT and FOXO1. Rescue experiments increasing AID expression in KI B cells restored CSR levels to those in wild-type B cells. Thus, mTOR plays an important immunoregulatory role in the germinal center, at least partially through AID signaling, in generating high affinity antibodies.
Introduction
The mechanistic target of rapamycin (mTOR, MTOR) regulates cell growth and metabolism through its activity as a serine-threonine kinase. MTOR forms two protein complexes, mTORC1 and mTORC2 which are involved in phosphorylating many downstream targets, including S6K, 4EBP1 and AKT (1, 2). Rapamycin and its analogs inhibit mTOR activity, are widely used as immunosuppressants during organ transplantation, and have been increasingly used to prevent graft versus host disease (GVHD) after bone marrow transplantation (3). mTOR inhibition is pleiotropic having differential effects on various immunocompetent cells (4–6). Many studies have focused on proliferation/activity of dendritic and T cell populations as the primary targets of immunosupression (3, 7, 8). In this study we chose to focus on B cells. We recently developed a potential mouse model of chronic immunosuppression by transcriptionally inactivating a knock-in (KI) allele of mTOR; spleens of these hypomorphs were disproportionately small relative to their total body weight and mTOR protein levels were reduced by 70%. Unexpectedly, we found several effects of this knock-in on B cell differentiation, migration and homeostasis, in addition to increases in induced Foxp3+ T regulatory cells (9). Similarly, rapamycin has also been shown to promote the expansion of Foxp3+ regulatory T cells after organ transplantation (10). In the knock-in mice, B cell proliferation was less impaired in response to LPS than to either anti-IgM or anti-CD40, suggesting that innate immune responses of the mTOR-deficient mice were more intact than their adaptive responses (9).
In this study, we examined the humoral immune responses of the mTOR KI mice to infection with Streptococcus pneumoniae, one of the most common bacterial infections arising, as a result of immunosuppression, in both marrow and solid organ transplant patients (11, 12), as well as in patients undergoing chemotherapy (13). Mediation of infection requires the formation of germinal centers (GC) within splenic follicles as an essential event in the generation of high-affinity, antibody-secreting plasma cells and memory B cells (14, 15). In this study, GC formation and GC B cell functions were compared between mTOR hypomorphs (KI) and wild-type (WT) littermates, as were their ability to produce high affinity antibody isotypes in response to the model T cell-dependent antigen nitrophenylacetyl chicken gamma globulin (NP-CGG) or S. pneumoniae infection. In addition, to address the role of B cells in these responses, we examined the humoral responses of conditional B cell knock-outs of mTOR (mTOR floxed hypomorphs were crossed to CD19cre mice (16)) immunized with NP-CGG.
Immunoglobulin somatic hypermutation (SHM) and class switch recombination (CSR) are the primary effectors of antibody diversity, and occur following stimulation of mature B cells by a cognate antigen within the GCs of peripheral lymphoid organs. SHM and CSR initiation requires activation-induced cytidine deaminase (Aicda; AID), which deaminates cytidine residues in DNA to produce uracil, thus generating U:G mis-matches (17–19). SHM introduces point mutations into heavy chain Ig genes, effectively producing antibodies with varying avidities for a given antigen, thereby increasing antibody diversity (20–22). CSR is an additional mechanism of antibody diversification by which switch regions within Ig heavy chain genes rearrange to express downstream constant regions corresponding to different Ig antibody classes (23–25); CSR is dependent upon cell division (26, 27). As CSR and SHM are obligatory for the production of high affinity antibodies, we performed both in vivo and ex vivo experiments to determine if these mechanisms are intact in our mTOR KI and KO mice.
Materials & Methods
Mice
Mice were bred in conventional facilities with food and water ad libitum. All animals were treated in accordance with the guidelines provided by the Animal Care and Use Committee of the National Cancer Institute for protocol numbers LG-009, LG-017 and LG-023. mTOR KI offspring carrying neo-mTOR (mTOR KI) were identified by PCR of tail DNA using forward (5′-CCTTGGCAGCTTTGAATTTGAAG-3′) and reverse (5′-CAGAGACAGGAGACGAAGAACAGG-3′) primers. 239 and 273 bp fragments were amplified from WT and KI mice, respectively as described (9). The mTOR KI mice were bred with β-actin cre mice and the resultant progeny deleted neo ubiquitously while retaining the BALB/c allele of mTOR. These KI neo−/− mice have the same amount of mTOR protein and make similar amounts of IgG antibody in response to antigen as WT mice (9). Conditional B cell knockout mice (mTOR KO) were generated by crossing CD19cre mice (16) with the KI neo−/−harboring mTORflox/flox (Figure S1A,B); the resultant progeny deleted the mTOR allele specifically in CD19+ B cells (KO mice: mTORfl/fl CD19cre/+; WT: mTOR+/+ CD19cre/+). mTOR KO offspring carrying the mTOR deletion in CD19+ B cells were identified by PCR of tail DNA using forward (5′-CAGCATCACTCTTGCCCTTCGAACCCTTGG-3′) and reverse (5′-CCAGCCTCTTCTCTGTTTCTACC-3′) primers. WT (1342 bps), floxed allele (1488 bp) and excised allele (840 bp) fragments were amplified from WT and KO mice, respectively as described (Fig. S1B) (9). Spleen weights from mTOR KO mice adjusted for total body weight were similar to that of WT mice (Fig. S1C). AID KO (Aicda−/−) mice backcrossed to BALB/c were originally obtained from Muramatu et al. (19, 28). AID KO mice and their wild-type littermates were aged in our conventional facility to assess their survival. One group of 17 AID KO mice were weaned onto bacon-flavored chow containing the antibiotic, metronidazole (138mg/kg).
Reagents
Pneumococcal surface protein A (PspA) was purified as described (29). Lipopolysaccharide (LPS) and rapamycin were purchased from Sigma-Aldrich. α-δ-dextran was purchased from Fina BioSolutions, LLC. Recombinant mouse IL-4 was purchased from PeproTech.
Immunizations
Mice (8–10 weeks old) were immunized intraperitoneally with 2 × 108 CFU heat-killed Pn14 (29) in saline or 100 ug of NP-CGG (Biosearch Technologies, Catalog #: N-5055) adsorbed on 25 μg of alum (Rehydragel HPA, Reheis). Control mice were injected with the same volume of saline. Mice injected with Pn14 were boosted 14 days after the first injection with 2 × 108 CFU per mouse. Measurements of anti-NP and anti-PspA Ig isotype titers were determined from sera of mice.
Preparation and infection of Pn (Streptococcus pneumoniae), capsular type 14
A frozen stock of Pn, capsular type 14 was thawed and plated on trypticase soy agar plus 5% sheep blood (TSA II, BD Diagnostic Systems) for 24h incubation in 37 °C under anaerobic conditions (BD BBL GasPak Plus Anaerobic System Envelopes with Palladium Catalyst); the isolated colonies were transferred to 15 ml of Bacto-Todd Hewitt Broth (BD Diagnostic Systems) for 18h without oxygen or shaking. Pn14 challenge titers were determined by10-fold serial dilution and plating onto the blood agar. Three-month-old mice were challenged with ~1–5×106 CFU/mouse of live Pn14 in a final volume of 200 μl of PBS by i.p. injection. After 14 days post-bacterial immunization, mice were bled to determine antibody titers. On day 15, the mice were challenged i.p. again with a high dose (1×108 CFU/mouse) of live Pn14; they were bled at 15 h post-challenge to determine bacteraemia levels. Survival was monitored for 15 additional days.
Measurement of serum Ag-specific Ig isotype titers and avidities
Serum samples were prepared at a 1/50,000 dilution for total IgG or at a 1/10,000 dilution for total IgM. Quantification of IgG and IgM was performed by ELISA, according to manufacturer’s protocols (Immunology Consultants Laboratory, Inc. Mouse IgG ELISA: E-90G and Mouse IgM ELISA: E-90M).
ELISA plates (Dynex immulon 4HBX) were coated with 5μg/ml (50 μl/well) of PspA, or 10 μg/ml (50μl/well) of NP-BSA (Biosearch Technologies, Cat#: N-5050-10) in PBS overnight at 4 °C. Plates were blocked with PBS plus 1% BSA for 1 hour at 37 °C. Two-fold dilutions of serum samples, starting at a 1/200 serum dilution, in PBS plus 0.5% BSA were then added for 1 hour at 37 °C and plates were washed three times with PBS plus 0.1% Tween 20. Alkaline phosphatase-conjugated polyclonal rat anti-mouse IgM and IgG isotype-specific antibodies (Southern Biotech) were then added and incubated at 37°C for 1 hour. Plates were washed three times with PBS plus 0.1% Tween 20. Substrate (p-nitrophenyl phosphate, disodium; Sigma-Aldrich) at 1 mg/ml in 0.1M glycine buffer (2 mM MgCI2, 1mM ZnCl2 (pH 10.4)) was then added for 30 min at room temperature for color development. Color was read at an absorbance of 405 nm on a FLUOstar Omega plate reader.
Avidity of anti-PspA antibodies was determined by modifying this immunoassay to include an 8-min wash with 6.5M urea (30). Percent avidity was calculated by dividing the optical density (OD) of the urea-washed samples by the OD of the samples not washed with urea to obtain the percentage of IgG bound after washing. The relative affinities of anti-NP Abs were determined using an ELISA with BSA coupled to NP at different ratios, respectively, NP4-BSA and NP14-BSA. The relative affinity of the antibodies was indicated by the level of Abs using NP4-BSA, which measures only high affinity anti-NP4-BSA versus NP14-BSA, which measures both high and low affinity anti-NP14 (31).
Immunohistochemical (IHC) labeling
Slides of spleen sections (prepared by Histoserv, Inc.) were first deparaffinized in xylene and rehydrated through a graded ethanol series. The slides were blocked for endogenous peroxidase with freshly prepared 30% hydrogen peroxide in methanol and blocked with 10% normal rat serum in 0.1%BSA in PBS. Slides were incubated with either Biotin labeled anti-CD45R/B220 (BD Bioscience) or Biotinylated Peanut Agglutinin (PNA) (Vector Laboratories), washed, and subsequently incubated with peroxidase-conjugated avidin-biotin complex (ABC) mix and DAB chromogen buffer (Vector Laboratories, Inc.). Finally, the slides were counter-stained with Hematoxylin Nuclear Counterstain (Vector Laboratories, Inc.) and dehydrated. The procedure of double immunolabeling with anti-CD3 (labeled with ABC conjugated with Alkaline Phosphatase, developed with VECTOR Blue chromogen, (AbD Serotec)) and anti-B220 (BD Bioscience) was similar and was performed by LASP, SAIC-Frederick, Inc., NCI-Frederick. Stained slides were scanned and germinal center area and number were measured using color deconvolution analysis software (Aperio Technologies, Inc., Vista, CA).
Cell purifications
CD43− B cells were purified from spleens using MACS kits (Miltenyi Biotec) following removal of RBCs with ACK lysis buffer. Cells were cultured in RPMI-1640 supplemented with 10% FBS, penicillin (100U/ml), streptomycin (100 μg/ml), 1% L-glutamine, and 2-β-mercaptoethanol (5×10−5M) (B cell culture media). WST-1 reagent (Roche 1644807) was used to measure cell viability/proliferation.
Flow cytometry analysis
All unlabeled antibodies, phycoerythrin (PE), fluorescein isothiocyanate (FITC), APC, or PerCP-conjugates were purchased from BD Pharmingen or eBioscience. Cells were separated from spleens or lymph nodes, and red blood cells removed using ACK lysis buffer. Stained cells were re-suspended in FACS buffer (PBS with 0.5% BSA, 2mM EDTA) to a final concentration of 107 cell/ml. 100 μl of cells were blocked with Fc blocker (anti-CD16/32) for 15 minutes (min) at 4 °C, incubated with single antibodies or an antibody cocktail at 4 °C for 30 min, and washed twice with FACS buffer. Cells were fixed with 400 μl FACS buffer containing 1% of paraformaldehyde and analyzed on a FACSCalibur with Flowjo8.7 and ModFit LT software.
Measurement of cell division by CFSE dilution
Cells were stained with 1 μM of carboxy-fluorescein diacetate, succinimidyl ester (Invitrogen, Vybrant CFDA-SE). After labeling, cells were washed two times with B cell culture medium. CFSE-loaded B cells were cultured in B cell culture medium for 72h at 0.5 × 106 cells/well in 6-well plates. Cells were analyzed on a FACSCalibur using Flowjo 8.7.
Apoptosis and cell cycle Assays
Purified B cells stimulated with LPS alone, LPS + IL4 or LPS + α–δ-Dextran for 48 h were stained with Annexin V and 7-AAD (BD Pharmingen™ Apoptosis Detection Kit) or PI/RNAase buffer (BD Pharmingen™ Cell cycle detection kit) using manufacturer’s suggested protocols. Cells were analyzed on a FACSCalibur using Flowjo8.7 and ModFit LT software.
Somatic Hypermutation analysis
B cells were prepared from spleen or lymph nodes, and RBCs were removed with ACK lysis buffer. GC cells from immunized mice were sorted using GL-7 (FITC), FAS (PE), and B220 (APC) (FACSDiva flow cytometer cell sorter). GC sorted cells (about 50,000 cells) were resuspended in 50 μl of lysis buffer (10mM Tris, 0.1mM EDTA, and 0.5mg/ml proteinase K), and incubated at 50 °C for 2.5 h, then protease K was inactivated at 95 °C for 10 minutes. The Ig-JH4 intronic region was amplified with primers V1: 5′-agcctgacatctgaggac-3′ and V2: 5′-tagtgtggaacattcctcac-3′ using 20μl of digested cell solution in a 50μl reaction for the first round of PCR, the region was amplified in the second round PCR by using 5 μl of reaction 1 with primers V3: 5′-ctgacatctgaggactctgc-3′ and V4: 5′-gctgtcacagaggtggtcctg-3′ (amplification program: first round PCR, 30 cycles of 30″ at 95 °C, 30″ at 55 °C, and 2′ at 72 °C; second round PCR, 30 cycles of 30″ at 95 °C, 30″ at 65 °C, and 2′ at 72 °C). Taq-PFU (19/1) was used for PCR. Switch μ mutations were detected by PCR amplification of a 650bp genomic DNA fragment from ex vivo B cells activated with LPS + IL4 (110 h) using the primers Sμ(B) (5′-GTAAGGAGGGACCCAGGCTAAG-3′) and Sμ(D) (5′-CAGTCCAGTGTAGGCAGTAGA-3′) at 95°C for 30s, 60°C for 30s, and 72°C for 30s. PCR products were purified from gel slices, ligated into TA vectors, and sequenced with M13 forward and reverse primers.
The data were analyzed with the web-based SHMTool (32). Mutations were counted by two separate methods to provide a more accurate estimate of point mutation frequency (Table 1A,B). A single B cell clone produces individual descendants each with a variant sequence that can potentially share common mutations with sequences from other members of the clone. This redundancy, which leads to an overestimate of mutation frequency, was corrected by counting mutations involving the same nucleotide change and position only once, therefore providing an underestimate of mutation frequency (as described in (33)). Thus, when comparing sequences, “non-unique” describes the mutations that are counted individually, regardless of commonality with other mutations in other sequences (Table 1A), and “unique” describes the mutations occurring at the same site and with the same nucleotide, thus grouped and counted only once (Table 1B) (SHMTool). WT and KI sequences were compared separately. The actual mutation frequency for either WT or KI lies between the extremes of the two methods ((33); SHMTool).
Table 1.
Somatic mutations in JH4 intronic sequences (403bp) from splenic GC B cells of WT or KI mice with NP-CGG challenge
| KI (NP-CGG) | WT (NP-CGG) | Chi-square Test | |
|---|---|---|---|
| Number of sequences | 82 | 77 | p-value
|
| Total length sequenced (bp) | 33046 | 31031 | KI vs WT |
| Unmutated Sequences (%) | 30/82, 36.6% | 13/77, 16.9% | |
| Number of deletions and insertions | 13 | 8 | |
| A. Total number of non-unique point mutations | 92 | 153 | |
| Mutation frequency per total sequences (per 100bp) | 0.32 | 0.52 | 0.0001 |
| Mutation frequency per mutated sequences (per 100bp) | 0.50 | 0.62 | 0.0264 |
| B. Total number of unique point mutations | 71 | 102 | |
| Mutation frequency per total sequences (per 100bp) | 0.25 | 0.35 | 0.0257 |
| Mutation frequency per mutated sequences (per 100bp) | 0.40 | 0.43 | 0.7227 |
Class Switch Recombination (CSR) analysis
CD43− resting B cells were obtained from spleens and lymph nodes using magnetic CD43 beads; CD19+ CD43− resting B cells were further purified from CD43− cells using magnetic CD19+ beads according to the manufacturer’s instructions (Miltenyi Biotec). The cells (0.5×106/well) were cultured in 6-well plates in 5 ml of B cell culture medium and incubated at 37°C for 72 h. To induce specific isotype switching, B cells were stimulated with 50 μg/ml LPS (Sigma L2880-25mg) plus either 20 ng/ml IL-4 (PeproTech, Inc., 214-14) for induction of IgG1 switching or 3 ng/ml α-δ-dextran for induction of IgG3 switching.
RNA preparation and real-time RT-PCR
Total RNA was isolated from cells with TRIzol Reagent (Invitrogen). cDNAs were made with TaqMan Reverse Transcription Reagents (Applied Biosystems: N808-0234), and real-time PCR was performed using SYBR Green PCR Master Mix (Part No: 4309155, ABI 7500) with the following primers: mTOR: forward primer: 5′-TCCTGCGCAAGATGCTCATC-3′, reverse primer:5′-TGTGCTCCAGCTCTGTCAGGA-3′; β-actin: forward primer: 5′-CTTCTTGGGTATGGAATCC-3′, reverse primer: 5′-GGCATAGAGGTCTTTACG-3′; AID: forward primer: 5′-GCCACCTTCGCAACAAGTCT-3′, reverse primer: 5′-CCGGGCACAGTCATAGCAC-3′; UNG: forward primer: 5′-TCATTGACAGGAAGCGTCACC-3′, reverse primer: 5′-GGAACCCTCTGTGCACCG-3′; Polη (DNA polymerase η): forward primer: 5′-CACATGGGTCAGTGCCACA-3′, reverse primer: 5′-AGCTTTCCTCCAAGACTTCGG -3′; Exo1: forward primer: 5′-CCATGGCCCACAAAGTAATAAAA-3′, reverse primer:5′AGCATCAGCTTCATACGGAGC-3′; Msh2: forward primer: 5′-ACCAAGTGAAAAAAGGTGTCTGTG-3′, reverse primer: 5′-CCTCGGGAAGTTAGCGAGC-3′; Msh6: forward primer 5′-GGCGCTTGTTCTGTAACTTCG-3′, reverse primer: 5′-CTGTAGTTAGCCAGGCACAGTAAGA-3′; RNAII: 5′-TGGGTTTGCCCTAATCCGT-3′, reverse primer: 5′-TGGTCTAGGCTCATTGCACTGA-3′;18S rRNA: forward primer: 5′-GCCGCTAGAGGTGAAATTCTTG-3′, reverse primer: 5′-CATTCTTGGCAAATGCTTTCG-3′.
PCR of germline transcripts was performed with the following primers to obtain the indicated product sizes: (μ) ImF and CmR, 245 bp; (γ3) Ig3F and Cg3R, 323 bp; (γ1) Ig1 and Cg1R, 429 bp; Post-switch transcripts were amplified using the following primer pairs: (γ3) ImF and Cg3R, 323 bp; (γ1) ImF and Cg1R, 353 bp; germline and postswitch transcripts were amplified by 30 or 35 cycles of PCR (19). PCR primers: ImF: 5′-CTCTGGCCCTGCTTATTGTTG-3′; Ig3F: 5′-TGGGCAAGTGGATCTGAACA-3′; Ig1: 5′-GGCCCTTCCAGATCTTTGAG-3′; CmR: 5′-GAAGACATTTGGGAAGGACTGACT-3′; Cg3R: 5′-CTCAGGGAAGTAGCCTTTGACA-3′; Cg1R: 5′-GGATCCAGAGTTCCAGGTCACT-3′.
Western blot analysis
Protein extracts from tissues and cultured cells were prepared with lysis buffer (50mM Tris-HCl, pH 7.4, 150mM NaCl, 1% NP-40, 1mM EDTA, 20mM beta glycerol phosphate, protease inhibitors (Roche), and phosphatase inhibitors (Santa Cruz)). Nuclear and cytoplasmic extracts were prepared using the NE-PER kit (Catalog #: 78833, Pierce) according to the manufacturer’s protocol. 20μg of total protein lysate or 10 μg of cytoplasmic/nuclear extracts were used for Western blot analysis. Membranes were incubated with AID (#4975), pAKT (#4058), pFOXO1/3a (#9464), FOXO1(#9462), FOXO3a(#9467) and β-actin (#4970L) (Cell Signaling) as well as UNG (# 3859, ProSci), HSP90 (SPC-104C, Stress Marq), and Histone H1 (SC-8030, Santa Cruz) antibodies.
Retroviral transduction of B cells
Retroviruses expressing AID-GFP or GFP were prepared by adapting previously described methods (34) in the following manner: PlatE cells were transfected with pMYs-mAID-IRES-EGFP or pMYs-IRES-EGFP plasmid by Lipofectamine 2000 (Invitrogen), and recombinant retrovirus was collected 24–36 h after transfection. The CD43− resting B cells isolated from spleens of WT or KI mice were pre-activated for 24 h with 0.5 μg/ml of anti-CD180, and then were transduced twice with retroviruses by ‘spin inoculation’ (650g for 90 min). Cells were cultured for an additional 3 days with 50 μg/ml of LPS, 20 ng/ml of IL-4, and 0.5 μg/ml of anti-CD180 before being analyzed for IgG1 cell surface expression by FACS.
Results
Germinal center formation is affected by mTOR expression
To evaluate the effects of reduced mTOR on GC formation, mTOR KI mice were either primed with NP-CGG (4-Hydroxy-3-nitrophenylacetyl hapten conjugated to chicken gamma globulin lysine), or primed and subsequently boosted with intact heat-killed Streptococcus pneumoniae capsular type 14 protein (Pn14) as described previously (9, 29). GCs were evaluated 14 days (d) and 21d post priming with NP-CGG and Pn14, respectively, using immunohistochemical (IHC) staining and analysis of cell surface markers by flow cytometry. The average number of GCs in spleens of KI mice was 3–5-fold lower than in WT spleens (Fig. 1A, S2A). Furthermore, both the percentage of GC cells within the GC and the composite GC area relative to the total area of B cell lymphoid follicles were reduced disproportionately in the KI mice (Fig. 1A, S2A). Consistent with the IHC findings, flow analyses showed the frequency of specific GC B cells (B220+, IgDlow, GL7+, Fas+ and B220+, IgDlow, CD38−, Fas+) was lower in spleens and lymph nodes of KI compared to WT mice (Fig. 1B, S2B). In contrast, the splenic and lymph node populations of CD11c+, CD21/35+ cells were similar between WT and KI mice that had received a prime/boost of Pn14 (Fig. S3A). In addition, CD3+ T cells were also abundant in the lymphoid follicles of both WT and KI mice (Fig. S3B).
Figure 1. Constitutive reductions in mTOR impair GC formation and decrease anti-NP antibodies in response to NP-CGG.
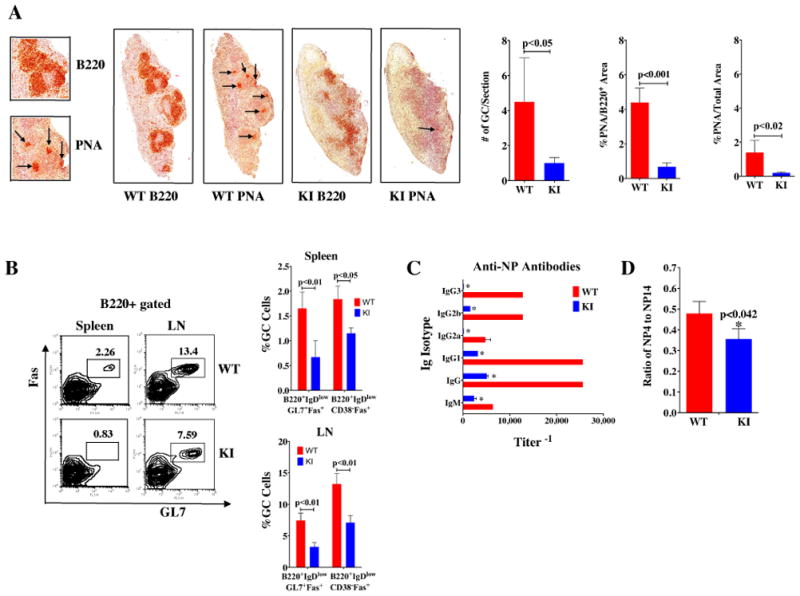
KI and WT (n = 5/group) mice were immunized i.p. with NP-CGG in Rehydragel. Spleens were collected on day 14 for IHC staining and FACS analysis. A) Splenic sections were stained with B220 or PNA. The numbers of GCs and the area of PNA staining was evaluated from scans of spleen sections stained with B220 or PNA using color deconvolution analysis software (Aperio Technologies). B) The cells from spleens and lymph nodes (LN) were stained with B220, GL7, Fas, CD38, and IgD antibodies. There were fewer GC B cells in KI than WT. Data are presented as mean ± SEM; p-values are indicated. C) Sera from KI and WT mice immunized with NP-CGG were collected on day 14 for measurement of Ag-specific IgM and IgG isotype antibody titers. Data are presented as mean ± SEM: *significance p<0.01. D) The relative affinities of anti-NP Abs were determined using an ELISA with BSA coupled to NP at different ratios, namely, NP4-BSA and NP14-BSA.
Reduced mTOR impairs IgG antibody affinity maturation
KI and WT mice were given priming doses of either NP-CGG or prime/boost doses of heat-killed Pn14 to assess their ability to mount humoral responses to antigens. Primary and secondary antigen-specific antibody titers were measured against NP-hapten or pneumococcal surface protein A (PspA) as previously described in (9) and (29), respectively. In these experiments, anti-NP and anti-PspA IgG antibody titers were lower in KI mice 14 and 21 days post receipt of priming doses (Fig. 1C, Fig. S2C,D). More specifically, the diminished capacity for generating PspA-specific IgG-isotype responses remained low a week after the boost immunization with S. pneumoniae, with the greatest deficits in IgG1 and IgG3 (6- and 10-fold, respectively) (Fig. S2D). Thus, constitutive reductions in mTOR expression caused deficits in specific antibody production that persisted even after secondary exposure to antigen.
To explore whether mTOR hypomorphic KI mice produce high-affinity antibodies, the avidity of the anti-NP antibodies were measured by the protocol described in Yamamoto et al. (31), and KI mice developed lower antigen affinity antibodies compared to WT littermates (Fig. 1D). The anti-PspA IgG antibodies were measured using a modified ELISA protocol (35) in which only higher affinity antibodies remained bound to PspA-coated microtiter plates after washing with 6.5M urea. mTOR KI mice did not produce detectable amounts of high affinity IgG to PspA until after the boost, while WT mice had readily measurable amounts as early as one week following the initial immunization (Fig. S2E). Although the boost induced some high affinity antibodies in KI mice, the degree of affinity maturation continued to lag behind that of WT (Fig. S2E). Thus, mTOR can regulate antibody maturation to antigenic challenge in response to either intact or haptenated antigens.
mTOR hypomorphic mice are more sensitive to live Streptococcus pneumoniae challenge
Protection against S. pneumoniae relies not only on innate immune responses, but also requires an intact adaptive response (36). To determine if mTOR plays a role in mediating infection and to assess whether impaired antibody responses in the mTOR KI mice might be biologically relevant, mice were challenged with a low dose (106 CFU) of live, encapsulated Pn14 and were re-challenged after 14d with a high dose (108 CFU) and followed for 15 days. Antibody titers to PspA 14d after the initial challenge, and before the high dose challenge were considerably lower in the mTOR KI than in WT mice (Fig. 2A). At 15 hours post-challenge with 108 CFU, only 1 out of 9 KI mice was bacteria-free, compared with 5 out of 10 WT mice; for the mice with detectable bacteremia at that time point, the average titer was slightly higher in the KI mice (Fig. 2B). All of the WT mice survived the high dose challenge, while only 60% of the KI mice survived (Fig. 2C).
Figure 2. Prior challenge of mTOR hypomorphic KI mice does not protect them from challenge with live Streptococcus pneumoniae (Pn14).

mTOR KI mice (n = 9) and wild-type littermates (n = 10) were immunized with 1–5×106 CFU live Pn14 and challenged at d14 with 1×108 CFU live Pn14/mouse. A) At day 14 before high dose challenge, KI mice had lower anti-IgG responses (*p<0.05, **p <0.01) than WT mice, B) 15 hours after high dose challenge, more KI mice were bacteremic than WT mice and C) 14 days after high dose challenge, KI mice had a higher rate of mortality than WT mice (p<0.05). The proportions of Ig sequences from WT and KI mice with defined numbers of mutations D,E) were determined. D) Somatic hypermutation (SHM) frequency in immunoglobulin JH 4 intron (Ig-JH4) sequences from WT and KI GC (B220+GL7+Fas+) splenic B cells isolated directly from mice immunized with NP-CGG. E) Mutation frequency in immunoglobulin switch μ (Sμ) region from WT and KI splenic CD43− resting B cells stimulated ex vivo with LPS and IL-4 for 120 hrs (IgG1 induction). The number highlighted in the center of the chart is the total number of sequences analyzed.
Constitutive reductions in mTOR impairs somatic hypermutation
The lack of high-affinity antibody responses in mTOR KI mice led us to hypothesize that the frequency of SHM, a driver of antibody diversity, may be reduced in KI germinal center B cells. To address this question, SHM frequency was measured in mice immunized with NP-CGG. We analyzed the IgH JH4-intronic sequence downstream of the rearranged VDJ region in GC B cells (B220+ GL7+ Fas+) sorted from splenocytes of NP-CGG immunized KI and WT mice. The frequency of SHM and pattern of point mutations were analyzed using SHMTool (32). The frequency of SHM was decreased in GC B cells from KI mice compared to WT littermates (Table 1, Fig. 2D). The non-unique mutation frequency/total sequences (per 100bp) was 0.52 in WT JH4 introns and lower at 0.32 in KI JH4 introns (p<0.001, Table 1A). These findings held true for the total number of unique point mutations as well (Table 1). Overall, the reduction in mutation frequency was due primarily to a paucity of mutated sequences in mTOR KI mice (Fig. 2D); 37% of sequences from KI splenic GC B cells were not mutated, while only 17% of sequences from WT cells fell in this category, representing a greater than 2-fold decrease in mutated JH4 sequences in KI B cells (Fig. 2D). Furthermore, while 6% of WT splenic GC B cell sequences had 6 mutations, none of the KI JH4 intron sequences had as many mutations (Fig. 2D).
The pattern of SHM point mutations was skewed in GC B cells from KI mice compared to WT (Table 2). The percentage of transversions from G to T or A to C was decreased for KI GC B cells (p< 0.05). Of the unique mutations, there were fewer G-T and C-A (12.5%) or T-G and AC (15.4%) transversions in KI B cells compared to WT (26.1% and 25%, respectively) (Table 2). Additionally, KI B cells had more (59.4%) transitions from G to A and C to T compared to WT (43.5%) (Table 2). Overall, there were fewer transversions and an increase in transitions seen in the mTOR deficient mice.
Table 2.
Pattern of nucleotide changes in JH4 intronic sequence from splenic GC B cells of WT and KI with NP-CGG challenge
| A. Non-unique Mutation Counts | GC:AT | trans: transv | within G/C
|
within A/T
|
||||
|---|---|---|---|---|---|---|---|---|
| trans
|
transv
|
trans
|
transv
|
|||||
| G/A C/T |
G/T C/A |
G/C C/G |
A/G T/C |
A/T T/A |
A/C T/G |
|||
| KI (NP-CGG) | 48.9:51.1 | 53.3:46.7 | 46.7 | 11.1 | 42.2 | 59.6 | 23.4 | 17.0 |
| WT (NP-CGG) | 47.1:52.9 | 47.6:52.4** | 45.8 | 23.6* | 30.6 | 49.4* | 24.7 | 25.9* |
|
| ||||||||
| B. Unique Mutation Counts | ||||||||
|
| ||||||||
| KI (NP-CGG) | 45.1:54.9 | 59.2:40.8 | 59.4 | 12.5 | 28.1 | 59.0 | 25.6 | 15.4 |
|
| ||||||||
| WT (NP-CGG) | 45.1:54.9 | 45.1:54.9** | 43.5* | 26.1* | 30.4 | 46.4* | 28.6 | 25.0* |
Note: p-value (Chi-square Test),
p<0.05;
p<0.01; trans: transition; transv: transversion
mTOR KI mice have fewer mutations in Sμ and defective immunoglobulin class switching to IgG1 ex vivo
The inability of mTOR KI mice to mount high-affinity IgG responses led us to ask whether their B cells were impaired in their ability to undergo class switch recombination. Class switching occurs by intrachromosomal deletion recombination within switch (S) regions upstream of each Ig heavy chain constant region and initiates in the Sμ segment (37). Frequent AID-dependent mutations occur at Sμ sequences during CSR (38–40). Ex vivo activated (LPS + IL-4) CD43− resting B cells from mTOR KI mice experienced a lower frequency of mutation (0.7×10−3) in their immunoglobulin Sμ region than WT mice (1.4×10−3) (Fig. 2E, Table 3).
Table 3.
Mutations in Sμ region (650bp) from splenic CD43− resting B cells of WT and KI mice with LPS and IL-4 induction
| KI | WT | Chi-square Test | |
|---|---|---|---|
| Number of sequences | 47 | 46 | p-value
|
| Total length sequenced (bp) | 30550 | 29900 | KI vs WT |
| Unmutated Sequences (%) | 32/47, 68.1% | 26/46, 56.5% | |
| Number of deletions and insertions | 1 | 15 | |
| A. Total number of non-unique point mutations | 20 | 26 | |
| Mutation frequency per total sequences (per 100bp) | 0.07 | 0.14 | 0.0125 |
| Mutation frequency per mutated sequences (per 100bp) | 0.22 | 0.32 | 0.1938 |
| B. Total number of unique point mutations | 18 | 25 | |
| Mutation frequency per total sequences (per 100bp) | 0.06 | 0.13 | 0.0073 |
| Mutation frequency per mutated sequences (per 100bp) | 0.20 | 0.31 | 0.1285 |
The lower mutation rates seen in Sμ sequences from the KI mice suggested that the ability of KI B cells to undergo CSR may also be impaired. The capability of KI mice to undergo CSR was determined by ex vivo stimulation of CD43− resting B cells as described (41). Compared with WT, fewer KI B cells stimulated with LPS and IL-4 expressed IgG1 (Fig. 3A), suggesting that the decreases in IgG antibody production may be due to defective CSR in the mTOR KI cells.
Figure 3. CSR frequency in mTOR hypomorphs and rapamycin treated WT cells.

A) CD43− resting B cells purified from spleens and LNs of KI and WT mice (age 5–12 wks) were stimulated with LPS and IL4 for 72 hours and cells were stained with B220 and IgG1 antibodies (n=9 for LNs, n=12 for spleens). B) CD43− cells from spleens of KI and WT mice (age 8–10 wks) were stained with CFSE, and then stimulated with LPS plus IL-4 (IgG1 induction) for 72 hours. The cells were stained with IgG1 (n = 6 samples) antibody and analyzed with FACS and FlowJo. Percentages of total cell number and IgG1+ cells in each division are presented. C–F) Rapamycin decreases CSR in CD43− B cells from wild-type mice. CD43− resting B cells purified from spleens of WT mice were stimulated (≥ 72hrs) with LPS and IL-4 to induce IgG1 switching over time (≥ 72hrs). C, D) Purified CD43− resting B cells were stained with CFSE, and then stimulated with LPS and IL-4. The cells were stained with IgG1 antibody. Cell proliferation was analyzed with CFSE staining (D), and percentages of total cell number and IgG1+ cells in each division are presented. FACS analyses were performed with PI-staining for cell cycle (E) or Annexin V and 7AAD staining for apoptosis (F) in CD43− resting B cells induced to switch to IgG1. Representative experiments are shown from two of three replicates (n=3). Cells were analyzed with FACS and FlowJo and data are presented as mean ± SEM; *significance p values (p<0.05) are indicated.
CSR has been linked to rates of B cell division (26, 27). Proliferation rates of B cells expressing IgG1 in KI mice were lower (Fig. 3B), probably resulting from cells undergoing G1 arrest (Fig. S4A) rather than apoptosis, as similar numbers of cells underwent apoptosis in the KI and WT mice (Fig. S4B). Analysis of CFSE staining over the course of at least 4 cell cycle divisions (Fig. 3B) indicated the percentage of cells within each cell cycle that switched to IgG1 expression was lower in the first and second cell divisions, with this difference persisting through the fourth division, likely due to less cells entering successive divisions (Fig. 3B). These results imply that IgG1 class switch was less efficient in mTOR KI B cells.
To further evaluate deficits in IgG switching in KI mice, the germline (GL) (pre-switch) and post-switch (PS) transcripts for IgG1 and IgG3 were measured by PCR (protocol adapted from (19)). These analyses confirmed that KI mice had no change in levels of germline μ, γ1, and γ3 transcripts, but lower levels of post-switch γ1 and γ3 transcripts (Fig. S4C,D).
Independent evaluation of the role of mTOR in CSR was gained by treating resting B cells from WT mice with rapamycin which preferentially inhibits mTORC1, and inhibits mTORC2 usually only after long-term treatment and in a cell type specific manner (42). IgG1 (Fig. 3C) switching was impaired in WT activated CD43− splenic B cells treated with low dose rapamycin (0.1nM), which had modest effects on cell viability and proliferation until the fifth cell division where KI cells were diminished with respect to WT (Fig. 3D), cell cycle arrest (Figs. 3E), or apoptosis (Figs. 3F). Together these findings confirm that mTOR can affect CSR through its affect on cell division/proliferation, but also suggest a possible role independent of proliferation.
Levels of CSR/SHM associated proteins are lower in mTOR KI mice
A number of DNA damage response/repair proteins, in addition to AID and uracil DNA glycosylase (UNG), are involved in SHM/CSR activities (24, 25). RT-PCR experiments using LPS stimulated B220+ cells examined the ratio of KI:WT mRNA levels for several genes implicated in CSR/SHM activity (Aicda, Ung, Pol η (polymerase eta, PolH), Exo1 (exonuclease 1), Msh2 (mutS homolog 2), and Msh6 (mutS homolog 6) in splenic B220+ cells. Following LPS stimulation, B220+ KI splenocytes were deficient only in Aicda (AID) mRNA levels relative to WT (Fig. 4A). AID protein levels were confirmed to be decreased in LPS-stimulated mTOR KI splenocytes relative to WT (Fig. 4B).
Figure 4. AID expression is lower in mTOR hypomorphic KI mice.
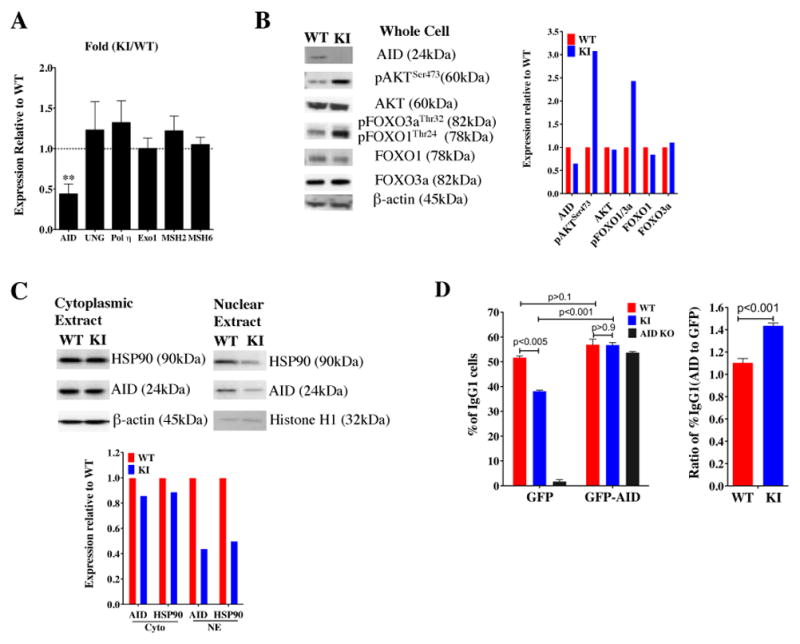
A) CD43− resting B cells from WT and KI spleens were stimulated with LPS and IL4 for 48 hours. mRNA expression of AID, UNG, Polη, ExoI, MSH2, and MSH6 were measured by real-time PCR. The results are displayed as relative fold changes of mRNA expression in KI compared with WT, upon normalization by 18S RNA (mean ± SD, n = 3). B) Western blots of AID signaling in CD43− resting B cells from WT and KI spleens stimulated with LPS and IL4 for 48 hours. C) Nuclear protein levels of HSP90 and its client protein, AID are lower in CD43− resting B cells from KI spleens stimulated with LPS and IL-4 for 48 hours. The fold change was normalized to the WT protein level, after normalization by β-actin. Western blots were repeated in at least two experiments. D) CD43− resting B cells isolated from spleens of WT or KI mice were infected with retroviruses expressing AID-GFP or GFP, and stimulated with LPS and IL-4 for 72 hours. CSR was measured by IgG1 cell surface expression. The percentage or ratio of IgG1+ cells from AID-GFP to GFP retrovirus infection was calculated in GFP+ cells. AID-KO mice were included as controls. Two independent experiments were in agreement.
Nuclear AID levels are low in mTOR KI cells
AID expression can be affected by both Forkhead transcription factors and the chaperone protein HSP90 (43, 44). Retroviral gene transfer studies (45, 46) have shown that constitutively active AKT in B cells can inactivate Forkhead transcription factor FOXO1 resulting in concomitant decreases in AID and suppression of CSR; in addition, phosphorylation of AKT at Ser473 is required for phosphorylation of FOXO1/3a (47). In our earlier study (9), LPS stimulated B cells from mTOR KI mice had increased levels of pAKTSer473 and in the current study, we have shown that B cells from mTOR KI mice have increased levels of pAKTSer473 and pFOXO1Thr24, concomitant with lower levels of AID in response to LPS and IL-4 (Fig. 4B). Recent studies have also shown that HSP90 can stabilize levels of AID in the cytoplasm and the nucleus (44). In contrast to cytoplasmic fractions, nuclear extracts from mTOR KI B cells stimulated with LPS and IL-4 had much lower levels of HSP90 and AID compared to WT B cells (Fig. 4C). Thus, lower AID protein levels in mTOR KI B cells may result from increases in phospho-AKT and -FOXO, in addition to reductions in nuclear HSP90.
Increasing AID expression rescues IgG1 switching in mTOR KI B cells
AID expression levels are directly proportional to the extent of CSR and SHM (41, 48). To determine if reintroduction of AID protein into mTOR hypomorphic KI cells could restore IgG1 switching, CD43− resting B cells were isolated from KI and WT mice, transduced with either GFP or AID-GFP retroviruses, and then stimulated with LPS and IL-4. As expected, introduction of the control GFP retrovirus expressing only GFP in KI resting B cells did not restore IgG1 switching to those levels seen in WT B cells, nor did GFP alone affect switching in AID knock-out (KO) B cells used as a control (Fig. 4D). When AID levels were increased in KI mice by the AID-GFP retrovirus, IgG1 switching was comparable between WT and KI cells and similar to AID KO cells transduced with the AID-GFP retrovirus (Fig. 4D). These data continue to support a role for mTOR in controlling AID levels.
Survival in AID knock-out mice is increased with prophylactic antibiotic treatment and filter-top cages
AID KO mice are known to experience a 100-fold expansion of anaerobic flora in their small intestine (49) and we experienced difficulty in maintaining a healthy population of these mice in a conventional (non-SPF) colony. As a result, we monitored the overall survival of AID KO mice and their wild-type littermates, with and without metronidazole, housed in a conventional facility over the course of ~2 years (Fig. S4D). Wild-type mice lived longer (~780d) than AID KO mice (240–360d) (Log-rank (Mantel-Cox) χ2 = 19.1, p < 0.0001). Furthermore, AID KO mice that were given feed containing the antibiotic and antiprotozoal, metronidazole, and housed in autoclaved, filter-topped cages lived longer (~360d) than conventionally maintained AID KOs (~240d) (Log-rank (Mantel-Cox) χ2 = 6.51, p < 0.01).
B cell specific mTOR knock-out (KO) mice are impaired in their germinal center responses
As mTOR levels were constitutively reduced in all cell types from birth in the mTOR (KI) hypomorphs, we sought to determine whether mice deleted for mTOR specifically in their B cell lineage (mTOR KO mice: mTORfl/flCD19Cre/+) (Fig. S1A) would exhibit similar defects in GC formation and antibody production. We did not find differences in B cell sub-populations in the bone marrow (Fig. S1D), or in the transitional T1 B cell subpopulation in the spleens (Fig. 5A), of KO versus WT (mTOR+/+CD19Cre/+) mice. In contrast, CD19+, mature, transitional T2 and marginal zone (MZ) (Fig. 5A–C) B cell populations were lower in spleens of KO mice compared to WT. Reductions in B cell populations in lymph nodes (LN) were also seen in mTOR KO mice (Fig. 5A–C). Similar to results obtained in mTOR KI mice, fewer GCs as assessed by IHC (Fig. 6A) and flow cytometry (Fig. 6B) formed in spleens of KO mice immunized with one dose of NP-CGG. These mTOR KO mice also produced fewer IgG isotypic antibodies, except IgG2a (Fig. 6C), and fewer high affinity anti-NP antibodies (Fig. 6D) compared to their WT littermates. Resting B cells, stimulated with LPS and IL-4, from the mTOR KO mice exhibited lower rates of CSR in ex vivo cultures (Fig. 6E). Furthermore, we have confirmed that B cells from mTOR KO mice, similar to their hypomorphic (KI) counterparts, have increased levels of pAKTSer473 and pFOXO1Thr24, concomitant with lower levels of AID (Fig. 6F) in response to LPS and IL-4. Thus, these experiments confirm that mTOR deletion in CD19+ cells alone can negatively affect GC formation, antibody production, CSR and establishment of B cell populations in the periphery.
Figure 5. B cell subpopulations are altered in the lymph node and spleens of B-cell specific KO mice.

A,B) Cells isolated from spleens and LNs of WT and KO mice were stained with antibodies to B220, CD23, and CD21, IgM and IgD. Flow cytometry analyses revealed that B cell subpopulations (mature, transitional T2, follicular and marginal zone B cells) were decreased in KO tissues. Stages of B cells were identified by the following cell surface markers: M: mature B cells (IgDhighCD21+IgM−B220+); T: transitional B cells (T1: B220+IgD−IgMhigh; T2: B220+IgDhighIgMhigh); follicular B cells (FO B: IgMlowIgDhighCD21intCD23+); marginal zone B cells (MZ B: IgMhighIgDlowCD21highCD23−). The age of the mice ranged from 8 to 12 weeks (n= 8). C) The percentage and size of CD19+ B cells in KO mice. D) FACs analysis of cells from spleens and lymph nodes (LN) stained with B220, GL7, Fas, CD38, and IgD antibodies. There were fewer GC B cells in KO than WT mice following NP-CGG priming. Data are presented as mean ± SEM; p-values are indicated.
Figure 6. mTOR KO in CD19+ B cells impairs GC formation and decreases anti-NP antibody response to NP-CGG.

KO (n=7) and WT (n = 5) mice were immunized i.p. with NP-CGG in Rehydragel. Spleens were collected on day 14 for IHC staining. A) Splenic sections were stained with B220 and PNA. The numbers of GCs and the area of PNA staining were evaluated from scans of spleen sections stained with B220 or PNA using color deconvolution analysis software (Aperio Technologies). B) FACs analysis of cells from spleens (SPL) and lymph nodes (LN) stained with B220, GL7, Fas, CD38 and IgD antibodies. C) Sera from KO and WT mice immunized with NP-CGG were collected on day 14 for measurement of Ag-specific IgM and IgG isotype antibody titers. Data are presented as mean ± SEM: significance * p<0.05 and ** p<0.01. D) The relative affinities of anti-NP Abs were determined using an ELISA with BSA coupled to NP at different ratios, namely, NP4-BSA and NP14-BSA. E) CD43− resting B cells purified from spleens and LNs of KO and WT mice (age 8–10 wks) were stimulated with LPS and IL-4 for 72 hours and cells were stained with CD19 and IgG1 antibodies (n=6). The cells were analyzed with FACS and FlowJo. Data are presented as mean ± SEM; p-values are indicated. F) CD43− CD19+ resting B cells from WT and KO LNs were stimulated with LPS and IL4 for 48 hours. WB analyses of protein expression in KO relative to WT mice. AID expression levels were lower while pAKTSer473 and pFOXO1Thr24 levels were higher in KI or KO mice relative to WT.
Discussion
mTOR controls cell growth, and is often dysregulated in several diseases, including cancer and immunosuppressive diseases (1, 2). Rapamycin analogs, specific inhibitors of mTOR kinase activity, have been used in the transplant setting to induce chronic immunosuppression to prevent rejection and GVHD (3, 11). Long-term survivors of organ and bone marrow transplantation are at increased risk for infections, particularly from encapsulated bacteria such as S. pneumoniae, which accounts for ~30% of community-acquired pneumonia, and is known to be a common infection in immunosuppressed individuals (11–13).
We recently showed that constitutive reductions in mouse mTOR gene expression are associated with a partial block in B cell development in the bone marrow, altered percentages of various B cell populations in the spleen, and smaller spleens (9). We now report that B cell specific reductions in mTOR impair germinal center formation, decrease the production of immunoglobulin gamma (IgG) isotypes in response to immunization and lead to a decrease in affinity maturation of antibodies in vivo. Remarkably, when challenged with an infection that elicits an antibody response, 40% of mTOR hypomorphic mice died compared with none of the WT counterparts. Therefore, these mTOR hypomorphic KI mice serve as a novel and clinically relevant model to evaluate infection in the presence of chronic immunosuppression.
The two processes, SHM and CSR, contributing to antibody gene diversity in mammalian cells were decreased in activated mTOR KI B cells. SHM patterns in KI mice were distinct from WT; there were more transitions from G-A to C-T base pairs, and fewer transversions (G/T; C/A and A/C; T/G) in KI B cells. Maul and Gearhart (2010) describe two potential pathways for processing U:G mismatches that are both initiated by AID activity (18). Specifically, mutations at the G-C base pair use UNG, APE, and the low fidelity polymerase Rev1, whereas mutations at the A-T base pair use the MSH2-MSH6 heterodimer, exonuclease 1, and the error prone polymerase Pol-η (18). Our findings in mTOR KI mice were consistent with lower Ung/UNG expression, but no significant change in Msh2/6. In addition, defects in KI B cells were further documented by ex vivo experiments demonstrating lower Sμ mutation and CSR frequencies in response to IL-4 and LPS. Lower IgG1 CSR frequencies were also observed in B cell specific KOs of mTOR.
The ability of differential mTOR expression to affect SHM and CSR identifies a new immunoregulatory role for mTOR. The results seen in the mTOR hypomorphic KI mice were similar to those obtained in AID haploinsufficent (AID+/−) mice; these mice also have impaired CSR, and reductions in both SHM and antibody affinity maturation (41, 48). Takizawa et al. (41) suggest that AID protein content may fall below a critical threshold in AID+/− B cells, potentially through mechanisms controlling nuclear AID, or due to compromised AID activity because of lower AID transcription.
Reduced AID levels in both our KI and B cell specific KO mice, particularly the lower AID levels seen in nuclear extracts from activated CD43− B cells from the KI mice, provide the link between mTOR expression and reductions in CSR/SHM and antibody affinity maturation, as AID is required for both CSR and SHM. In fact, CSR to IgG1 could be restored to WT levels in mTOR hypomorphic KI B cells following transduction with AID-GFP retrovirus.
Reductions in CSR/SHM and antibody affinity maturation ultimately affect antibody diversification, which can interfere with the ability of the immune system to respond to microbial challenge and recognize specific antigens. Indeed, under conventional conditions, the survival of AID KO mice was much lower than that of their WT littermates; prophylactic antibiotic treatment of the AID KO mice increased their survival.
Our studies suggest that suppression of AID activity via rapamycin may contribute to the impairment of humoral immune responses, and provide further rationale for potential immunization against microbial agents prior to immunosuppressive treatment. Immunizations have typically been given simultaneous with or after cessation of immunosuppressive drugs (50, 51). The improved survival of AID KO mice treated prophylactically with antibiotics suggests that the co-administration of antibiotics with rapamycin might decrease the risk of opportunistic streptococcal infections. In addition, these findings may have relevance in other settings, specifically the use of mTOR inhibitors as anti-cancer agents (2). In this situation, increased AID has been shown to promote both tumor progression (41, 52–55) and drug resistance (56). Our studies have confirmed that mTOR inhibition can down-regulate AID as reported perhaps by the phosphorylation of both AKT and FOXO proteins and the amount of nuclear HSP90 (43). Thus, fine-tuning mTOR inhibitors with appropriate dosing regimens may help restore normal AID levels or activities needed for the establishment of GC reactions and genesis of high affinity antibodies while effectively regulating the AID mutator phenotype during tumor progression and/or drug resistance.
Supplementary Material
Acknowledgments
The authors thank Douglas Lowy, Crystal Mackall, Ron Gress, Dan Fowler, Howard Young, Glenn Merlino, Stuart Yuspa, Michael Potter, Richard Robinson, Val Bliskovsky, Kaori Sakakibara and anonymous reviewers for their comments on the manuscript. We thank Alexander Kovalchuk for advice on SHM sequence analysis and Makiko Takizawa for cloning AID coding sequences into the pMYs-IRES-GFP vector. We also thank Stefan Kuchen-Brandes and Arito Yamane for their advice and assistance with AID rescue experiments. AID KO mice were kindly provided by Michael Potter.
Footnotes
The Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, and the Uniformed Services University of the Health Sciences supported this work. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the U.S. Government.
References
- 1.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cutler C, Antin JH. Sirolimus for GVHD prophylaxis in allogeneic stem cell transplantation. Bone Marrow Transplant. 2004;34:471–476. doi: 10.1038/sj.bmt.1704604. [DOI] [PubMed] [Google Scholar]
- 4.Xu X, Ye L, Araki K, Ahmed R. mTOR, linking metabolism and immunity. Semin Immunol. 2012;24:429–435. doi: 10.1016/j.smim.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saemann MD, Remuzzi G. Time to rethink immunosuppression by mTOR inhibitors? Nature Reviews. 2009;5:611–612. doi: 10.1038/nrneph.2009.168. [DOI] [PubMed] [Google Scholar]
- 6.Limon JJ, Fruman DA. Akt and mTOR in B Cell Activation and Differentiation. Front Immunol. 2012;3:228. doi: 10.3389/fimmu.2012.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Page AJ, Ford ML, Kirk AD. Memory T-cell-specific therapeutics in organ transplantation. Curr Opin Organ Transplant. 2009;14:643–649. doi: 10.1097/MOT.0b013e328332bd4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh K, Kozyr N, Stempora L, Kirk AD, Larsen CP, Blazar BR, Kean LS. Regulatory T Cells Exhibit Decreased Proliferation but Enhanced Suppression After Pulsing with Sirolimus. Am J Transplant. 2012 doi: 10.1111/j.1600-6143.2011.03963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang S, Readinger JA, DuBois W, Janka-Junttila M, Robinson R, Pruitt M, Bliskovsky V, Wu JZ, Sakakibara K, Patel J, Parent CA, Tessarollo L, Schwartzberg PL, Mock BA. Constitutive reductions in mTOR alter cell size, immune cell development, and antibody production. Blood. 2011;117:1228–1238. doi: 10.1182/blood-2010-05-287821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu L, Qian XF, Rao JH, Wang XH, Zheng SG, Zhang F. Rapamycin promotes the expansion of CD4(+) Foxp3 (+) regulatory T cells after liver transplantation. Transpl Proc. 2010;42:1755–1757. doi: 10.1016/j.transproceed.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 11.Fortun J, Martin-Davila P, Pascual J, Cervera C, Moreno A, Gavalda J, Aguado JM, Pereira P, Gurgui M, Carratala J, Fogueda M, Montejo M, Blasco F, Bou G, Torre-Cisneros J. Immunosuppressive therapy and infection after kidney transplantation. Transpl Infect Dis. 2010;12:397–405. doi: 10.1111/j.1399-3062.2010.00526.x. [DOI] [PubMed] [Google Scholar]
- 12.Kulkarni S, Powles R, Treleaven J, Riley U, Singhal S, Horton C, Sirohi B, Bhagwati N, Meller S, Saso R, Mehta J. Chronic graft versus host disease is associated with long-term risk for pneumococcal infections in recipients of bone marrow transplants. Blood. 2000;95:3683–3686. [PubMed] [Google Scholar]
- 13.Reich G, Mapara MY, Reichardt P, Dorken B, Maschmeyer G. Infectious complications after high-dose chemotherapy and autologous stem cell transplantation: comparison between patients with lymphoma or multiple myeloma and patients with solid tumors. Bone Marrow Transplant. 2001;27:525–529. doi: 10.1038/sj.bmt.1702822. [DOI] [PubMed] [Google Scholar]
- 14.Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control systems and decision making for antibody production. Nat Immunol. 2010;11:681–688. doi: 10.1038/ni.1900. [DOI] [PubMed] [Google Scholar]
- 15.Moser K, Wong DM, Manz RA. Antibody memory: a question of life or death beyond the germinal center. Immunol Cell Biol. 2011;89:164–166. doi: 10.1038/icb.2010.159. [DOI] [PubMed] [Google Scholar]
- 16.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longerich S, Basu U, Alt F, Storb U. AID in somatic hypermutation and class switch recombination. Curr Opin Immunol. 2006;18:164–174. doi: 10.1016/j.coi.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Maul RW, Gearhart PJ. AID and somatic hypermutation. Adv Immunol. 2010;105:159–191. doi: 10.1016/S0065-2776(10)05006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 20.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 21.Li Z, Woo CJ, Iglesias-Ussel MD, Ronai D, Scharff MD. The generation of antibody diversity through somatic hypermutation and class switch recombination. Genes Dev. 2004;18:1–11. doi: 10.1101/gad.1161904. [DOI] [PubMed] [Google Scholar]
- 22.Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF, Scharff MD. The biochemistry of somatic hypermutation. Annu Rev Immunol. 2008;26:481–511. doi: 10.1146/annurev.immunol.26.021607.090236. [DOI] [PubMed] [Google Scholar]
- 23.Kracker S, Durandy A. Insights into the B cell specific process of immunoglobulin class switch recombination. Immunol Lett. 2011;138:97–103. doi: 10.1016/j.imlet.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 24.Pan-Hammarstrom Q, Zhao Y, Hammarstrom L. Class switch recombination: a comparison between mouse and human. Adv Immunol. 2007;93:1–61. doi: 10.1016/S0065-2776(06)93001-6. [DOI] [PubMed] [Google Scholar]
- 25.Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasbold J, Corcoran LM, Tarlinton DM, Tangye SG, Hodgkin PD. Evidence from the generation of immunoglobulin G-secreting cells that stochastic mechanisms regulate lymphocyte differentiation. Nat Immunol. 2004;5:55–63. doi: 10.1038/ni1016. [DOI] [PubMed] [Google Scholar]
- 27.Rush JS, Liu M, Odegard VH, Unniraman S, Schatz DG. Expression of activation-induced cytidine deaminase is regulated by cell division, providing a mechanistic basis for division-linked class switch recombination. Proc Natl Acad Sci U S A. 2005;102:13242–13247. doi: 10.1073/pnas.0502779102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kovalchuk AL, duBois W, Mushinski E, McNeil NE, Hirt C, Qi CF, Li Z, Janz S, Honjo T, Muramatsu M, Ried T, Behrens T, Potter M. AID-deficient Bcl-xL transgenic mice develop delayed atypical plasma cell tumors with unusual Ig/Myc chromosomal rearrangements. J Exp Med. 2007;204:2989–3001. doi: 10.1084/jem.20070882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snapper CM. Differential regulation of protein- and polysaccharide-specific Ig isotype production in vivo in response to intact Streptococcus pneumoniae. Curr Protein Pept Sci. 2006;7:295–305. doi: 10.2174/138920306778017972. [DOI] [PubMed] [Google Scholar]
- 30.Polack FP, Hoffman SJ, Crujeiras G, Griffin DE. A role for nonprotective complement-fixing antibodies with low avidity for measles virus in atypical measles. Nat Med. 2003;9:1209–1213. doi: 10.1038/nm918. [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto M, Nojima T, Hayashi K, Goitsuka R, Furukawa K, Azuma T, Kitamura D. BASH-deficient mice: limited primary repertoire and antibody formation, but sufficient affinity maturation and memory B cell generation, in anti-NP response. Int Immunol. 2004;16:1161–1171. doi: 10.1093/intimm/dxh116. [DOI] [PubMed] [Google Scholar]
- 32.Maccarthy T, Roa S, Scharff MD, Bergman A. SHMTool: a webserver for comparative analysis of somatic hypermutation datasets. DNA Repair (Amst) 2009;8:137–141. doi: 10.1016/j.dnarep.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell. 2004;16:163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 34.Kitamura T, Koshino Y, Shibata F, Oki T, Nakajima H, Nosaka T, Kumagai H. Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp Hematol. 2003;31:1007–1014. [PubMed] [Google Scholar]
- 35.Delgado MF, Coviello S, Monsalvo AC, Melendi GA, Hernandez JZ, Batalle JP, Diaz L, Trento A, Chang HY, Mitzner W, Ravetch J, Melero JA, Irusta PM, Polack FP. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat Med. 2009;15:34–41. doi: 10.1038/nm.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colino J, Snapper CM. Dendritic cell-derived exosomes express a Streptococcus pneumoniae capsular polysaccharide type 14 cross-reactive antigen that induces protective immunoglobulin responses against pneumococcal infection in mice. Infect Immun. 2007;75:220–230. doi: 10.1128/IAI.01217-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrader CE, Bradley SP, Vardo J, Mochegova SN, Flanagan E, Stavnezer J. Mutations occur in the Ig Smu region but rarely in Sgamma regions prior to class switch recombination. EMBO J. 2003;22:5893–5903. doi: 10.1093/emboj/cdg550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dunnick W, Hertz GZ, Scappino L, Gritzmacher C. DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Res. 1993;21:365–372. doi: 10.1093/nar/21.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, Redon C, Ried T, Bonner WM, Honjo T, Nussenzweig MC, Nussenzweig A. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagaoka H, Muramatsu M, Yamamura N, Kinoshita K, Honjo T. Activation-induced deaminase (AID)-directed hypermutation in the immunoglobulin Smu region: implication of AID involvement in a common step of class switch recombination and somatic hypermutation. J Exp Med. 2002;195:529–534. doi: 10.1084/jem.20012144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takizawa M, Tolarova H, Li Z, Dubois W, Lim S, Callen E, Franco S, Mosaico M, Feigenbaum L, Alt FW, Nussenzweig A, Potter M, Casellas R. AID expression levels determine the extent of cMyc oncogenic translocations and the incidence of B cell tumor development. J Exp Med. 2008;205:1949–1957. doi: 10.1084/jem.20081007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janes MR, Fruman DA. Immune regulation by rapamycin: moving beyond T cells. Sci Signal. 2009;2:pe25. doi: 10.1126/scisignal.267pe25. [DOI] [PubMed] [Google Scholar]
- 43.Baracho GV, Miletic AV, Omori SA, Cato MH, Rickert RC. Emergence of the PI3-kinase pathway as a central modulator of normal and aberrant B cell differentiation. Curr Opin Immunol. 2011;23:178–183. doi: 10.1016/j.coi.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Orthwein A, Patenaude AM, Affarel B, Lamarre A, Young JC, Di Noia JM. Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J Exp Med. 2010;207:2751–2765. doi: 10.1084/jem.20101321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dengler HS, Baracho GV, Omori SA, Bruckner S, Arden KC, Castrillon DH, DePinho RA, Rickert RC. Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nat Immunol. 2008;9:1388–1398. doi: 10.1038/ni.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Omori SA, Cato MH, Anzelon-Mills A, Puri KD, Shapiro-Shelef M, Calame K, Rickert RC. Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity. 2006;25:545–557. doi: 10.1016/j.immuni.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 47.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 48.Sernandez IV, V, de Yebenes G, Dorsett Y, Ramiro AR. Haploinsufficiency of activation-induced deaminase for antibody diversification and chromosome translocations both in vitro and in vivo. PLoS One. 2008;3:e3927. doi: 10.1371/journal.pone.0003927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fagarasan S, Muramatsu M, Suzuki K, Nagaoka H, Hiai H, Honjo T. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science. 2002;298:1424–1427. doi: 10.1126/science.1077336. [DOI] [PubMed] [Google Scholar]
- 50.Meerveld-Eggink A, van der Velden AM, Ossenkoppele GJ, van de Loosdrecht AA, Biesma DH, Rijkers GT. Antibody response to polysaccharide conjugate vaccines after nonmyeloablative allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2009;15:1523–1530. doi: 10.1016/j.bbmt.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 51.Willcocks LC, Chaudhry AN, Smith JC, Ojha S, Doffinger R, Watson CJ, Smith KG. The effect of sirolimus therapy on vaccine responses in transplant recipients. Am J Transplant. 2007;7:2006–2011. doi: 10.1111/j.1600-6143.2007.01869.x. [DOI] [PubMed] [Google Scholar]
- 52.Albesiano E, Messmer BT, Damle RN, Allen SL, Rai KR, Chiorazzi N. Activation-induced cytidine deaminase in chronic lymphocytic leukemia B cells: expression as multiple forms in a dynamic, variably sized fraction of the clone. Blood. 2003;102:3333–3339. doi: 10.1182/blood-2003-05-1585. [DOI] [PubMed] [Google Scholar]
- 53.Endo Y, Marusawa H, Kou T, Nakase H, Fujii S, Fujimori T, Kinoshita K, Honjo T, Chiba T. Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology. 2008;135:889–898. 898 e881–883. doi: 10.1053/j.gastro.2008.06.091. [DOI] [PubMed] [Google Scholar]
- 54.Greeve J, Philipsen A, Krause K, Klapper W, Heidorn K, Castle BE, Janda J, Marcu KB, Parwaresch R. Expression of activation-induced cytidine deaminase in human B-cell non-Hodgkin lymphomas. Blood. 2003;101:3574–3580. doi: 10.1182/blood-2002-08-2424. [DOI] [PubMed] [Google Scholar]
- 55.Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, Honjo T, Nussenzweig A, Nussenzweig MC. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004;118:431–438. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 56.Klemm L, Duy C, Iacobucci I, Kuchen S, von Levetzow G, Feldhahn N, Henke N, Li Z, Hoffmann TK, Kim YM, Hofmann WK, Jumaa H, Groffen J, Heisterkamp N, Martinelli G, Lieber MR, Casellas R, Muschen M. The B cell mutator AID promotes B lymphoid blast crisis and drug resistance in chronic myeloid leukemia. Cancer Cell. 2009;16:232–245. doi: 10.1016/j.ccr.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.