

Abstract
Stress reduces fertility, but the mechanisms mediating this are not understood. For a successful pregnancy, placental trophoblast stem cells (TSCs) in the implanting embryo proliferate and then a subpopulation differentiates to produce hormones. Normally, differentiation occurs when inhibitor of differentiation 2 (ID2) protein is lost in human and mouse placental stem cells. We hypothesize that stress enzyme-dependent differentiation occurs in association with insufficient TSC accumulation. We studied a well-defined model where TSC differentiation requires ID2 loss. The loss of ID2 derepresses the promoter of chorionic somatomammotropin hormone 1 (CSH1), the first hormone after implantation. Csh1 mRNA is known to be induced in stressed TSCs. In this study, we demonstrate that AMP-activated protein kinase (PRKAA1/2, aka AMPK) mediates the stress-induced proteasome-dependent loss of ID2 at high stress levels. At very low stress levels, PRKAA1/2 mediates metabolic adaptation exemplified by the inactivation of acetyl coA carboxylase by phosphorylation without ID2 loss. At the highest stress levels, irreversible TSC differentiation as defined by ID2 loss and slower cell accumulation occurs. However, lower stress levels lead to reversible differentiation accompanied by metabolic adaptation. These data support the hypothesis that PRKAA1/2 mediates preparation for differentiation that is induced by stress at levels where a significant decrease in cell accumulation occurs. This supports the interpretation that enzyme-mediated increases in differentiation may compensate when insufficient numbers of stem cells accumulate.
Introduction
The most problematic period in mammalian development is that of embryonic implantation into the uterus, when most of the (70%) loss of human embryos occurs (Cross et al. 1994). The preimplantation blastocyst is composed of stem cells that are determined, but not yet terminally differentiated for parenchymal functions. The implanting mouse and human embryo must begin differentiated functions that include invasion to contact the maternal blood supply and production of essential placental hormones (Roberts et al. 1996, Cross et al. 2002).
The genetic program for mediating trophoblast stem cell (TSC) differentiation to hormone-secreting trophoblast giant cells (TGCs) has recently come into view. Different transcription factor subsets must be upregulated and downregulated to mediate differentiation that produces the first mouse placental hormone, chorionic somatomammotropin hormone 1 (CSH1). A primary driver of differentiation is the upregulation of tissue-restricted basic helix loop helix transcription factors such as heart and neural crest derivatives expressed 1 (HAND1), which directly interacts with the CSH1 promoter and activates it (Cross et al. 2002). This interaction can occur only after the loss of inhibitor of differentiation 2 (ID2), the dominant negative inhibitor of HAND1/TCF2A dimerization (Cross et al. 2002). In mouse TSC differentiation, ID2 is lost; HAND1 binds and activates the CSH1 promoter. Similarly in humans, cytotrophoblast stem cell differentiation to an invasive phenotype requires the loss of ID2 (Janatpour et al. 2000).
In the mouse, CSH1 is produced by three related genes (AKA, Prl3d1/2/3; Wiemers et al. 2003). CSH1 is a part of the maternal recognition of pregnancy secretory phenotype, synthesized by some TGC sub-lineages. CSH1 first appears in maternal blood within 2 days of implantation (Colosi et al. 1987, 1988, Ogren et al. 1989). ID2 is present during preimplantation development (Cross et al. 1995), and presumably the loss of ID2 and gain of HAND1 drives mouse placental differentiation. This occurs when a small subpopulation of TSCs differentiates into parietal TGC, leaving a large stem cell population prepared for later differentiation into several other differentiated placental lineages (Simmons et al. 2007).
Developmentally relevant stimuli in vitro and in vivo are necessary and sufficient to mediate normal, early TSC differentiation to parietal TGC. These include leukemia inhibitory factor (LIF; Takahashi et al. 2003), retinoic acid (RA; Yan et al. 2001), the nonphysiological ligand diethylstilbestrol, which drives differentiation by blocking estrogen-related receptor-β (Tremblay et al. 2001), and fibroblast growth factor 4 (FGF4) removal (Tanaka et al. 1998). For a review, see Cross et al. (2002). FGF4 removal in vitro or migration away or loss of contact with FGF4 sources in vivo is associated with normal TSC differentiation (Chai et al. 1998, Rassoulzadegan et al. 2000, Uy et al. 2002).
Enzymes mediating cellular stress such as PRKAA1/2 (also known as protein kinase, AMP-activated, α1/2 catalytic subunit) have not been studied in preimplantation embryos, but are required for hormonal and stress-induced oocyte maturation (LaRosa & Downs 2006, Chen & Downs 2008) and are expressed during the preimplantation development (Wang et al. 2004a). PRKAA1/2 acts as an energy sensor, sensing an increased AMP/ATP ratio, and is known to regulate substrates that mediate metabolic activity, such as phosphorylation of acetyl coA carboxylase (ACACA, aka ACC) at Ser79 (Hardie & Hawley 2001). This inactivates fatty acid synthesis and favors the shift to ATP production through catabolism of carbon not used in anabolic processes. PRKAA1/2 has been implicated in the benzopyrene-induced ID2 loss in TSCs (Xie et al. 2010) and we hypothesize that this enzyme is a general sensor of stress in TSCs.
Hyperosmolar stress that activates the maximal levels of stress-activated protein kinase/jun kinase (MAPK9/MAPK8/9) causes TSC to differentiate into Hand1/Stra13/Csh1 mRNA-positive parietal TGCs, while suppressing GCM1 and TPBPA-positive chorionic/syncytiotrophoblasts and spongiotrophoblasts respectively (Liu et al. 2009). A similar finding was reported for Map3k4 null mutant stem cells and wild-type TSCs that required MAPK8/9 to suppress GCM1 and TPBPA (Abell et al. 2009). In the hyperosmolar cellular stress model, the production of de novo Csh1 mRNA would require the loss of ID2 protein; however, Id2 mRNA was not decreased during 24 h of hyperosmolar stress (Liu et al. 2009).
In this study, we show that ID2 protein loss is induced by hyperosmolar cellular stress in a proteasome-dependent manner. The ID2 loss was reversible at lower stress levels, but at higher stress levels ID2 loss was irreversible. In addition, PRKAA1/2-dependent metabolic substrates were regulated at low stress levels where ID2 was not regulated, but ID2 was regulated at higher levels of stress that also significantly decreased TSC accumulation.
Results
Stress induces loss of ID2 that is dependent on PRKAA1/2 and proteasome function, but is not dependent on MAPK1/2/5 or MAPK8/9
MAPK8/9 regulates many aspects of the stress response of preimplantation embryos and their derived TSC (reviewed in Rappolee (2007) and Rappolee et al. (2010a, 2010b)), so we anticipated that MAPK8/9 would also regulate ID2. Hyperosmolar stress induces TSC differentiation (Liu et al. 2009) that requires ID2 loss in humans and mice (Cross et al. 1995, Janatpour et al. 2000), and thus we tested for stress-induced loss of ID2 in mouse TSCs, and whether this was MAPK8/9 dependent. Since previous studies had established that 400 mM sorbitol induced highest MAPK8/9 activity and function in TSCs and embryos (Xie et al. 2007, 2008, Zhong et al. 2007) we began our studies in mouse TSC using 400 mM sorbitol and we used levels of MAPK8/9 and MAPK1/2/5 inhibitors we previously determined to have highest inhibition of enzyme phosphorylation at the lowest level of inhibitor (Wang et al. 2004b, Xie et al. 2006a, 2007, Zhong et al. 2007; data not shown). Stress induced an 80% loss of ID2 in TSC that was not MAPK8/9 dependent (sensitive to MAPK8/9 inhibitor DJNKl1, Fig. 1A) nor was it MAPK1/3 dependent (sensitive to MAPK2K1/2/5 inhibitor U0126, Supplementary Figure 1, see section on supplementary data given at the end of this article).
Figure 1.

Effect of hyperosmolar stress on ID2 protein is independent of MAPK8/9 in TSC, but dependent on PRKAA1/2 in TSC and E3.5 mouse blastocysts. (A) TSCs were cultured in 0 or 400 mM sorbitol with or without the MAPK8/9 inhibitor DJNKI1 (1 μM) for 20 h. Asterisks indicate that the MAPK8/9 inhibitor LJNKL1 has no significant effect on ID2 levels. (B) TSCs were cultured in 0 or 400 mM sorbitol without or with compound C at 1 μM (inhibitor of PRKAA1/2) or SB203580 at 5 μM (inhibitor of MAPK11/14) for 20 h. (C) Embryos were cultured in 0 or 400 mM sorbitol without or with compound C for 2 h. (D) TSCs were incubated with 2 mM AICAR for 0–2 h or with 400 mM for 2 h. (E) TSCs were incubated for 2 h with or without 200 mM sorbitol, with or without 10 μM of the proteasome inhibitor MG132. After treatments, equal amounts of protein were examined with western blots using ID2 and ACTB antibody or amido black. Histograms show the ratio of ID2/ACTB or ID2/amido black band intensity. Error flags are S.D.s for triplicate experiments for TSC or duplicate experiments for embryos.
There are 510 protein kinases shared in the human and mouse kinomes with 8 homologues specific to humans and 25 specific to mice (Caenepeel et al. 2004). However, a small subset of protein kinases in the kinome typically respond to stress stimuli and are not isolated in focus-forming assays, suggesting that these are good candidates for mediating responses to hyperosmolar stress (Rappolee et al. 2010b). We assayed several kinases that typically respond to stress, and identified PRKAA1/2 as the enzyme that mediates stress-induced ID2 loss in mouse TSC as 90% of ID2 loss was reversed by PRKAA1/2 inhibitors compound C or AraA in TSC or the stage of embryo from which TSCs were derived (Fig. 1B and C, data not shown). The levels of PRKAA1/2 inhibitors and agonists used were previously established to have the maximal effect at the lowest possible dose (Xie et al. 2010; data not shown). Band doublets at 13 and 16 kDa were previously reported for ID2 in human cytotrophoblasts (Janatpour et al. 2000) and were detected here in mouse TSC. However, in the E3.5 mouse blastocysts only the 13 kDa was observed. Since the ID2-to-ACTB density ratios for TSCs and embryos were similar this suggests that the missing band is not due to insufficient protein loading, but is due to some intrinsic difference between ID2 regulation in TSCs and embryos. The PRKAA1/2 agonist 5-amino-4-imidazole-carboxamide riboside (AICAR) activates PRKAA1/2 Thr172 (Fig. 1D) and induces ID2 loss to about 50% of baseline ID2; however, the amount of loss induced by AICAR was not as great as the 70–90% loss induced by 400 mM sorbitol in the same experiments (Fig. 1D, data not shown). The dose of AICAR used was previously shown to produce maximal AMPK activity in mouse TSC (data not shown). PRKAA1/2 Thr172 phosphorylation is the key event necessary for, and indicative of, enzyme activity (Stein et al. 2000). ID2 loss in mouse TSC was reversed 92% by proteasome inhibitors (MG132 or lactacystin; Fig. 1E; Supplementary Figure 2, see section on supplementary data given at the end of this article). The doses of MG132 and lactacystin were established to have maximal effect on stress-induced ID2 loss at the lowest dose possible (data not shown). Thus, stress-induced ID2 protein loss was largely dependent on PRKAA1/2 and almost entirely dependent on proteasome function.
We knew that hyperosmolar stress induced Csh1 mRNA (Liu et al. 2009) and that Csh1 promoter activation required the loss of ID2 protein. Thus, we hypothesized that PRKAA1/2 would have a kinetic response commensurate with its role in mediating stress-induced loss of ID2.
We found that ID2 loss and PRKAA1/2 Thr172 phosphorylation showed interesting kinetics in response to 200 mM sorbitol (Fig. 2A–D). Much of the earlier work on hyperosmolar stress in mouse TSCs and embryos suggested that 400 mM sorbitol not only produced maximal enzyme activation and homeostatic and developmental effects, but also created high levels of morbidity. In other experiments, we showed that 200 mM sorbitol had very robust effects in mediating differentiation but with much less morbidity (data not shown). Thus, we have performed some of the studies here using 200 mM sorbitol. In E3.5 embryos, TSCs, and embryonic stem cells (ESCs), peak induction of PRKAA1/2 Thr172 occurred at 10 min and subsided by 30–120 min (Fig. 2A, C, and E). There was a twofold increase in PRKAA1/2 Thr172 in E3.5 embryos and TSCs and a sixfold peak induction in ESC. ID2 loss displayed kinetics in embryos and TSCs complementary to the PRKAA1/2 Thr172 kinetics. ID2 loss was well underway by the 10 min peak for PRKAA1/2, and ID2 loss was maximal by the time PRKAA1/2 Thr172 returned to baseline. ID2 loss did not continue after PRKAA1/2 Thr172 had returned to the baseline. Thus, the rapid kinetics of PRKAA1/2 induction and return to the baseline and the simultaneous baseline of maximal loss for ID2 were consistent with the PRKAA1/2 dependence of ID2 loss.
Figure 2.

Rapid transient activation of phosphorylated PRKAA1/2 Thr172 in TSC, ESC, and embryos occurs simultaneously with rapid loss of ID2 protein in TSCs and embryos. Embryos (A and B), TSCs (C and D), and ESC (E) were cultured under optimal conditions, sorbitol was added for 0–90/120 min, and cells and embryos were lysed, proteins fractionated by SDS–PAGE and immunoblotted for PRKAA1/2 Thr172 or ID2 and reprobed for actin (ACTB) or amido black as a loading control. Histograms below each immunoblot show the PRKAA1/2 Thr172 normalized to the loading control and this ratio is set to 1 for unstressed cells/embryos. Y error bars show S.D. of triplicate experiments in TSC and ESC or duplicate experiments in embryos.
We hypothesized that significant decreases in TSC accumulation and induction of molecular mechanisms mediating differentiation would occur at similar hyperosmolar stress dose thresholds as had previously been noted for benzopyrene stress (Xie et al. 2010). At 2 h of stress, ID2 had reached a low baseline (Fig. 2), and at this time point significant dose-dependent ID2 loss was occurring at high sorbitol doses ≥200 mM sorbitol. Significant decreases in cell accumulation occurred at high doses at ≥100 mM sorbitol (Fig. 3A and B; P values ≤ 0.05, ANOVA, and Duncan’s post hoc test). Both ID2 loss and PRKAA1/2 activation were ended by 2 h. In contrast to the developmental substrate ID2, significant phosphorylation of the homeostatic substrate ACACA Ser79 occurred at very low stress doses, 12.5–100 mM (Fig. 3B; P values ≤0.05, ANOVA, and Duncan’s post hoc test). Similar to ID2 loss, ACACA phosphorylation is PRKAA1/2 dependent (Supplementary Figure 3, see section on supplementary data given at the end of this article). Thus, PRKAA1/2 activation occurs over a broad range of doses (Fig. 3), resulting in a broad dose range of early phosphorylation of homeostatic substrate ACACA. By 2 h phosphorylation of PRKAA1/2 and ACACA has decreased to the baseline at high doses. However, persistent loss of ID2 occurs by 2–24 h and is longer only at high doses ≥200 mM sorbitol.
Figure 3.

Hyperosmolar stress activates PRKAA1/2 over a wide dose range, but ACACA Ser79 is phosphorylated at low doses and ID2 loss and decrease in TSC accumulation occur at high doses. (A) TSCs were plated and cultured for 24 h in media without sorbitol and with 12.5, 25, 50, and 100 mM sorbitol and then counted. (B) TSCs were cultured with the indicated doses of sorbitol for 2 h, fractionated by SDS–PAGE, and immunoblotted and stained for antigens using polyclonal antibodies. All experiments were repeated three times and histograms show X±S.E.M. Superscript ‘a’ indicates significant changes for 0 mM sorbitol stress for cell counts and proteins, and superscript ‘b’ indicates no significant difference for the same unstressed TSCs. Significance was determined by ANOVA followed by Duncan’s post hoc tests. For proteins, the earliest unstressed time point shown was set to 1 and served as a denominator to normalize the experimental values from stressed TSCs.
ID2 loss depended on the developmental stage of the TSC as well as the dose of sorbitol. In unstressed cells, a small subpopulation of (1–5%, data not shown) TSC underwent default differentiation, lost ID2, and increased the nuclear size through endoreduplication (Tanaka et al. 1998). Similarly, default differentiation was observed in a few cells with larger nuclei and less ID2 as in Fig. 4A. After 30 h, 400 mM sorbitol caused a total loss of ID2 in cells with small and medium-sized nuclei (Fig. 4E), but 100 mM sorbitol spared ID2 loss in cells with small nuclei, while causing loss of ID2 in other cells with medium-sized nuclei (Fig. 4C). Thus, 100 mM sorbitol affected ID2 loss in cells that may have started differentiation, but a 400 mM dose is dominant, causing ID2 loss even in cells with small nuclei that are the least differentiated TSCs.
Figure 4.

Hyperosmolar stress leads to destruction of ID2 protein in nearly all cells at high doses (400 mM), but lower doses (100 mM) spared ID2 in TSCs with small nuclei. TSCs were cultured in 0 mM (A and B), 100 mM (C and D), or 400 mM (E and F) sorbitol for 30 h and then fixed and stained for ID2 using indirect immunocytochemistry. In (G and H), 400 mM sorbitol-treated TSCs were probed without primary antibody; (B, D, F, and H) are the Hoechst-stained nuclei corresponding to cells in (A, C, E, and G) respectively. The orange arrow and arrowhead show the position of the protein corresponding to its nucleus of a giant cell (>2000 arbitrary units), red arrows and arrowheads show the protein and medium-sized nuclei (1000–2000 arbitrary units), and purple arrows and arrowheads show the protein and small TSC nuclei (100–999 arbitrary units). Histogram shows the mean intensity of ID2/intensity of Hoechst to normalize depth of nuclei, Y error flags are S.D., and numbers in bars show number of nuclei counted. Statistical comparisons show, in unstressed TSCs, the decrease in ID2 intensity from small to medium (a) and medium to giant nuclei (b) are each significant (P<0.005). The decreases in ID2 intensity in medium-sized nuclei from unstressed to 100 mM (c) and in small nuclei from unstressed or 100–400 mM (d) are significant (P<0.001). This is a representative experiment from three similar experiments.
Interestingly, although MAPK11/14 inhibitors partially blocked stress-induced ID2 loss at 400 mM sorbitol (Fig. 1B), active MAPK11/14 inhibitors caused ID2 loss in TSC at 100 mM sorbitol whereas inactive inhibitor analogues had no effect (Supplementary Figure 4, see section on supplementary data given at the end of this article).
Stress induces reversible ID2 loss at 100–200 mM sorbitol, but ID2 loss was irreversible at 400 mM sorbitol
The residual high levels of Id2 mRNA after 24 h of hyperosmolar stress suggested that ID2 protein might be re-synthesized if stress were to subside. After 24 h, 400 mM sorbitol was removed or continued for another 24 h. ID2 was 63% lower and CSH1 89% higher after the removal of sorbitol than they had been 24 h earlier (Fig. 5A and B), suggesting that differentiation did not reverse at high stress level.
Figure 5.
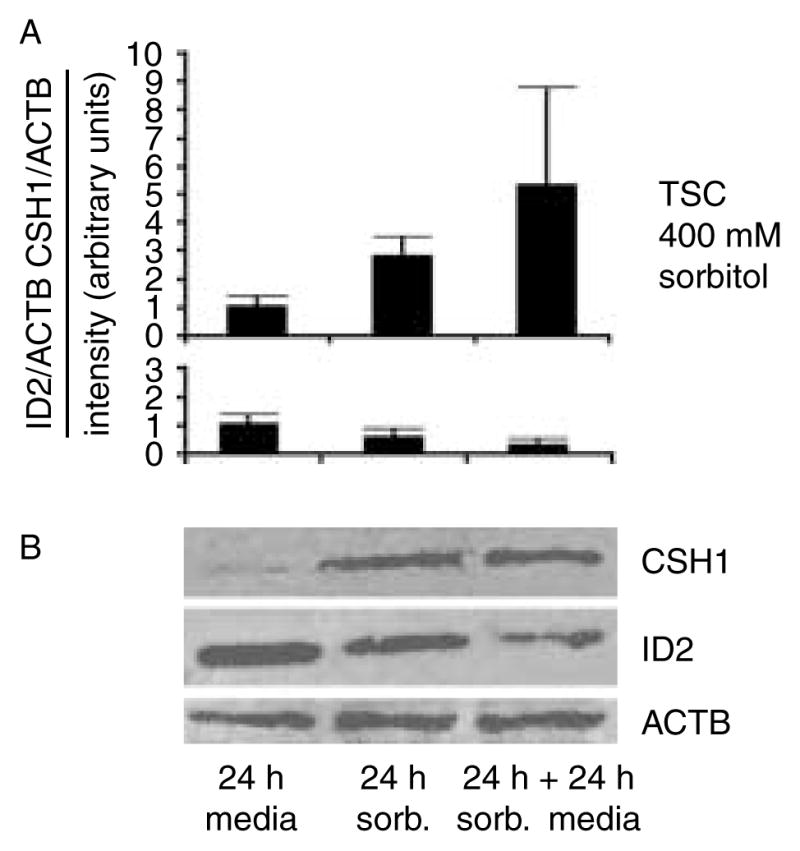
CSH1 increases and ID2 decreases in 24 h after 400 mM sorbitol is removed from cultured TSCs. (A) TSCs were cultured for 24 h without or with 400 mM sorbitol, or cultured with sorbitol for 24 h followed by 24 h without sorbitol, cells were lysed, proteins were fractionated by SDS–PAGE and immunoblotted for CSH1, ID2, and reprobed for actin (ACTB) as a loading control. (B) Histograms below each immunoblot show either CSH1 or ID2 normalized to the loading control and this ratio is set to 1 for unstressed cells/embryos. Y error bars show S.D. of triplicate experiments.
After an initial 24 h, 100–200 mM sorbitol was removed or continued for another 24 h. For 100 and 200 mM sorbitol, cell accumulation rates increased 100% after the removal of sorbitol (Fig. 6A), and ID2 protein increased 70% (Fig. 6B and C) compared with cells that continued with sorbitol. Thus, stress induces a reversible state of differentiation at lower doses of sorbitol, but at a higher dose commitment to differentiation does not reverse.
Figure 6.

Cell accumulation rates and ID2 increase 24 h after 100 or 200 mM sorbitol is removed from cultured TSCs. (A) TSCs were cultured without sorbitol or with 100 or 200 mM sorbitol for 24 h, and then sorbitol was removed or continued. TSCs were counted using Trypan blue at 0, 24, and 48 h. The constant stress groups were designated by unchanged color for 0–48 h at 100 mM (yellow) and 200 mM (light purple). The blue line represents a removal of 200 mM sorbitol in 24–48 h and the dark purple line represents a removal of 100 mM sorbitol in 24–48 h. (B) TSCs were cultured with or without 100 or 200 mM sorbitol for 24 h, then sorbitol was removed or continued, and cell lysates were fractionated by SDS–PAGE and probed for ID2 and reprobed for ACTB. (C) Histogram shows the ratio of ID2/ACTB band intensity. Error flags are S.D.s for triplicate experiments. In (A) and (C), arrows show the comparison of 48 h with sorbitol and 24 h of sorbitol followed by 24 h without it. In (A), * and ** show the significant (P<0.01 and P<0.001 respectively) increase in cell number when 200 and 100 mM sorbitol were removed for the second 24 h. The top ** indicate a significant decrease in cell number from 0 to 100 mM sorbitol. In (C), * and ** show the significance (P<0.01 and P<0.001 respectively) when 200 or 100 mM sorbitol was removed for the second 24 h.
Discussion
Summary
After implantation, survival of the conceptus requires differentiation of a TSC subpopulation to produce a set of endocrine hormones exemplified by CSH1. Cellular (hyperosmolar) stress is sufficient to induce Csh1 mRNA (Liu et al. 2009), the first placental hormone detected in maternal blood within 36 h after implantation (Ogren et al. 1989) and a hallmark of normal differentiation. However, without the activity of PRKAA1/2 or proteasomes, stressed TSCs are largely unable to destroy ID2 protein (Fig. 1), an event necessary to produce CSH1 (Cross et al. 2002).
We recently reported that benzopyrene induces ID2 protein loss at higher levels of benzopyrene-mediated stress that also significantly reduce cell TSC accumulation rates (Xie et al. 2010). In the benzopyrene stress study, there was a substantial lower-dose range where PRKAA1 phosphorylation and activation occurred but ID2 was not significantly decreased. Thus, benzopyrene and hyperosmolar cellular stress share a low-dose range where ID2 is not affected and cell accumulation rates are not affected and a high-dose range where cell accumulation is affected and ID2 is lost in an AMPK-dependent way.
We used hyperosmolar stress because it is a standard stressor used in adult somatic cells, and because it quickly and robustly induces stress responses in all cellular, oocyte, and embryo models (Rappolee 2007, Rappolee et al. 2010b). Hyperosmolar stress has also been used to study sperm function (McCarthy et al. 2010), oocyte function (LaRosa & Downs 2006), and preimplantation embryo function by other laboratories (Steeves & Baltz 2005, Bell et al. 2009). We have previously shown that cellular stress leads to slower first trimester HTR/SV40neo placental cell accumulation through decreased BrdU incorporation into DNA, decreased fraction of cells >2N and <4N, and increased apoptosis of HTR cells and embryos largely through MAPK8/9 activity (Xie et al. 2006a, 2007, Zhong et al. 2007). In addition, ID2 loss and cell accumulation decreases occur at similar high threshold doses of stress, whereas PRKAA1/2-dependent ACACA phosphorylation occurs at lower doses below this threshold. It is interesting to note that at low doses of benzopyrene where AMPK was induced and ID2 was not affected, ACACA was also not phosphorylated. We anticipate that AMPK function at low benzopyrene doses will include some homeostatic function, but it is clear that sorbitol and benzopyrene stress are not similar to the types of homeostatic mechanisms that are induced in the low-dose range.
ID2 protein loss and Csh1 mRNA induction is not a unique exemplar of stress-induced TSC differentiation as stress induces a global mRNA response including many genes that mediate differentiation (Liu et al. 2009). Stress induces genes for other placental hormones such as proliferin and prolactin-like protein-M (Wiemers et al. 2003), and other differentiation-mediating transcription factors such as stimulated by RA (STRA)13, Gata-binding (GATA)2, and hairy/enhancer of split 1 (Cross et al. 2002). Thus, differentiation is a global response of stressed TSCs, not an anomaly of the induction of ID2 loss tested in this study.
Enzyme kinetics is associated with the role PRKAA1/2
One surprise was that stress-induced MAPK8/9 was not necessary for and had no role in stress-induced ID2 loss. We sought to understand whether enzyme–substrate kinetics might be a reason for the function of PRKAA1/2 in stress-induced ID2 loss. The kinetics of ID2 loss and PRKAA1/2 are complementary. The rapid PRKAA1/2 Thr172 activity peak and return to baseline fit the kinetics of ID2 loss in embryos and TSCs. However, the short activation period of PRKAA1/2 would not be sufficient for long-term maintenance of transcription factors such as HAND1 that are needed for induction and maintenance of the differentiated state and Csh1 promoter activation (Cross et al. 2002). We hypothesize that stress enzymes with long activation periods such as MAPK8/9 maintain transcription factors such as HAND1 that maintain differentiation (data not shown). Conversely, the loss of transcription factors blocking differentiation may be mediated by fast-acting enzymes such as PRKAA1/2. These hypotheses are currently being tested and preliminary evidence supports an association of substrates that fit the longevity of enzyme activity.
Hyperosmolar stress-induced ID2 loss is proteasome dependent, but AICAR-induced PRKAA1/2 is not sufficient for high levels of ID2 loss
Stress-induced ID2 loss requires proteasome activity. Proteasome dependence suggests that ID2 loss is not an artifact of ID2 epitope change and loss of detection by antibodies. High levels of PRKAA1/2 activation by AICAR without stress are not sufficient for the magnitude of rapid, stress-induced loss of ID2 approaching levels induced by high levels of sorbitol. This suggests that additional pathways required for higher levels of ID2 loss are activated by sorbitol, which are not activated by the PRKAA1/2 agonist AICAR or mediated by PRKAA1/2 alone.
How might this lead to diseases of placental insufficiency? If sufficient high doses of stress lasted long enough, then irreversible differentiation of TSC would occur, which would severely deplete stem cell pools. This might lead to early spontaneous miscarriage if insufficient placental hormone is produced to sustain the corpus luteum, or sufficient placental hormone might be produced to sustain the corpus luteum, but insufficient TSCs may be left to increase CSH1 production or to mediate later essential placental functions. Two recent reviews suggested that preeclampsia and intrauterine growth retardation begin with deficiencies in decision making in peri-implantation and early first trimester placental stem cells (Huppertz 2008, Roberts & Hubel 2009). In humans, first trimester conceptus loss has been associated with chemical pregnancies that produce sufficient human chorionic gonadotropin (hCG) early on, but where the later hCG production attenuates prior to spontaneous abortion late in the first and early in the second trimester (Johnson et al. 1993). Another possibility is that sublethal stress may have more subtle effects such as changing the potency of the stem cells so that they are misprogrammed to interpret later developmental cues. Recently, it was shown that Map3k4−/− mutant TSCs are much more invasive than wild-type TSCs (Abell et al. 2009). These mutant, cultured TSCs also have enhanced differentiation to GCM1-positive lineages and decreased TGC. These data suggest that MAP3K4-dependent MAPK8/9 or MAPK11/14 lead to an imbalanced differentiation that may potentially be pathogenic in vivo. Abell et al. performed their studies with TSCs cultured in FGF4 and 20% oxygen, a combination that activates substantial MAPK8/9 (Y Xie & S Zhou, unpublished observations); however, MAPK8/9 mediates suppression of GCM1 and enhances HAND1 at both 20 and 2% oxygen. A substantial goal of future research should test whether the fundamental peri-implantation stress responses characterized in this study can contribute to diseases of placental insufficiency in the mouse model.
Conclusions
The work presented in this study suggests that hyperosmolar cellular stress, as well as benzopyrene stress, induces ID2 protein loss in a proteasome- and PRKAA1/2-dependent manner. The induction of PRKAA1/2 at low stress levels, which mediates dependent ACACA metabolic regulation and the PRKAA1/2-dependent induction of ID2 protein loss at higher doses, suggests a two-stage regulation of TSCs by PRKAA1/2. At low stress a cellular adaptation and survival strategy is induced, but at high stress, where cell accumulation is significantly reduced, an organismal survival is induced that requires that stem cells differentiate to produce essential developmental proteins and programs. The studies using benzopyrene as a stressor and AICAR as an AMPK inducer suggest caution in interpreting these results. The quality of the stress is important and leads to different qualities of low-dose homeostatic responses and high-dose developmental responses.
Materials and Methods
Embryo and stem cell media were obtained as described earlier (Xie et al. 2007, Zhong et al. 2007). The primary antibodies for total ID2 and HAND1 were from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and Chemicon (Temecula, CA, USA), antibodies for phosphorylated PRKAA1/2 Thr172, and the metabolic substrate of PRKAA1/2, phosphorylated ACACA Ser79, were from Cell Signaling (Danvers, MA, USA) and antibodies for CSH1 were from Chemicon. An additional ID2 antibody was purchased from Zymed (South San Francisco, CA, USA). PRKAA1/2 inhibitors Compound C (Zhou et al. 2001) and AraA (Musi et al. 2001) and PRKAA1/2 agonist AICAR (Sullivan et al. 1994) were purchased from Calbiochem (San Diego, CA, USA). MAPK11/14 (p38MAPK) inhibitors were 505 126 purchased from Calbiochem and SB203580, SB220025, and the inactive MAPK11/14 inhibitor SB202474 purchased from Alexis (Enzo Life Sciences, Plymouth Meeting, PA, USA). Proteasome inhibitors MG132 and lactacystin were purchased from Calbiochem and Alexis respectively. Germ line competent E3-D3 ESCs were purchased from American Type Tissue Collection (Rockville, MD, USA), and cultured with embryo-tested fetal bovine serum (FBS) purchased from Hyclone (Logan, UT, USA) and leukemia inhibitory factor (LIF), which was purchased from Millipore (Billerica, MA, USA).
Collection and culture conditions for mouse embryos
Standard techniques were used for obtaining mouse embryos (Hogan et al. 2002, Xie et al. 2006a). Female MF-1 mice were mated with C57BL/6J×SJL/J F1 hybrid males. Noon of the day following coitus was considered day E0.5. Embryos were obtained at the early blastocyst stage (E3.5). The animal used protocols were approved by the Wayne State University Animal Investigation Committee. Embryos were preloaded with inhibitors for 2 h and then subjected to sorbitol at the doses and durations indicated in the figures, figure legends, and Results section. In a typical experiment, 15 females produced 300 embryos and these were collected from groups of five females killed at a time. Embryos from each group of five females were grouped together and cultured in KSOMaa until all embryos had been collected. Since it requires about 2 h for the embryos to adapt to KSOMaa, we grouped all the embryos collected from all females for 2 h after the last group of five females. We then stimulated and randomized groups of 100 embryos for each time point or inhibitor test. Inhibitors had been optimized for dose previously (Xie et al. 2006a, 2006b, 2007, 2010, Zhong et al. 2007).
Cell lines and culture conditions
SV40/large T-transformed human trophoblast cell line and mouse TSC were obtained and cultured as described earlier (Graham et al. 1993, Tanaka et al. 1998). ES-D3 ESC (Doetschman et al. 1985) were cultured with 10% FBS and 1000 U/ml LIF as previously described (Robertson 1987).
Indirect immunocytochemistry
Indirect immunocytochemistry was performed as described previously (Liu et al. 2004, Wang et al. 2004b, Xie et al. 2005). To analyze stress-induced CSH1 gain and ID2 loss, randomly chosen photomicrographs were taken at the microscope to insure that objective analysis of a high sample size was performed. Attempts to use fluorescence-activated cell sorter (FACS) to associate nuclear size with ID2 expression failed when cells processed immunocytochemically fragmented during trypsinization and fractionation by FACS. Photomicrographs were analyzed for intensity, cytolocalization, and lineage localization of proteins using Photodex CompuPic 6.1, and formatted using Adobe Photoshop 6.0 as previously described (Liu et al. 2004, Wang et al. 2004b, Xie et al. 2005). FITC intensity measurement and comparison were performed with SimplePCI DNN software as previously described (Liu et al. 2004, Wang et al. 2004b, Xie et al. 2005). Nuclear sizes of TSC were determined using Image J software (http://rsb.info.nih.gov/ij/), and modified our use in previous reports (Liu et al. 2004, Xie et al. 2007). We analyzed randomly acquired micrographs of TSC in monolayer after culture at 0, 100, and 400 mM sorbitol for 30 h. Three general sizes of nuclei were observed, small (100–999 arbitrary units), medium (1000–1999 arbitrary units), and giant (over 2000 arbitrary units). Small nuclei also had smaller internuclear distances than medium or giant nuclei, but we have used arbitrary units of the nuclear area in the analysis here.
Western blot analysis
Cells were lysed, protein was determined using BCA assay, and cell lysates were fractionated by electrophoresis, transferred to nitrocellulose membranes, and developed as previously described (Xie et al. 2005).
Statistical analysis
The data in this study were representative of three independent biological experiments and indicated as mean±S.D. or S.E.M. as indicated. Statistical significance between different treated samples was calculated as done earlier using one-way ANOVA (SPSS 13.0; SPSS Inc., Chicago, IL, USA), followed by post hoc Duncan tests where applicable (Xie et al. 2007, Zhong et al. 2007).
Supplementary Material
Acknowledgments
Funding
This research was supported by grants from the National Institute of Child Health and Human Development, NIH (R01 HD40972A (D A Rappolee), 1R03HD061431-01), and NASA (NRA, NAG 2-150309).
We thank Mike Kruger for advice on statistical analysis and Fangfei Wang for excellent technical assistance. We are also indebted to Dr Michael Diamond and Dr Zena Werb for helpful discussion and criticisms of the manuscript.
Footnotes
This is linked to the online version of the paper at http://dx.doi.org/10.1530/REP-10-0268.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Abell AN, Granger DA, Johnson NL, Vincent-Jordan N, Dibble CF, Johnson GL. Trophoblast stem cell maintenance by fibroblast growth factor 4 requires MEKK4 activation of Jun N-terminal kinase. Molecular and Cellular Biology. 2009;29:2748–2761. doi: 10.1128/MCB.01391-08. 10.1128/MCB.01391-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CE, Lariviere NM, Watson PH, Watson AJ. Mitogen-activated protein kinase (MAPK) pathways mediate embryonic responses to culture medium osmolarity by regulating Aquaporin 3 and 9 expression and localization, as well as embryonic apoptosis. Human Reproduction. 2009;24:1373–1386. doi: 10.1093/humrep/dep010. 10.1093/humrep/dep010. [DOI] [PubMed] [Google Scholar]
- Caenepeel S, Charydczak G, Sudarsanam S, Hunter T, Manning G. The mouse kinome: discovery and comparative genomics of all mouse protein kinases. PNAS. 2004;101:11707–11712. doi: 10.1073/pnas.0306880101. 10.1073/pnas.0306880101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai N, Patel Y, Jacobson K, McMahon J, McMahon A, Rappolee DA. FGF is an essential regulator of the fifth cell division in preimplantation mouse embryos. Developmental Biology. 1998;198:105–115. doi: 10.1006/dbio.1997.8858. 10.1016/S0012-1606(98)80031-6. [DOI] [PubMed] [Google Scholar]
- Chen J, Downs SM. AMP-activated protein kinase is involved in hormone-induced mouse oocyte meiotic maturation in vitro. Developmental Biology. 2008;313:47–57. doi: 10.1016/j.ydbio.2007.09.043. 10.1016/j.ydbio.2007.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosi P, Talamantes F, Linzer DI. Molecular cloning and expression of mouse placental lactogen I complementary deoxyribonucleic acid. Molecular Endocrinology. 1987;1:767–776. doi: 10.1210/mend-1-11-767. 10.1210/mend-1-11-767. [DOI] [PubMed] [Google Scholar]
- Colosi P, Ogren L, Southard JN, Thordarson G, Linzer DI, Talamantes F. Biological, immunological, and binding properties of recombinant mouse placental lactogen-I. Endocrinology. 1988;123:2662–2667. doi: 10.1210/endo-123-6-2662. 10.1210/endo-123-6-2662. [DOI] [PubMed] [Google Scholar]
- Cross JC, Werb Z, Fisher SJ. Implantation and the placenta: key pieces of the development puzzle. Science. 1994;266:1508–1518. doi: 10.1126/science.7985020. 10.1126/science.7985020. [DOI] [PubMed] [Google Scholar]
- Cross JC, Flannery ML, Blanar MA, Steingrimsson E, Jenkins NA, Copeland NG, Rutter WJ, Werb Z. Hxt encodes a basic helix-loop-helix transcription factor that regulates trophoblast cell development. Development. 1995;121:2513–2523. doi: 10.1242/dev.121.8.2513. [DOI] [PubMed] [Google Scholar]
- Cross JC, Anson-Cartwright L, Scott IC. Transcription factors underlying the development and endocrine functions of the placenta. Recent Progress in Hormone Research. 2002;57:221–234. doi: 10.1210/rp.57.1.221. 10.1210/rp.57. 1.221. [DOI] [PubMed] [Google Scholar]
- Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R. The in vitro development of blastocyst-derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. Journal of Embryology and Experimental Morphology. 1985;87:27–45. [PubMed] [Google Scholar]
- Graham CH, Hawley TS, Hawley RG, MacDougall JR, Kerbel RS, Khoo N, Lala PK. Establishment and characterization of first trimester human trophoblast cells with extended lifespan. Experimental Cell Research. 1993;206:204–211. doi: 10.1006/excr.1993.1139. 10.1006/excr.1993.1139. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. BioEssays. 2001;23:1112–1119. doi: 10.1002/bies.10009. 10.1002/bies.10009. [DOI] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Constantini F, Lacy B. Manipulating the Mouse Embryo: A Laboratory Manual, Appendix A. Cold Spring Harbor: Cold Spring Harbor Laboratory; 2002. [Google Scholar]
- Huppertz B. Placental origins of preeclampsia: challenging the current hypothesis. Hypertension. 2008;51:970–975. doi: 10.1161/HYPERTENSIONAHA.107.107607. 10.1161/HYPERTENSIO-NAHA.107.107607. [DOI] [PubMed] [Google Scholar]
- Janatpour MJ, McMaster MT, Genbacev O, Zhou Y, Dong J, Cross JC, Israel MA, Fisher SJ. Id-2 regulates critical aspects of human cytotrophoblast differentiation, invasion and migration. Development. 2000;127:549–558. doi: 10.1242/dev.127.3.549. [DOI] [PubMed] [Google Scholar]
- Johnson MR, Riddle AF, Grudzinskas JG, Sharma V, Collins WP, Nicolaides KH. The role of trophoblast dysfunction in the aetiology of miscarriage. British Journal of Obstetrics and Gynaecology. 1993;100:353–359. doi: 10.1111/j.1471-0528.1993.tb12979.x. 10.1111/j.1471-0528.1993.tb12979.x. [DOI] [PubMed] [Google Scholar]
- LaRosa C, Downs SM. Stress stimulates AMP-activated protein kinase and meiotic resumption in mouse oocytes. Biology of Reproduction. 2006;74:585–592. doi: 10.1095/biolreprod.105.046524. 10.1095/biolreprod.105.046524. [DOI] [PubMed] [Google Scholar]
- Liu J, Puscheck EE, Wang F, Trostinskaia A, Barisic D, Maniere G, Wygle D, Zhong W, Rings EH, Rappolee DA. Serine–threonine kinases and transcription factors active in signal transduction are detected at high levels of phosphorylation during mitosis in preimplantation embryos and trophoblast stem cells. Reproduction. 2004;128:643–654. doi: 10.1530/rep.1.00264. 10.1530/rep.1.00264. [DOI] [PubMed] [Google Scholar]
- Liu J, Xu W, Sun T, Wang F, Puscheck E, Brigstock D, Wang QT, Davis R, Rappolee DA. Hyperosmolar stress induces global mRNA responses in placental trophoblast stem cells that emulate early post-implantation differentiation. Placenta. 2009;30:66–73. doi: 10.1016/j.placenta.2008.10.009. 10.1016/j.placenta.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MJ, Baumber J, Kass PH, Meyers SA. Osmotic stress induces oxidative cell damage to rhesus macaque spermatozoa. Biology of Reproduction. 2010;82:644–651. doi: 10.1095/biolreprod.109.080507. 10.1095/biolreprod.109.080507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musi N, Hayashi T, Fujii N, Hirshman MF, Witters LA, Goodyear LJ. AMP-activated protein kinase activity and glucose uptake in rat skeletal muscle. American Journal of Physiology. Endocrinology and Metabolism. 2001;280:E677–E684. doi: 10.1152/ajpendo.2001.280.5.E677. [DOI] [PubMed] [Google Scholar]
- Ogren L, Southard JN, Colosi P, Linzer DI, Talamantes F. Mouse placental lactogen-I: RIA and gestational profile in maternal serum. Endocrinology. 1989;125:2253–2257. doi: 10.1210/endo-125-5-2253. 10.1210/endo-125-5-2253. [DOI] [PubMed] [Google Scholar]
- Rappolee DA. Impact of transient stress and stress enzymes on development. Developmental Biology. 2007;304:1–8. doi: 10.1016/j.ydbio.2006.12.032. 10.1016/j.ydbio.2006.12.032. [DOI] [PubMed] [Google Scholar]
- Rappolee DA, Awonuga AO, Puscheck EE, Zhou S, Xie Y. Benzopyrene and experimental stressors cause compensatory differentiation in placental trophoblast stem cells. Systems Biology in Reproductive Medicine. 2010a;56:168–183. doi: 10.3109/19396360903431638. 10.3109/19396360903431638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappolee DA, Xie Y, Zhou S, Awonuga A, Abdallah M, Elshmoury M, Slater JA, Puscheck EE. Interpreting the stress response of early mammalian embryos and their stem cells. International Review of Cell and Molecular Biology. 2010b doi: 10.1016/B978-0-12-386043-9.00002-5. in press. [DOI] [PubMed] [Google Scholar]
- Rassoulzadegan M, Rosen BS, Gillot I, Cuzin F. Phagocytosis reveals a reversible differentiated state early in the development of the mouse embryo. EMBO Journal. 2000;19:3295–3303. doi: 10.1093/emboj/19.13.3295. 10.1093/emboj/19.13.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JM, Hubel CA. The two stage model of preeclampsia: variations on the theme. Placenta. 2009;30 (Supplement A):S32–S37. doi: 10.1016/j.placenta.2008.11.009. 10.1016/j.placenta.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RM, Xie S, Mathialagan N. Maternal recognition of pregnancy. Biology of Reproduction. 1996;54:294–302. doi: 10.1095/biolreprod54.2.294. 10.1095/biolre-prod54.2.294. [DOI] [PubMed] [Google Scholar]
- Robertson EJ. A Practical Approach. ch 3. Oxford, UK: IRL Press; 1987. Teratocarcinomas and Embryonic Stem Cells. [Google Scholar]
- Simmons DG, Fortier AL, Cross JC. Diverse subtypes and developmental origins of trophoblast giant cells in the mouse placenta. Developmental Biology. 2007;304:567–578. doi: 10.1016/j.ydbio.2007.01.009. 10.1016/j.ydbio.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Steeves CL, Baltz JM. Regulation of intracellular glycine as an organic osmolyte in early preimplantation mouse embryos. Journal of Cellular Physiology. 2005;204:273–279. doi: 10.1002/jcp.20284. 10.1002/jcp.20284. [DOI] [PubMed] [Google Scholar]
- Stein SC, Woods A, Jones NA, Davison MD, Carling D. The regulation of AMP-activated protein kinase by phosphorylation. Biochemical Journal. 2000;345:437–443. 10.1042/0264-6021:3450437. [PMC free article] [PubMed] [Google Scholar]
- Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Letters. 1994;353:33–36. doi: 10.1016/0014-5793(94)01006-4. 10.1016/0014-5793(94)01006-4. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Carpino N, Cross JC, Torres M, Parganas E, Ihle JN. SOCS3: an essential regulator of LIF receptor signaling in trophoblast giant cell differentiation. EMBO Journal. 2003;22:372–384. doi: 10.1093/emboj/cdg057. 10.1093/emboj/cdg057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Kunath T, Hadjantonakis AK, Nagy A, Rossant J. Promotion of trophoblast stem cell proliferation by FGF4. Science. 1998;282:2072–2075. doi: 10.1126/science.282.5396.2072. 10.1126/science.282.5396.2072. [DOI] [PubMed] [Google Scholar]
- Tremblay GB, Kunath T, Bergeron D, Lapointe L, Champigny C, Bader JA, Rossant J, Giguere V. Diethylstilbestrol regulates trophoblast stem cell differentiation as a ligand of orphan nuclear receptor ERRβ. Genes and Development. 2001;15:833–838. doi: 10.1101/gad.873401. 10.1101/gad.873401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uy GD, Downs KM, Gardner RL. Inhibition of trophoblast stem cell potential in chorionic ectoderm coincides with occlusion of the ectoplacental cavity in the mouse. Development. 2002;129:3913–3924. doi: 10.1242/dev.129.16.3913. [DOI] [PubMed] [Google Scholar]
- Wang QT, Piotrowska K, Ciemerych MA, Milenkovic L, Scott MP, Davis RW, Zernicka-Goetz M. A genome-wide study of gene activity reveals developmental signaling pathways in the preimplantation mouse embryo. Developmental Cell. 2004a;6:133–144. doi: 10.1016/s1534-5807(03)00404-0. 10.1016/S1534-5807(03)00404-0. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wang F, Sun T, Trostinskaia A, Wygle D, Puscheck E, Rappolee DA. Entire mitogen activated protein kinase (MAPK) pathway is present in preimplantation mouse embryos. Developmental Dynamics. 2004b;231:72–87. doi: 10.1002/dvdy.20114. 10.1002/dvdy.20114. [DOI] [PubMed] [Google Scholar]
- Wiemers DO, Shao LJ, Ain R, Dai G, Soares MJ. The mouse prolactin gene family locus. Endocrinology. 2003;144:313–325. doi: 10.1210/en.2002-220724. 10.1210/en.2002-220724. [DOI] [PubMed] [Google Scholar]
- Xie Y, Sun T, Wang QT, Wang Y, Wang F, Puscheck E, Rappolee DA. Acquisition of essential somatic cell cycle regulatory protein expression and implied activity occurs at the second to third cell division in mouse preimplantation embryos. FEBS Letters. 2005;579:398–408. doi: 10.1016/j.febslet.2004.10.109. 10.1016/j.febslet.2004.10.109. [DOI] [PubMed] [Google Scholar]
- Xie Y, Puscheck EE, Rappolee DA. Effects of SAPK/JNK inhibitors on preimplantation mouse embryo development are influenced greatly by the amount of stress induced by the media. Molecular Human Reproduction. 2006a;12:217–224. doi: 10.1093/molehr/gal021. 10.1093/molehr/gal021. [DOI] [PubMed] [Google Scholar]
- Xie Y, Wang F, Zhong W, Puscheck E, Shen H, Rappolee DA. Shear stress induces preimplantation embryo death that is delayed by the zona pellucida and associated with stress-activated protein kinase-mediated apoptosis. Biology of Reproduction. 2006b;75:45–55. doi: 10.1095/biolreprod.105.049791. 10.1095/biolreprod.105.049791. [DOI] [PubMed] [Google Scholar]
- Xie Y, Zhong W, Wang Y, Trostinskaia A, Wang F, Puscheck EE, Rappolee DA. Using hyperosmolar stress to measure biologic and stress-activated protein kinase responses in preimplantation embryos. Molecular Human Reproduction. 2007;13:473–481. doi: 10.1093/molehr/gam027. 10.1093/molehr/gam027. [DOI] [PubMed] [Google Scholar]
- Xie Y, Liu J, Proteasa S, Proteasa G, Zhong W, Wang Y, Wang F, Puscheck EE, Rappolee DA. Transient stress and stress enzyme responses have practical impacts on parameters of embryo development, from IVF to directed differentiation of stem cells. Molecular Reproduction and Development. 2008;75:689–697. doi: 10.1002/mrd.20787. 10.1002/mrd.20787. [DOI] [PubMed] [Google Scholar]
- Xie Y, Abdallah ME, Awonuga AO, Slater JA, Puscheck EE, Rappolee DA. Benzo(a)pyrene causes PRKAA1/2-dependent ID2 loss in trophoblast stem cells. Molecular Reproduction and Development. 2010;77:533–539. doi: 10.1002/mrd.21178. 10.1002/mrd.21178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Tanaka S, Oda M, Makino T, Ohgane J, Shiota K. Retinoic acid promotes differentiation of trophoblast stem cells to a giant cell fate. Developmental Biology. 2001;235:422–432. doi: 10.1006/dbio.2001.0300. 10.1006/dbio.2001.0300. [DOI] [PubMed] [Google Scholar]
- Zhong W, Xie Y, Wang Y, Lewis J, Trostinskaia A, Wang F, Puscheck EE, Rappolee DA. Use of hyperosmolar stress to measure stress-activated protein kinase activation and function in human HTR cells and mouse trophoblast stem cells. Reproductive Sciences. 2007;14:534–547. doi: 10.1177/1933719107307182. 10.1177/1933719107307182. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. Journal of Clinical Investigation. 2001;108:1167–1174. doi: 10.1172/JCI13505. 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.