

Abstract
Health care has become increasingly information intensive. The advent of genomic data, integrated into patient care, significantly accelerates the complexity and amount of clinical data. Translational research in the present day increasingly embraces new biomedical discovery in this data-intensive world, thus entering the domain of “big data.” The Electronic Medical Records and Genomics consortium has taught us many lessons, while simultaneously advances in commodity computing methods enable the academic community to affordably manage and process big data. Although great promise can emerge from the adoption of big data methods and philosophy, the heterogeneity and complexity of clinical data, in particular, pose additional challenges for big data inferencing and clinical application. However, the ultimate comparability and consistency of heterogeneous clinical information sources can be enhanced by existing and emerging data standards, which promise to bring order to clinical data chaos. Meaningful Use data standards in particular have already simplified the task of identifying clinical phenotyping patterns in electronic health records.
Keywords: clinical data representation, big data, genomics, health information technology standards
The notion of big data currently captures our imagination, although it defies simple characterization. Pundits claim that big data is that class of problems that can barely be solved today, and that could not be solved yesterday; this raises obvious questions of context. More usefully, a recent textbook on the topic asserts that “big data refers to things one can do at a larger scale that cannot be done at a smaller one, to extract new insights or create new forms of value, in ways that change markets, organizations, the relationship between citizens and governments, and more.”1 More disturbingly, that same text goes on to challenge conventional notions of inference and scientific proof, suggesting that “society will need to shed some of its obsession for causality in exchange for simple correlations: not knowing why but only what. This overturns centuries of established practices and challenges our most basic understanding of how to make decisions and comprehend reality.” Although data volume alone may not defy scientific criteria for belief, we assert that in translational research one confronts notions of data complexity that extend beyond those seen in most big data problems. However, the hope to manage such big data complexity can emerge from principles of comparability and consistency, manifest through standard formats and vocabularies.
In this review, we provide some context for big data, outline some relevant experiences from our Electronic Medical Records and Genomics (eMERGE) consortium,2,3 address some general aspects of translational data such as those used within eMERGE, highlight a particularly promising development in computer science (Hadoop),4 and propose a general strategy around comparable and consistent data to mitigate heterogeneity and misclassification.
Astronomy, physics, and the origins of big science
There once was a time when a single person, with what we might consider today a “cheap telescope,” could raise that instrument skyward and shake the foundations of science and philosophy overnight. Arguably, this was the effect of Galileo’s modest observations, with repercussions to the present day. Yet from a data volume perspective, Galileo’s journals—distilled to their computable essence—barely register on any modern scale.
Astronomy is one of the original big science disciplines, requiring sizeable teams, substantial physical resources well beyond “cheap telescopes,” and accumulating vastly more data that can be easily interpreted. The most recent release of the Sloan Digital Sky Survey contains ~1 million fields of 3 million pixels and catalogs more than 1.2 billion objects, including 200 million galaxies, and nearly 2 million detailed spectra about many of these objects.5 Processing these objects manually is clearly not a scalable task.
Correspondingly, the “gold foil” tabletop experiments of Rutherford, demonstrating the nuclear structure of the atom,6 involved a paltry amount of data by modern standards yet transformed our understanding of quantum physics. This contrasts strikingly with recent phenomena surrounding the discovery of the Higgs boson. That effort perhaps epitomizes current models for big data, involving as it did 600 institutions, 10,000 scientists, 800 trillion collisions, and 200 petabytes of data. That is 2 × 1017 bytes of data—bordering on an astronomical number in its own right.
THE eMERGE CONSORTIUM AND BALANCING PHENOTYPING
The large-scale generation of data has accelerated in the genomics world by many orders of magnitude, attributable to advances in chip-based genotyping and next-generation sequencing (NGS) methods. However, the recognition that the rate-limiting step for genotype-to-phenotype associations was resoundingly on the phenotyping side prompted the National Human Genome Research Institute to propose the eMERGE consortium.2,3 Initially a cooperative agreement among five academic medical centers, the eMERGE consortium balanced biobanking and genotyping with the development of a scalable capacity to execute “high-throughput phenotyping” of patient cohorts using electronic medical records.7 To ensure reproducibility and portability of these algorithms across medical centers, the consortium embraced their mapping to health information technology (HIT) standards,8 conformant with Meaningful Use specifications.9 As a consequence, the consortium was able to demonstrate the reproducible identification of disease-specific research cohorts using portable algorithms across medical centers.10
On the genomics side, the eMERGE effort encountered challenges in merging genotyping data from multiple organizations, particularly into a single record11 for the database of Genotypes and Phenotypes (dbGaP).12 These problems included temporal and platform differences, manifest in surprising and under-documented heterogeneity in genotyping files. That heterogeneity included format differences (single sample versus PLINK files), strand orientation issues, sample and maker checks, and batch effects. Although recognizing and accommodating these differences were possible, such corrections required substantial quality control efforts and data normalization. As Zuvich and colleagues11 amply outline, the consequences of incorrect data analyses and interpretation are predictable unless great care and substantial quality control are invested in creating “big data” aggregations.
In both the clinical and the genomics domains, eMERGE epitomizes what is becoming a typical translational research framework in 2013, invoking big data principles. Furthermore, eMERGE has demonstrated the importance of data normalization and harmonization in both domains as prerequisites to valid data inferencing across aggregated data sources.
eMERGE is also spearheading the integration of genomic data into clinical practice,13 in partnership with the Pharmacogenomics Research Network.14 Among the largest challenges confronted by both research consortia is the absence of reliable and robust nomenclatures for genomic findings, particularly in NGS data.15 It is perhaps in the distillation of large genomic data sources to practical application in patient care16 that the eMERGE consortium has put forth its most important demonstration.
THE NATURE OF TRANSLATIONAL DATA
Phenotype data
Nigam Shah from Stanford has outlined an interesting perspective on big data,17 pointing out that data can be shallow (few rows) but very broad (many columns), deep (the opposite), or both. Harkening back to the Higgs boson, those data are unquestionably “big” (order 1017); however, they comprise mostly parameters about mass, direction, and energy. A more extreme case might be financial data, whose most critical parameter is the decimal point. It readily becomes apparent that data that are both broad and deep pose a third dimension of complexity or texture to already large data sets, and that granularity of that texture can in itself constitute big data. These correspond to the well-accepted notions of volume (breadth and/ or depth) and variety (what we are calling texture) recognized as the hallmarks of big data by the computer science community,18 although this community adds their third dimension of velocity (speed of acquisition).
Clinical phenotyping data are big data of our third kind—intrinsically complex, fraught with heterogeneity, and amply having the potential for enormous depth (many records). A single patient may have many thousands of unique attributes, each of which may have arbitrarily repeated measures. Most eMERGE clinical sites host millions of such records; aggregated across the United States, there are hundreds of millions of electronic health records (EHRs). Although larger data sets clearly exist, they rarely exhibit the complexity or texture of clinical data. Indeed, characterizing the clinical characteristics of patients is so fraught with complexity, the National Institutes of Health/National Heart, Lung, and Blood Institute released a grant opportunity last year19 whose sole purpose was to overlay some order to the cacophony of phenotype descriptions amassed in a relatively small database (dbGaP).12
A recent JAMA viewpoint article suggested that the routine application of big data to health care was inevitable.20 Specifically, those inevitable opportunities are: (i) capacity to generate new knowledge, (ii) knowledge dissemination, (iii) translation of personalized medicine into clinical practice, and (iv) transformation of health care by empowering patients through direct information and knowledge delivery. In any event, the growing availability of big data, and our accelerating capacity to intelligently leverage it, will clearly transform what most now acknowledge as the most information-intensive human discipline—health care.
Data sources
Clinical genetic/genomic, public health, and direct-to-consumer testing laboratories
Big data sources include genetic/genomic data collected during the course of patient care, public health reporting, and research. The most complex is patient care data and is represented in several forms ranging from free-text narratives, synoptic reporting (templated text reports), structured findings (e.g., Logical Observation Identifiers Names and Codes (LOINC) qualified findings and metadata), to coded data. Guidelines for molecular genetic reporting provide a general format for narrative molecular reports;21–23 however, reporting checklists (e.g., College of American Pathologists checklists) provide a more robust standard for synoptic reporting.24 The HL7 Clinical Genomics Workgroup has extended established laboratory reporting standards, commonly used for other clinical laboratory tests, to inclusion of structured genetic findings, references, and interpretations, as well as the narrative report.25,26 In the very early stages of adoption, as reporting systems advance more quickly than receiving systems, a common first step is inclusion of these vocabulary guidelines in molecular reports.
Data translated from the EHR into public health reporting systems (e.g., BioSense, ref. 27; notifiable diseases, ref. 28) aggregate into big data on a population scale. Increasingly, these data are highly structured and amenable to algorithmic normalization,29 although in some cases (such as cancer registries) they may be manually curated by registrars trained in reviewing the patient chart and summarizing findings and outcomes from the EHR.30,31
There remain several important gaps within clinical genomics standards for health care, including (i) coding of important cancer biomarkers and association with causal DNA variants and (ii) standards for transmission of genomics data within the health-care environment.15 As the field moves to using high-throughput NGS to perform genetic-based clinical tests, robust concept mapping from observed DNA sequence variation to reported biomarkers is critical. Proactive work in this area will further translational research and improve clinical and public health reporting. In addition, standardization of genomic data representation was identified as a gap by the Centers for Disease Control and Prevention-sponsored Next Generation Sequencing: Standardization of Clinical Testing Working Group.32 In follow-up, a federally mediated workgroup including representatives from the Centers for Disease Control and Prevention, the National Center for Biotechnology Information (NCBI), the National Institute of Standards and Technology, and the Food and Drug Administration, as well as experts from the clinical laboratory, bioinformatics, and health-care IT standards community, has been formed to develop a clinical grade VCF/gVCF (variant call format)1 file format in preparation for broad adoption of NGS. In conjunction with this effort, the HL7 Clinical Genomics Workgroup is developing an implementation guide for the transmission of VCF/gVCF within standard health-care IT messages. This work should be completed by the end of 2013.
The present trend toward consumer-driven health care highlights the importance of comparable and consistent clinical genomics standards, such as VCF/gVCF, in the direct-to-consumer (DTC) genetic testing marketplace. The rapidly falling cost of gene sequencing will result in large amounts of patient-controlled genomic data from disparate DTC sources that patients will demand be integrated into their EHRs. Recently, clinical providers could point to the absence of Clinical Laboratory Improvement Amendments certification, which provides quality control standards for sample handling, laboratory process, and analytic methods, in these DTC genomic labs; however, as more DTC companies generate Clinical Laboratory Improvement Amendments-certified results, resistance from the medical world will fade, and clinical import and use of DTC data will become more mainstream. Nevertheless, the Clinical Laboratory Improvement Amendments do not address the problem of data comparability or consistency from a representation or format perspective. Data from DTC sources will come in the range of approximately 1 million variants from companies such as 23andme.com; to efforts such as openSNP. org, which claim they are “crowdsourcing genome-wide association studies;” to the Personal Genome Project, which gathers “volunteers who are willing to share their genome sequence… with the research community and the general public.”33 The reuse of the DTC data for clinical practice or research will be more practical if those data are distributed in predicable, well-documented, and ideally standardized machinable formats.
BIG DATA IN NGS STUDIES
Personalized medicine intends to optimize clinical decisions about a patient’s care by using all available data, including genomic data.34 With the use of genomic data efficiently generated by NGS in clinical practice, a key challenge is how to manage and retrieve the high-volume genetic data appropriately.
Whole genome/exome sequencing: a source of big data
NGS makes it possible to rapidly compare the genetic content among samples and identify germline and somatic variants of interest, such as single-nucleotide variants, short insertions and deletions, copy-number variants, and other structural variations. NGS technologies can quickly generate the sequence of a whole genome or can be more targeted using an approach called exome sequencing. Exome sequencing focuses specifically on generating reads from known coding regions. In contrast to whole-genome sequencing, exome sequencing is a more cost-effective approach that can detect single-nucleotide variants or short insertion and deletion variants in coding regions and provide sufficient information for many research needs.
Because NGS generates high-resolution genomic data35,36 much more efficiently, researchers in many fields have turned to NGS to identify particular features of the genome that contribute to specific phenotypes. With the declining cost of sequencing and the ongoing discovery of disease genes, NGS is making its way into clinical laboratories. Although there is some use in infectious disease testing, most applications have been in diagnostic testing for hereditary disorders and, more recently, therapeutic decision making for somatic cancers.37 The use of NGS technologies to move from testing single genes or small panels of genes to large multigene disease-targeted panels38 is a logical first step for the clinical application of these technologies. This approach allows geneticists to increase clinical sensitivity for many existing tests and to continue to investigate the substantial contribution of unique and rare variants to understanding these diseases, which can be assayed only through sequencing.
HADOOP AND ITS MODULES: A COMPUTATIONAL FRAMEWORK FOR BIG DATA
Rarely, computing encounters a fundamental shitf in the way it conceptualizes problems and approaches. This arguably occurred with the advent of compiled languages, object-oriented constructs, relational databases, and network computing. The advent of big data has spawned what many regard as a notable, fundamental shift in computing, generally classified under “share nothing architectures”39 and popularized by companies such as Tandem and Teradata. Quite simply, this architectural design supports nearly infinite scaling because the independent and self-sufficient component parts of the system “share nothing” with the others, such as file systems or memory.
Apache Hadoop4 is an open-source framework that most dramatically extends “share nothing” designs, thus allowing for the distributed processing of large data sets across clusters of computers using simple programming models. Hadoop owes much of its heritage to proprietary architectures developed by Google to accommodate and index humankind’s knowledge. Google published the underlying principles of these efforts, called MapReduce,40 which hierarchically breaks tasks into smaller, independently achievable components, and the Google File System,41 which permits redundant, fault-tolerant computing and storage over commodity computing. Hadoop leverages these by creating its own distributed database architecture, HBase (derived from descriptions of the Google File System), and implementing MapReduce. Therefore, like Google, Hadoop is designed to scale up from a single server to thousands of machines, each offering local computation and storage. Furthermore, it is highly fault tolerant because it uses redundant storage and coordinated task scheduling; thus, it can accommodate very-large-scale systems from inexpensive, commodity hardware rather than custom supercomputers or even enterprise class computer hardware.
The key innovation of Hadoop is twofold: (i) it brings computing to the data through task assignment, rather than bringing data to an analytic environment; and (ii) it creates analytic schema from raw data at read time through the MapReduce40 model, rather than requiring traditional “extract–transform–load” preprocessing against a fixed and typically lossy data transform.
Between open-source and commodity hardware, Hadoop has fundamentally changed the economics of big data processing and is widely deployed in the biosciences and genom-ics. Consistent with other models of distributed computing, Hadoop also enjoys orders of magnitude improvement in computing time, enabling problems that would ordinarily take days or weeks to complete in seconds or minutes. Therefore, Hadoop has become the dominant big data solution for NGS analysis. NGS is generating high-volume data and requiring computationally extensive analysis; that analysis would be prohibitively expensive without Hadoop and its family of extensions.
Growth of Hadoop and its applications
As Hadoop grows more stable with new releases, researchers are becoming more comfortable using it (Figure 1). New related software extends Hadoop’s applicability to other areas of bioinformatics. For example, Mahout42 (a Hadoop machine learning library) can be used for classification (the automatic labeling of data) and clustering (forming groups of similar data within a larger data set), both useful in bioinformatics. The Hadoop and MapReduce approach is also being explored for automated reasoning and rule engines, which have tremendous potential. For instance, IBM’s Watson on Jeopardy has already used Hadoop to preprocess large unstructured data sets for automated reasoning.
Figure 1. The number of papers in PubMed on big data topics relevant to translational research.
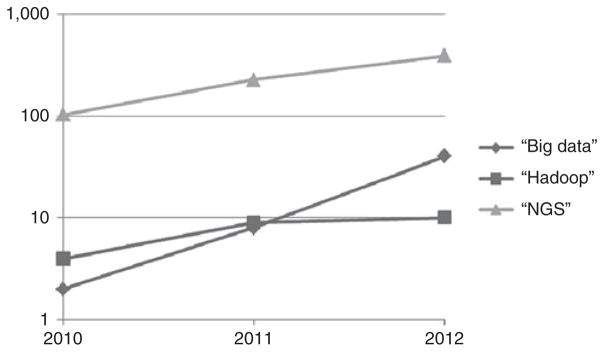
NGS, next-generations equencing.
The community around Hadoop is developing and increasing researchers’ confidence. The wealth of related software and growing availability of support make Hadoop the open-source solution of choice, and new related projects are on the way. Hadoop is already a key to delivering on the promise of bioinformatics. Because of its ability to store and process complex data of almost any kind, Hadoop provides a platform that makes it easier to integrate and analyze not just nucleotide sequences but also other clinical data. Combining diverse scientific data on Hadoop provides a huge opportunity for new approaches to understanding molecular function in gene activation and disease pathways.
Hadoop applications in NGS
In the world of NGS, there are a few Hadoop-based frameworks available, such as Cloudburst43 and Crossbow,44 that leverage Hadoop to perform “read mapping” (approximate string matching for taking a DNA sequence from the sequencer and figuring out where in a known genome it came from). Myrna34 and Eoulsan38 do the same but also extend the workflow to quantifying gene expression and identifying differentially expressed genes based on the sequences. Contrail45 does Hadoop-based de novo assembly. These are essentially MapReduce implementations of existing software. Contrail offers an opportunity for research groups that do not have access to random access memory–rich computers to perform assembly on commodity clusters.
In addition, there are the projects that attempt to build a Hadoop infrastructure for NGS. These include Seal,46 which provides “map-reducification” for a number of common NGS operations; Hadoop-BAM,47 which is a library for processing BAM files based on Hadoop, a common sequence alignment format; and SeqPig,48 which is a library with import and export functions to allow common genomic sequence formats to be used in a high-level programming environment for MapReduce called Pig.49 Some of us (M.H. and S.M.L.) are also developing a reliable association analysis toolset, which is built on the Hadoop and HBase framework, to analyze genetic variants in NGS.
COMPARABLE AND CONSISTENT DATA
Standards as a basis for “high-texture” big data analytics
Progress on the road toward integrating big data—both high-volume genomic findings and heterogeneous clinical observations—into practical clinical protocols and standard health-care delivery requires that providers, HIT vendors, federated knowledge resources, and patients can ultimately depend upon those data being comparable and consistent. Absent comparability, the data are more or less by definition not able to support inferencing of any scalable kind, such as automated clinical decision support. Without consistency, users of complex biomedical data will have to spend added resources transforming the data into usable and predictable formats that use interoperable semantics (vocabularies and value sets). There are two viable solutions to address heterogeneous data: (i) defining a “common representation” and transforming all data into that common interlingua, or (ii) adopting standards at the point of data generation to obviate the costs and confusion that often emerge from data transformation. In this section, we give an overview of the status and promise of many clinical data standards used in health care today.
Data standards for clinical genomic information
Below are the current standards for representation of genetic test results, associated metadata, and optional interpretation. Specifications for this vocabulary are currently detailed in the HL7 Version 2 Implementation Guide for Genetic Variation (detailed below).25 However, these vocabularies are in the process of being extended for whole-genome or exome sequencing within the context of a broad set of clinical workflow scenarios, under the “Clinical Sequencing” project.
Reference sequences
To date, genetic data are reported as differences from a reference sequence or assumed “wild-type.” Currently, mutations are required to be defined in the context of a NCBI genomic or transcriptional reference sequence in RefSeq.50 However, many common genetic biomarkers are reported without a reference sequence, further necessitating the coding of somatic biomarkers.
Gene/mutation/variant/biomarker
Gene symbols are expressed as Human Gene Nomenclature Committee symbols, which also catalog historical symbols that may still be in use. Variants are expressed according to Human Genome Variation Society51 nomenclature standards, and cytogenetic variants are represented using the International System for Human Cytogenetic Nomenclature. dbSNP52 and COSMIC53 identifiers may also be included as additional information.
As mentioned, representation of biomarkers remains a gap. In the January 2013 meeting of the HL7 Clinical Genomics Workgroup with stakeholders from the HL7 Image Integration, Anatomic Pathology, and Laboratory workgroups, the work-group approved coding tumor-specific clinical grade biomarkers in LOINC and providing these codes to NCBI for inclusion in several NCBI databases,54 including MedGen,55 for links to corresponding DNA changes in dbSNP,52 dbVAR,56 ClinVar,57 and tests in the genetic test repository.58 A clinical partner (e.g., the College of American Pathologists) still needs to commit to this project, to provide biomarkers. This is a short-term solution, as the NCBI would extend its databases for biomarkers.
Genetic interpretation: clinical reporting
DNA variants with clinical implications “fit” into one or more clinical scenarios: (i) drug metabolism, (ii) drug efficacy, (iii) confirmatory diagnosis, (iv) predictive risk, (v) drug toxicity, (vi) prognosis, (vii) diagnosis or subtyping. Currently, LOINC codes support the reporting of the first four scenarios and extensions for the remainder are in progress. RxNORM59 and SNOMED CT60 are used for the coding of the interpreted drug or disease context.
Coded references to external knowledge include Online Mendelian Inheritance in Man,61 PubMed,62 PharmGKB (Pharmacogenomics Knowledge Base),63 and http://www.ClinicalTrials.gov.64
HL7 Version 2 messaging and the genetic variation model
The genetic variation model specifies the structure and semantics for the transmission of genetic findings from single- or multiple-gene testing using laboratory methods such as single-nucleotide polymorphism probes, genotyping, and sequencing. This may include NGS, if test results are reported for a gene panel.
Because US laboratories use HL7 Version 2 to transmit clinical test results, and this standard is used to implement Meaningful Use requirements, it followed to extend the standard and create a Version 2 Genetic Variation implementation guide for the clinical environment. With the implementation of this data messaging model, genetic test results flow from the genetic testing laboratory into the EHR as structured data.
The implementation guide describes how to construct a data message for genetic test results using many of the standards previously mentioned (LOINC, Human Gene Nomenclature Committee, Human Genome Variation Society, etc.). The guide is titled HL7 Version 2 Implementation Guide: Clinical Genomics; Fully LOINC-Qualified Genetic Variation Model, Release 2 (US Realm).25 Example messages in the guide include genetic disease analysis, pharmacogenomic-based drug metabolism, and drug efficacy. The Release 2 update to the guide includes the ability to transmit large data sets for tumor profiling and sequencing.
HL7 Version 2 messaging and cytogenetics
Closely based on the Genetic Variation guide, a Version 2 cytogenetic implementation guide is available for reporting of structural variants. Currently, cytogenetic data are reported in text-based narrative form and require development of a structured data model around the International System for Human Cytogenetic Nomenclature in order to make the data available for outcomes analysis and other secondary usage. The guide is titled HL7 Version 2 Implementation Guide: Clinical Genomics; Fully LOINC-Qualified Cytogenetics Model, Release 1.65
Genetic test report for clinical document architecture
A clinical document architecture document is both human and computer readable. A clinical document architecture–based genetic test report document specification and implementation guide has been developed. Genetic testing methods are diverse and span testing for known germline mutations, to full sequencing of genes in tumor tissues in a search for somatic variations in cancer cells, to gene expression testing in clinical care. As a consequence of that diversity, the clinical document architecture genetic test report offers report formats on detailed but easy-to-understand interpretations of the test results, along with clinical recommendations, test information, and references.26
Meaningful Use as a framework for electronic medical record phenotyping standards
In counterpoint to genomic standards are those surrounding the representation of clinical observations in electronic formats, such as EHRs. There has been no lack of thoughtful and even useful clinical HIT standards over the past several decades, the most ubiquitous and successful arising from HL7. However, adoption was not uniform, and more significantly, adaptations—while adding greater expressiveness and flexibility—perniciously eroded interoperability (the capability of electronically exchanging health records while ensuring that people and machines, such as decision support engines, could faithfully understand them at the receiving end).
The emergence of Meaningful Use9 3 years ago has dramatically transformed the clinical data landscape in the United States. Although most visibly driving the adoption of EHRs among clinical providers, it has more quietly created the de jure HIT standards for health information exchange between and among providers. These exchange standards have had every EHR vendor in the country focusing almost exclusively on implementing and supporting the clinical standards mandated by Meaningful Use. As recently as 5 years ago, sessions on the topic of data standards would attract small and somewhat irrelevant devotees at major HIT meetings; today standards sessions constitute core plenaries with overflow capacity, framing the basis for strategic conversations with health-care CEOs, providers, and those providing HIT support.
The specifics of Meaningful Use HIT standards do not introduce radically new formats or vocabularies; rather, in phase II they sanction the use of one, and only one, vocabulary or format for clinical information. Examples include the universal requirement to represent drug ordering data using the RxNORM59 standard, rather than proprietary drug knowledge codes, unreliable National Drug Code numbers that would change with the number of pills in a bottle, or the cacophony of trade and generic names used throughout health care. Other endorsed standards include many HL7 structures, SNOMED60 terms for problems lists, and LOINC codes66 for laboratory tests, rather than the perverse fashion for every clinical laboratory to invent its own idiosyncratic codes for things as ordinary as a serum glucose measurement.
The consequence of this sea change is that EHR data is asymptotically approaching the practical goal of comparable and consistent data. Although the granularity and detail that clinicians need and patients expect to make reliable inferences from clinical data remain elusive, the advent of scalable and practical applications of big data in the clinical domain has accelerated and continues as progressive versions of Meaningful Use continue to refine clinical data standard specifications.
DISCUSSION
We have presented the emerging importance of large-scale data in health care and biological studies. We have pointed to our experience in the eMERGE consortium, and also to some transformative developments in computer science leading to affordable methods for big data processing. Nevertheless, we must emphasize the crucial importance of data comparability and consistency.
Halevy, Norvig, and Pereira, in their influential “The Unreasonable Effectiveness of Data,”67 contrast two approaches: elaborately curated rules and schema, or simply trusting to inferencing useful knowledge from more data. Their premise, and in fairness experience, is that more data yield more accurate answers. Although this is arguably true for many machine learning problems, where more training data always seem to help, this may not always be true in biology and medicine. The misclassification problem, if badly distributed, can move controls into the case column, which simultaneously erodes statistical power through a bias toward the null and will also underestimate effect estimates. Indeed, one wonders if a major contributor to the well described but unexpected tendency for genomic associations with disease to be surprisingly weak in the face of clear inheritance patterns68 may be attributable in part to larger misclassification of case/control status than is often suspected. Even Peter Norvig, in many of his presentations, acknowledges that having more data is not always good, particularly if the incremental data are “dirty” or malformed. He uses examples around machine translation where adding Internet-sourced data full of misspellings or nongrammatical language to a well-curated corpus will make the training data bigger but in many cases provably not better.
We propose that more is not always better, if that more is “dirty” in the sense Norvig implies. Specifically, new biomedical inferences will have hugely more power and accuracy if we aim big data methods at information that shares names and values; we do not want to waste analytic resources “discovering” that renal cancer behaves similarly to kidney cancer, when an ontological assertion (such as a standard terminology) used in the beginning could render those terms comparably and consistently.
CONCLUSION
Health care has become a profoundly information-intensive industry. In parallel, the capacity and capability of modern computing infrastructures continues to accelerate nearly beyond any human capacity to fully leverage the opportunities. The juxtaposition of health care and computing can only result in their joint application, which promises to dramatically enhance the effectiveness and efficiency of health care. However, the flood of big data that may power this transformation remains fraught with noncomparable and inconsistent renderings. Although this may be a mere annoyance in many analytic problem spaces, patients and providers cannot be content with “the right answer” appearing somewhere on an information retrieval listing, akin to a Google search. The safest and most promising application of big data in health care will be driven by clinical and genomic data that are generated with or transformed into standards-based representations, to ensure comparability and consistency. There is convergence in both the genomic and the clinical phenotyping worlds, driven by the application of genomics to clinical practice and Meaningful Use. Together, these promise to make good on the promise of higher quality and lower cost health care in an information-driven industry.
Acknowledgments
We are grateful for the grant support in part from the National Human Genome Research Institute as eMERGE consortium members, specifically U01-HG06379 (Mayo Clinic) and U01-HG006389 (Marshfield Clinic).
Footnotes
DISCLOSURE
The authors declare no conflict of interest.
References
- 1.Mayer-Schönberger V, Cukier K. Big Data: A Revolution That Will Transform How We Live, Work, and Think. Houghton Mifflin Harcourt; Boston, MA: 2013. p. 242. [Google Scholar]
- 2.The eMERGE Network. [Accessed 20 January 2010];2010 https://www.mc.vanderbilt.edu/victr/dcc/projects/acc/index.php/Main_Page.
- 3.McCarty CA, Chisholm RL, Chute CG, et al. eMERGE Team. The eMERGE Network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med Genomics. 2011;4:13. doi: 10.1186/1755-8794-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Apache Foundation. [Accessed 9 April 2013];Hadoop. http://hadoop.apache.org/
- 5.Sloan Digital Sky Survey. [Accessed 2 April 2013];The Scope of the Ninth SDSS Data Release (DR9) http://www.sdss3.org/dr9/scope.php.
- 6.Geiger H, Marsden E. On a diffuse reflection of the α-particles. Proc Royal Soc Series A. 1909;82:495–500. [Google Scholar]
- 7.Kullo IJ, Fan J, Pathak J, Savova GK, Ali Z, Chute CG. Leveraging informatics for genetic studies: use of the electronic medical record to enable a genome-wide association study of peripheral arterial disease. J Am Med Inform Assoc. 2010;17:568–574. doi: 10.1136/jamia.2010.004366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pathak J, Wang J, Kashyap S, et al. Mapping clinical phenotype data elements to standardized metadata repositories and controlled terminologies: the eMERGE Network experience. J Am Med Inform Assoc. 2011;18:376–386. doi: 10.1136/amiajnl-2010-000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blumenthal D, Tavenner M. The “meaningful use” regulation for electronic health records. N Engl J Med. 2010;363:501–504. doi: 10.1056/NEJMp1006114. [DOI] [PubMed] [Google Scholar]
- 10.Kho AN, Pacheco JA, Peissig PL, et al. Electronic medical records for genetic research: results of the eMERGE consortium. Sci Transl Med. 2011;3:79re1. doi: 10.1126/scitranslmed.3001807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuvich RL, Armstrong LL, Bielinski SJ, et al. Pitfalls of merging GWAS data: lessons learned in the eMERGE network and quality control procedures to maintain high data quality. Genet Epidemiol. 2011;35:887–898. doi: 10.1002/gepi.20639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.National Center for Biotechnology Information. Database of Genotypes and Phenotypes (dbGaP) 2013 http://www.ncbi.nlm.nih.gov/gap.
- 13.Manolio TA, Chisholm RL, Ozenberger B, et al. Implementing genomic medicine in the clinic: the future is here. Genet Med. 2013;15:258–267. doi: 10.1038/gim.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.NIH Pharmacogenomics Research Network. [Accessed April 2012]; http://www.nigms.nih.gov/Research/FeaturedPrograms/PGRN.
- 15.Chute CG, Kohane IS. Genomic medicine, health information technology, and patient care. JAMA. 2013;309:1467–1468. doi: 10.1001/jama.2013.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Starren J, Williams MS, Bottinger EP. Crossing the omic chasm: a time for omic ancillary systems. JAMA. 2013;309:1237–1238. doi: 10.1001/jama.2013.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shah NH. Translational bioinformatics embraces big data. Yearb Med Inform. 2012;7:130–134. [PMC free article] [PubMed] [Google Scholar]
- 18.Computing Community Consortium. Challenges and Opportunities with Big Data. 2012 http://www.cra.org/ccc/files/docs/init/bigdatawhitepaper.pdf.
- 19.NIH National Heart Lung and Blood Institute. [Accessed 9 April 2013];PFINDR: Phenotype Finder IN Data Resources: A tool to support cross-study data discovery among NHLBI genomic studies RFA-HL-11–020. 2011 http://grants.nih.gov/grants/guide/rfa-files/RFA-HL-11-020.html.
- 20.Murdoch TB, Detsky AS. The inevitable application of big data to health care. JAMA. 2013;309:1351–1352. doi: 10.1001/jama.2013.393. [DOI] [PubMed] [Google Scholar]
- 21.Gulley ML, Braziel RM, Halling KC, et al. Molecular Pathology Resource Committee, College of American Pathologists. Clinical laboratory reports in molecular pathology. Arch Pathol Lab Med. 2007;131:852–863. doi: 10.5858/2007-131-852-CLRIMP. [DOI] [PubMed] [Google Scholar]
- 22.Lubin IM, Caggana M, Constantin C, et al. Ordering molecular genetic tests and reporting results: practices in laboratory and clinical settings. J Mol Diagn. 2008;10:459–468. doi: 10.2353/jmoldx.2008.080050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheuner MT, Hilborne L, Brown J, Lubin IM members of the RAND Molecular Genetic Test Report Advisory Board. A report template for molecular genetic tests designed to improve communication between the clinician and laboratory. Genet Test Mol Biomarkers. 2012;16:761–769. doi: 10.1089/gtmb.2011.0328. [DOI] [PubMed] [Google Scholar]
- 24.Baskovich BW, Allan RW. Web-based synoptic reporting for cancer checklists. J Pathol Inform. 2011;2:16. doi: 10.4103/2153-3539.78039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fully LOINC-Qualified Genetic Variation Model, Release 2, 2013. Health Level Seven International; Ann Arbor, MI: HL7 Version 2 Implementation Guide: Clinical Genomics. [Google Scholar]
- 26.HL7 Implementation Guide for CDA ® Release 2: Genetic Testing Report (GTR), DSTU Release 1, 2013. Health Level Seven International; Ann Arbor, MI: [Google Scholar]
- 27.Bradley CA, Rolka H, Walker D, Loonsk J. BioSense: implementation of a National Early Event Detection and Situational Awareness System. MMWR Morb Mortal Wkly Rep. 2005;54(suppl):11–19. [PubMed] [Google Scholar]
- 28.Gichoya J, Gamache RE, Vreeman DJ, Dixon BE, Finnell JT, Grannis S. An evaluation of the rates of repeat notifiable disease reporting and patient crossover using a health information exchange-based automated electronic laboratory reporting system. AMIA Annu Symp Proc. 2012;2012:1229–1236. [PMC free article] [PubMed] [Google Scholar]
- 29.Rea S, Pathak J, Savova G, et al. Building a robust, scalable and standards-driven infrastructure for secondary use of EHR data: the SHARPn project. J Biomed Inform. 2012;45:763–771. doi: 10.1016/j.jbi.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.North American Association of Central Cancer Registries Inc. Standards for Cancer Registries Volume V: Pathology Laboratory Electronic Reporting. 2011:310. http://www.naaccr.org/LinkClick.aspx?fileticket=Po1eQNqGQF8%3D&tabid=136&mid=476.
- 31.Houser SH, Colquitt S, Clements K, Hart-Hester S. The impact of electronic health record usage on cancer registry systems in Alabama. Perspect Health Inf Manag. 2012;9:1f. [PMC free article] [PubMed] [Google Scholar]
- 32. [Accessed 13 April 2013];Next Generation Sequencing: Standardization of Clinical Testing (Nex-StoCT) Working Groups. http://www.cdc.gov/osels/lspppo/Genetic_Testing_Quality_Practices/Nex-StoCT.html.
- 33.Pandey A. A piece of my mind. Preparing for the 21st-century patient. JAMA. 2013;309:1471–1472. doi: 10.1001/jama.2012.116971. [DOI] [PubMed] [Google Scholar]
- 34.Langmead B, Hansen KD, Leek JT. Cloud-scale RNA-sequencing differential expression analysis with Myrna. Genome Biol. 2010;11:R83. doi: 10.1186/gb-2010-11-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abecasis GR, Altshuler D, Auton A, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clarke L, Zheng-Bradley X, Smith R, et al. 1000 Genomes Project Consortium. The 1000 Genomes Project: data management and community access. Nat Methods. 2012;9:459–462. doi: 10.1038/nmeth.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rehm HL. Disease-targeted sequencing: a cornerstone in the clinic. Nat Rev Genet. 2013;14:295–300. doi: 10.1038/nrg3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jourdren L, Bernard M, Dillies MA, Le Crom S. Eoulsan: a cloud computing-based framework facilitating high throughput sequencing analyses. Bioinformatics. 2012;28:1542–1543. doi: 10.1093/bioinformatics/bts165. [DOI] [PubMed] [Google Scholar]
- 39.Stonebraker M. The case for shared nothing. Database Eng. 1986;9:4–9. [Google Scholar]
- 40.Dean J, Ghemawat S. MapReduce: Simplified Data Processing on Large Clusters in OSDI’04. Sixth Symposium on Operating System Design and Implementation 2004; San Francisco, CA, USA. [Google Scholar]
- 41.Ghemawat S, Gobioff H, Leung S-T. The Google File System. 19th Symposium on Operating Systems Principles: SOSP’032003; Lake George, Bolton Landing, NY, USA. [Google Scholar]
- 42.Apache Foundation. [Accessed 9 Apr 2013];Mahout. http://mahout.apache.org/
- 43.Schatz MC. CloudBurst: highly sensitive read mapping with MapReduce. Bioinformatics. 2009;25:1363–1369. doi: 10.1093/bioinformatics/btp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langmead B, Schatz MC, Lin J, Pop M, Salzberg SL. Searching for SNPs with cloud computing. Genome Biol. 2009;10:R134. doi: 10.1186/gb-2009-10-11-r134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schatz M, Chambers J, Gupta A, et al. Contrail: Assembly of Large Genomes using Cloud Computing. http://sourceforge.net/apps/mediawiki/contrail-bio/index.php?title=Contrail.
- 46.CRS4 - Center for Advanced Studies Research and Development: Sardinia. [Accessed 9 April 2013];Seal. http://biodoop-seal.sourceforge.net/index.html.
- 47.Niemenmaa M, Kallio A, Schumacher A, Klemelä P, Korpelainen E, Heljanko K. Hadoop-BAM: directly manipulating next generation sequencing data in the cloud. Bioinformatics. 2012;28:876–877. doi: 10.1093/bioinformatics/bts054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.SeqPig. [Accessed 9 April 2013]; http://sourceforge.net/projects/seqpig/
- 49.Apache Foundation. [Accessed 19 June 2013];Pig. http://pig.apache.org/
- 50.NCBI Reference Sequence Database (RefSeq) http://www.ncbi.nlm.nih.gov/refseq/
- 51.Nomenclature for the description of sequence variants. http://www.hgvs.org/mutnomen/
- 52.Database of Single Nucleotide Polymorphisms (dbSNP) http://www.ncbi.nlm.nih.gov/projects/SNP/
- 53.Catalogue of Somatic Mutations in Cancer (COSMIC) http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/
- 54.CG Working Group Meeting Minutes. http://wiki.hl7.org/index.php?title=CG_Working_Group_Meeting_Minutes.
- 55.MedGen: Human Medical Genetics database. http://www.ncbi.nlm.nih.gov/medgen.
- 56.dbVAR: database of genomic structural variation. http://www.ncbi.nlm.nih.gov/dbvar/
- 57.ClinVar: Clinical Variant database. http://www.ncbi.nlm.nih.gov/clinvar/
- 58.NCBI’s Genetic Test Repository (GTR) http://www.ncbi.nlm.nih.gov/gtr/
- 59.RxNORM: Normalized names for clinical drugs. doi: 10.1136/amiajnl-2011-000116. http://www.nlm.nih.gov/research/umls/rxnorm/ [DOI] [PMC free article] [PubMed]
- 60.SNOMED Clinical Terms® (SNOMED CT®) http://www.nlm.nih.gov/research/umls/Snomed/snomed_main.html.
- 61.Online Mendelian Inheritance in Man (OMIM) http://www.ncbi.nlm.nih.gov/omim.
- 62.PubMed. http://www.ncbi.nlm.nih.gov/pubmed.
- 63.The Pharmacogenomics Knowledge Base (PharmGKB) http://www.pharmgkb.org/
- 64.ClinicalTrials.gov. http://clinicaltrials.gov/
- 65.Fully LOINC-Qualified Cytogenetics Model, Release 1. Health Level Seven International; Ann Arbor, MI: HL7 Version 2 Implementation Guide: Clinical Genomics. [Google Scholar]
- 66.Logical Observation Identifiers Names and Codes (LOINC®) [Accessed 15 April 2013]; http://loinc.org/
- 67.Halevy A, Norvig P, Pereira F. The unreasonable effectiveness of data. IEEE Intelligent Syst. 2009;24(2):8–12. [Google Scholar]
- 68.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]